
Learning Parsimonious Classification Rules from Gene Expression Data Using Bayesian Networks with Local Structure


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bayesian Rule Learning
2.1.1. Bayesian Rule Learning-Global Structure Search (BRL-GSS)
2.1.2. Bayesian Rule Learning-Local Structure Search (BRL-LSS)
| Algorithm 1: Bayesian Local Structure Search |
 |
2.2. Experimental Design
2.2.1. Classification Algorithms
2.2.2. Dataset
2.2.3. Evaluation
3. Results and Discussion
3.1. Case Study
4. Future Work
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bigbee, W.L.; Gopalakrishnan, V.; Weissfeld, J.L.; Wilson, D.O.; Dacic, S.; Lokshin, A.E.; Siegfried, J.M. A multiplexed serum biomarker immunoassay panel discriminates clinical lung cancer patients from high-risk individuals found to be cancer-free by CT screening. J. Thorac. Oncol. 2012, 7, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Ganchev, P.; Malehorn, D.; Bigbee, W.L.; Gopalakrishnan, V. Transfer learning of classification rules for biomarker discovery and verification from molecular profiling studies. J. Biomed. Inform. 2011, 44, S17–S23. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Ganchev, P.; Ranganathan, S.; Bowser, R. Rule Learning for Disease-Specific Biomarker Discovery from Clinical Proteomic Mass Spectra. In International Workshop on Data Mining for Biomedical Applications; Springer: Berlin/Heidelberg, Germay, 2006; pp. 93–105. [Google Scholar]
- Ranganathan, S.; Williams, E.; Ganchev, P.; Gopalakrishnan, V.; Lacomis, D.; Urbinelli, L.; Newhall, K.; Cudkowicz, M.E.; Brown, R.H.; Bowser, R. Proteomic profiling of cerebrospinal fluid identifies biomarkers for amyotrophic lateral sclerosis. J. Neurochem. 2005, 95, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, H.; An, J.; Darko, S.; Lustgarten, J.L.; Jaffa, M.; Gopalakrishnan, V.; Lacomis, D.; Cudkowicz, M.; Bowser, R. Discovery and verification of amyotrophic lateral sclerosis biomarkers by proteomics. Muscle Nerve 2010, 42, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Williams, E.; Ranganathan, S.; Bowser, R.; Cudkowic, M.E.; Novelli, M.; Lattazi, W.; Gambotto, A.; Day, B.W. Proteomic data mining challenges in identification of disease-specific biomarkers from variable resolution mass spectra. In Proceedings of the SIAM Bioinformatics Workshop, Lake Buena Vista, FL, USA, 7–10 October 2004; Volume 10.
- Gopalakrishnan, V.; Lustgarten, J.L.; Visweswaran, S.; Cooper, G.F. Bayesian rule learning for biomedical data mining. Bioinformatics 2010, 26, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.H.; Gopalakrishnan, V.; Kasi, P.M.; Zeng, X.; Malhotra, U.; Balasubramanian, J.; Visweswaran, S.; Sun, M.; Flint, M.S.; Davison, J.M.; et al. Evaluation of a 4-protein serum biomarker panel biglycan, annexin-A6, myeloperoxidase, and protein S100-A9 (B-AMP) for the detection of esophageal adenocarcinoma. Cancer 2014, 120, 3902–3913. [Google Scholar] [CrossRef] [PubMed]
- Fürnkranz, J.; Widmer, G. Incremental reduced error pruning. In Proceedings of the 11th International Conference on Machine Learning (ML-94), New Brunswick, NJ, USA, 10–13 July 1994; pp. 70–77.
- Cohen, W.W. Fast effective rule induction. In Proceedings of the Twelfth International Conference on Machine Learning, Tahoe City, CA, USA, 9–12 July1995; pp. 115–123.
