
Cultivation and Imaging of S. latissima Embryo Monolayered Cell Sheets Inside Microfluidic Devices
 , , and
, , and 
Abstract
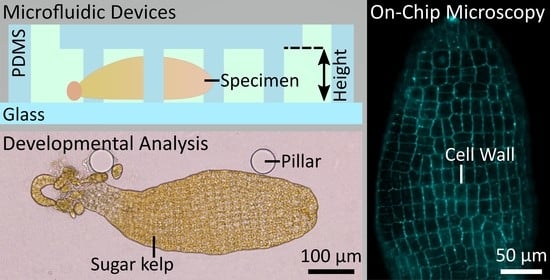
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
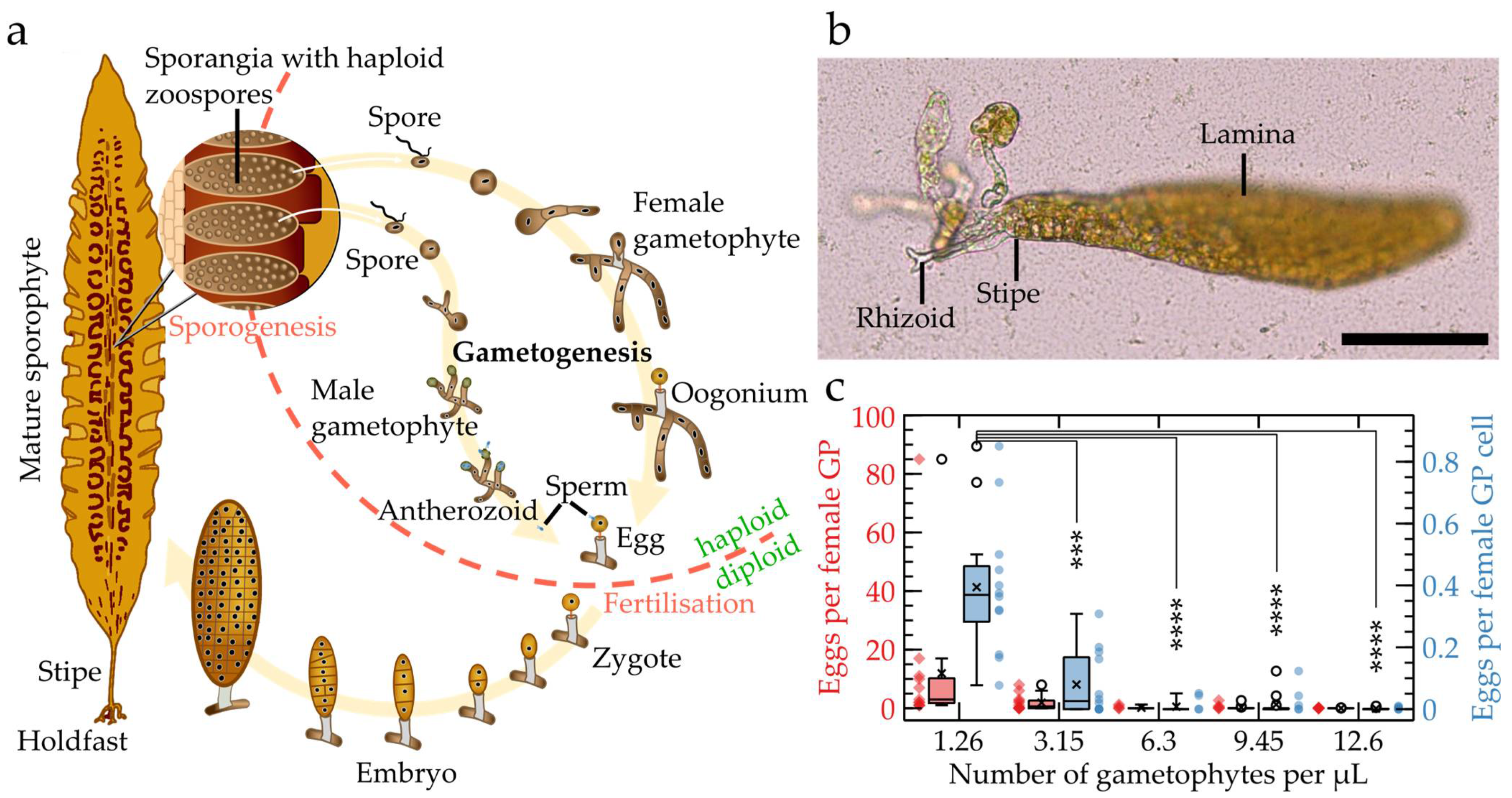
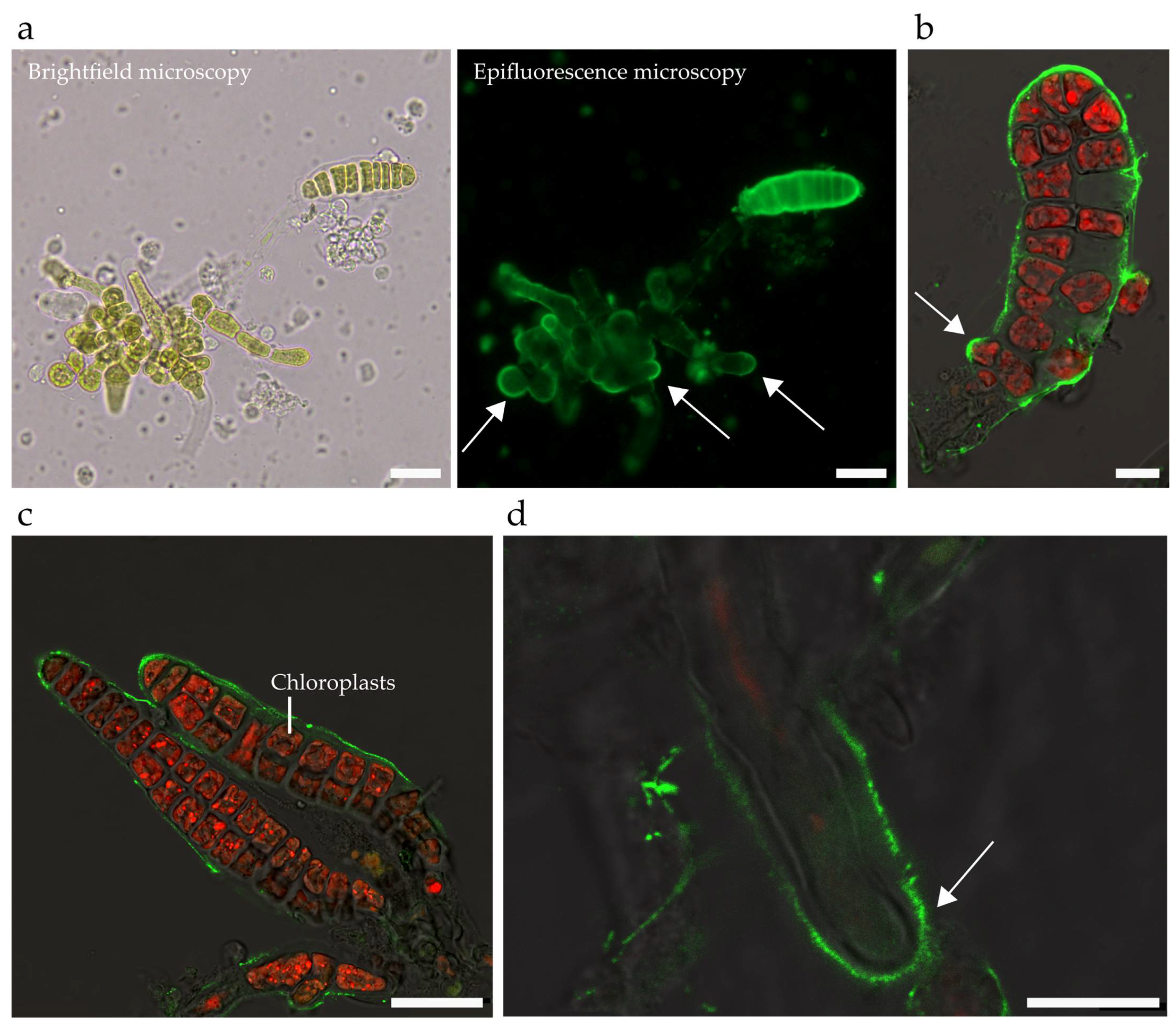
2.1. Characteristics of S. latissima Development and Growth