- Quinlan, J.R. C4.5: Programs for Machine Learning; Elsevier: San Mateo, CA, USA, 2014. [Google Scholar]
- Neapolitan, R.E. Learning Bayesian Networks; Pearson: Upper Saddle River, NJ, USA, 2004. [Google Scholar]
- Cooper, G.F.; Herskovits, E. A Bayesian method for the induction of probabilistic networks from data. Mach. Learn. 1992, 9, 309–347. [Google Scholar] [CrossRef]
- Aronis, J.M.; Provost, F.J. Increasing the Efficiency of Data Mining Algorithms with Breadth-First Marker Propagation. In Proceedings of the Third International Conference on Knowledge Discovery and Data Mining, Menlo Park, CA, USA, 14–17 August 1997; pp. 119–122.
- Friedman, N.; Goldszmidt, M. Learning Bayesian Networks with Local Structure. In Learning in Graphical Models; Springer: Dordrecht, The Netherlands, 1998; pp. 421–459. [Google Scholar]
- Witten, I.H.; Frank, E.; Hall, M.A.; Pal, C.J. Data Mining: Practical Machine Learning Tools and Techniques; Morgan Kaufmann: Cambridge, MA, USA, 2016. [Google Scholar]
- Yeoh, E.J.; Ross, M.E.; Shurtleff, S.A.; Williams, W.K.; Patel, D.; Mahfouz, R.; Behm, F.G.; Raimondi, S.C.; Relling, M.V.; Patel, A.; et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002, 1, 133–143. [Google Scholar] [CrossRef]
- Gravendeel, L.A.; Kouwenhoven, M.C.; Gevaert, O.; de Rooi, J.J.; Stubbs, A.P.; Duijm, J.E.; Daemen, A.; Bleeker, F.E.; Bralten, L.B.; Kloosterhof, N.K.; et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009, 69, 9065–9072. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Li, C.; Higgins, J.P.; Van De Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Beer, D.G.; Kardia, S.L.; Huang, C.C.; Giordano, T.J.; Levin, A.M.; Misek, D.E.; Lin, L.; Chen, G.; Gharib, T.G.; Thomas, D.G.; et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 2002, 8, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, N.; Oka, M.; Yamada-Okabe, H.; Nishida, M.; Maeda, Y.; Mori, N.; Takao, T.; Tamesa, T.; Tangoku, A.; Tabuchi, H.; et al. Oligonucleotide microarray for prediction of early intrahepatic recurrence of hepatocellular carcinoma after curative resection. Lancet 2003, 361, 923–929. [Google Scholar] [CrossRef]
- Hedenfalk, I.; Duggan, D.; Chen, Y.; Radmacher, M.; Bittner, M.; Simon, R.; Meltzer, P.; Gusterson, B.; Esteller, M.; Raffeld, M.; et al. Gene-expression profiles in hereditary breast cancer. N. Engl. J. Med. 2001, 344, 539–548. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar]
- Soneson, C.; Delorenzi, M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinform. 2013, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- Lustgarten, J.L.; Visweswaran, S.; Gopalakrishnan, V.; Cooper, G.F. Application of an efficient Bayesian discretization method to biomedical data. BMC Bioinform. 2011, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, J.B.; Visweswaran, S.; Cooper, G.F.; Gopalakrishnan, V. Selective Model Averaging with Bayesian Rule Learning for Predictive Biomedicine. AMIA Summits Transl. Sci. Proc. 2014, 2014, 17–22. [Google Scholar] [PubMed]
- Wong, K.C.; Zhang, Z. SNPdryad: Predicting deleterious non-synonymous human SNPs using only orthologous protein sequences. Bioinformatics 2014, 30, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Stemmer, M.; Thumberger, T.; del Sol Keyer, M.; Wittbrodt, J.; Mateo, J.L. CCTop: An intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS ONE 2015, 10, e0124633. [Google Scholar] [CrossRef] [PubMed]
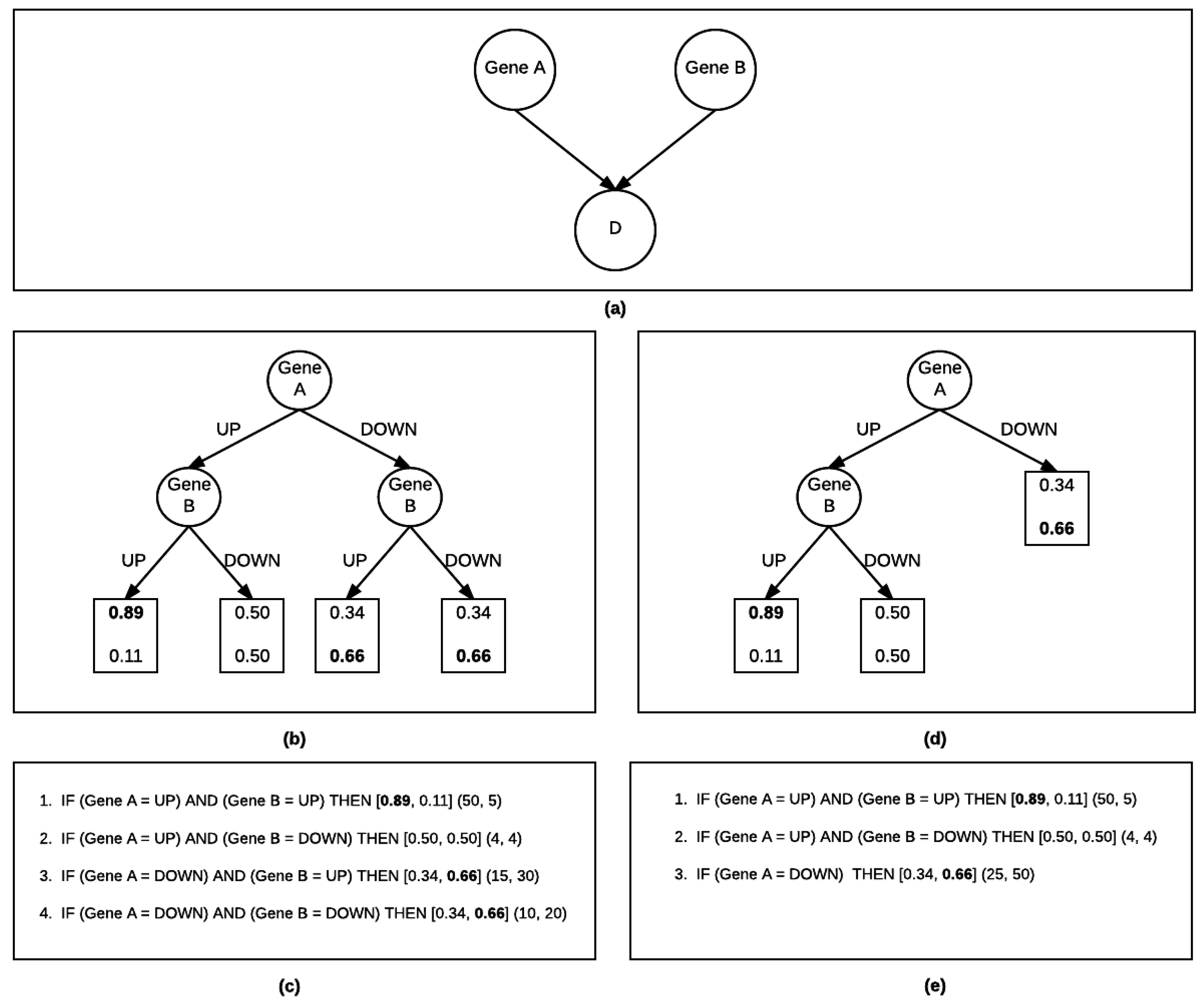



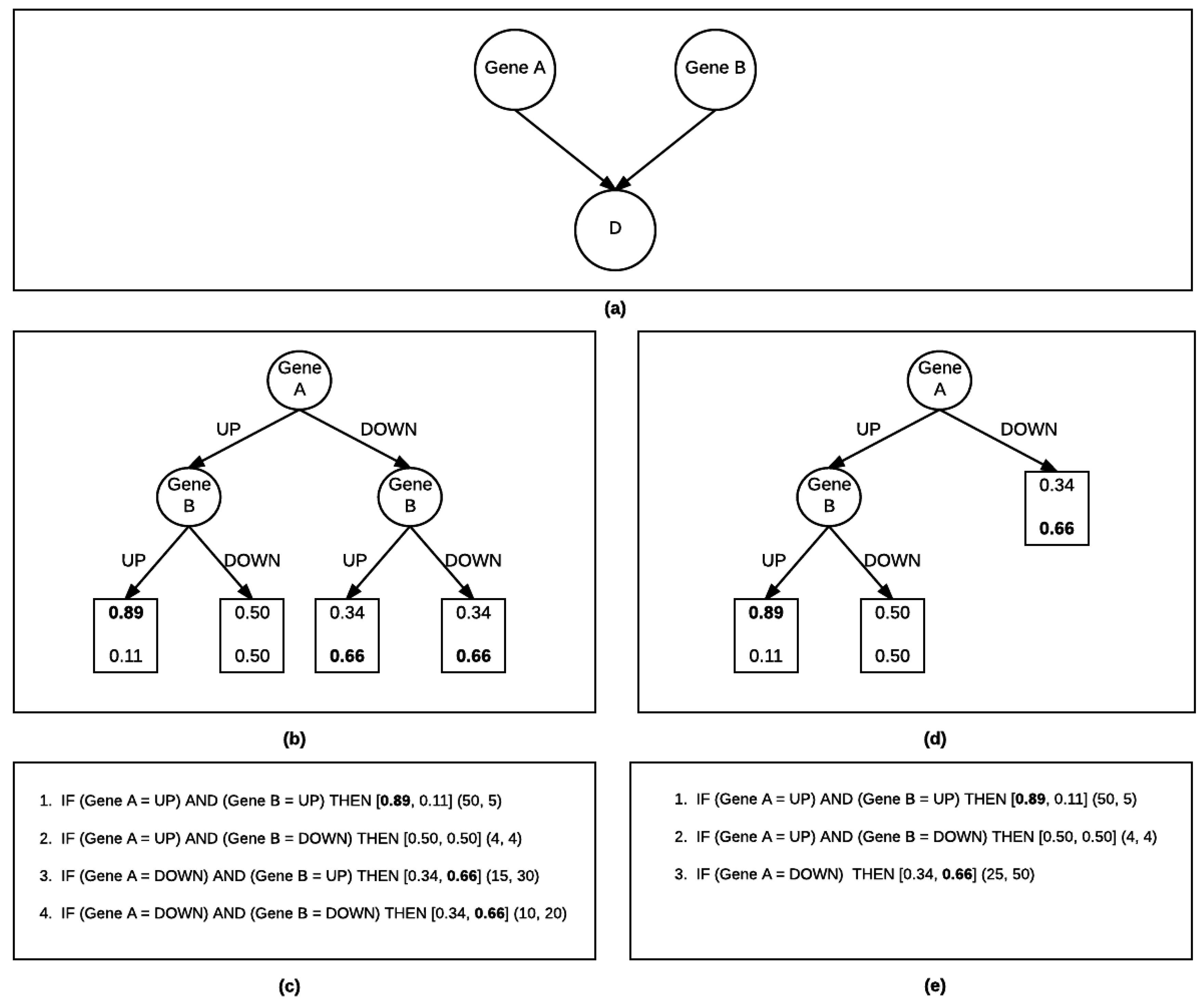{kind=link}
{kind=link}
{kind=link}
| Data ID | # Instances | # Features | Class Distribution | Source |
|---|---|---|---|---|
| 1 | 249 | 12,625 | (201, 48) | [17] |
| 2 | 175 | 6019 | (159, 16) | [18] |
| 3 | 103 | 6940 | (62, 41) | [19] |
| 4 | 100 | 6019 | (76, 24) | [20] |
| 5 | 96 | 5481 | (75, 21) | Dr. Kaminski |
| 6 | 86 | 5372 | (69, 17) | [21] |
| 7 | 72 | 7129 | (47, 25) | [22] |
| 8 | 63 | 5481 | (52, 11) | Dr. Kaminski |
| 9 | 60 | 7129 | (40, 20) | [23] |
| 10 | 36 | 7464 | (18, 18) | [24] |
| Data ID | Average AUC | Average Accuracy | ||||
|---|---|---|---|---|---|---|
| BRL-GSS | BRL-LSS | C4.5 | BRL-GSS | BRL-LSS | C4.5 | |
| 1 | 0.864 | 0.809 | 0.821 | 73.08 | 74.63 | 69.92 |
| 2 | 0.596 | 0.570 | 0.528 | 90.33 | 85.16 | 83.46 |
| 3 | 0.948 | 0.936 | 0.929 | 90.36 | 90.18 | 93.09 |
| 4 | 0.694 | 0.807 | 0.469 | 73.00 | 80.00 | 61.00 |
| 5 | 0.815 | 0.921 | 0.807 | 86.56 | 92.67 | 88.44 |
| 6 | 0.497 | 0.540 | 0.732 | 72.22 | 67.22 | 85.14 |
| 7 | 0.898 | 0.877 | 0.877 | 85.71 | 84.64 | 87.50 |
| 8 | 0.857 | 0.847 | 0.807 | 92.38 | 90.71 | 90.71 |
| 9 | 0.463 | 0.494 | 0.594 | 55.00 | 60.00 | 61.67 |
| 10 | 0.950 | 0.950 | 0.950 | 94.17 | 94.17 | 94.17 |
| Average | 0.758 | 0.775 | 0.751 | 81.28 | 81.94 | 81.51 |
| SEM | 0.06 | 0.05 | 0.05 | 3.96 | 3.62 | 3.98 |
| Data ID | Average Number of Rules | Average Number of Features | ||||
|---|---|---|---|---|---|---|
| BRL-GSS | BRL-LSS | C4.5 | BRL-GSS | BRL-LSS | C4.5 | |
| 1 | 307.10 | 18.00 | 13.30 | 8.00 | 15.70 | 25.60 |
| 2 | 105.60 | 6.50 | 5.30 | 6.50 | 5.40 | 9.60 |
| 3 | 5.40 | 3.90 | 2.90 | 2.30 | 2.70 | 4.70 |
| 4 | 33.60 | 6.10 | 6.50 | 4.80 | 4.70 | 11.90 |
| 5 | 7.60 | 3.30 | 2.50 | 2.60 | 2.00 | 4.00 |
| 6 | 42.40 | 6.30 | 5.30 | 5.10 | 4.30 | 9.40 |
| 7 | 4.40 | 3.10 | 2.80 | 2.10 | 2.10 | 4.60 |
| 8 | 3.20 | 2.60 | 2.00 | 1.60 | 1.60 | 3.00 |
| 9 | 24.80 | 5.80 | 5.10 | 4.40 | 4.20 | 9.10 |
| 10 | 2.00 | 2.00 | 2.00 | 1.00 | 1.00 | 3.00 |
| Average | 53.61 | 5.76 | 4.77 | 3.84 | 4.37 | 8.49 |
| SEM | 29.89 | 1.46 | 1.08 | 0.72 | 1.34 | 2.15 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lustgarten, J.L.; Balasubramanian, J.B.; Visweswaran, S.; Gopalakrishnan, V. Learning Parsimonious Classification Rules from Gene Expression Data Using Bayesian Networks with Local Structure. Data 2017, 2, 5. https://doi.org/10.3390/data2010005
Lustgarten JL, Balasubramanian JB, Visweswaran S, Gopalakrishnan V. Learning Parsimonious Classification Rules from Gene Expression Data Using Bayesian Networks with Local Structure. Data. 2017; 2(1):5. https://doi.org/10.3390/data2010005
Chicago/Turabian StyleLustgarten, Jonathan Lyle, Jeya Balaji Balasubramanian, Shyam Visweswaran, and Vanathi Gopalakrishnan. 2017. "Learning Parsimonious Classification Rules from Gene Expression Data Using Bayesian Networks with Local Structure" Data 2, no. 1: 5. https://doi.org/10.3390/data2010005
APA StyleLustgarten, J. L., Balasubramanian, J. B., Visweswaran, S., & Gopalakrishnan, V. (2017). Learning Parsimonious Classification Rules from Gene Expression Data Using Bayesian Networks with Local Structure. Data, 2(1), 5. https://doi.org/10.3390/data2010005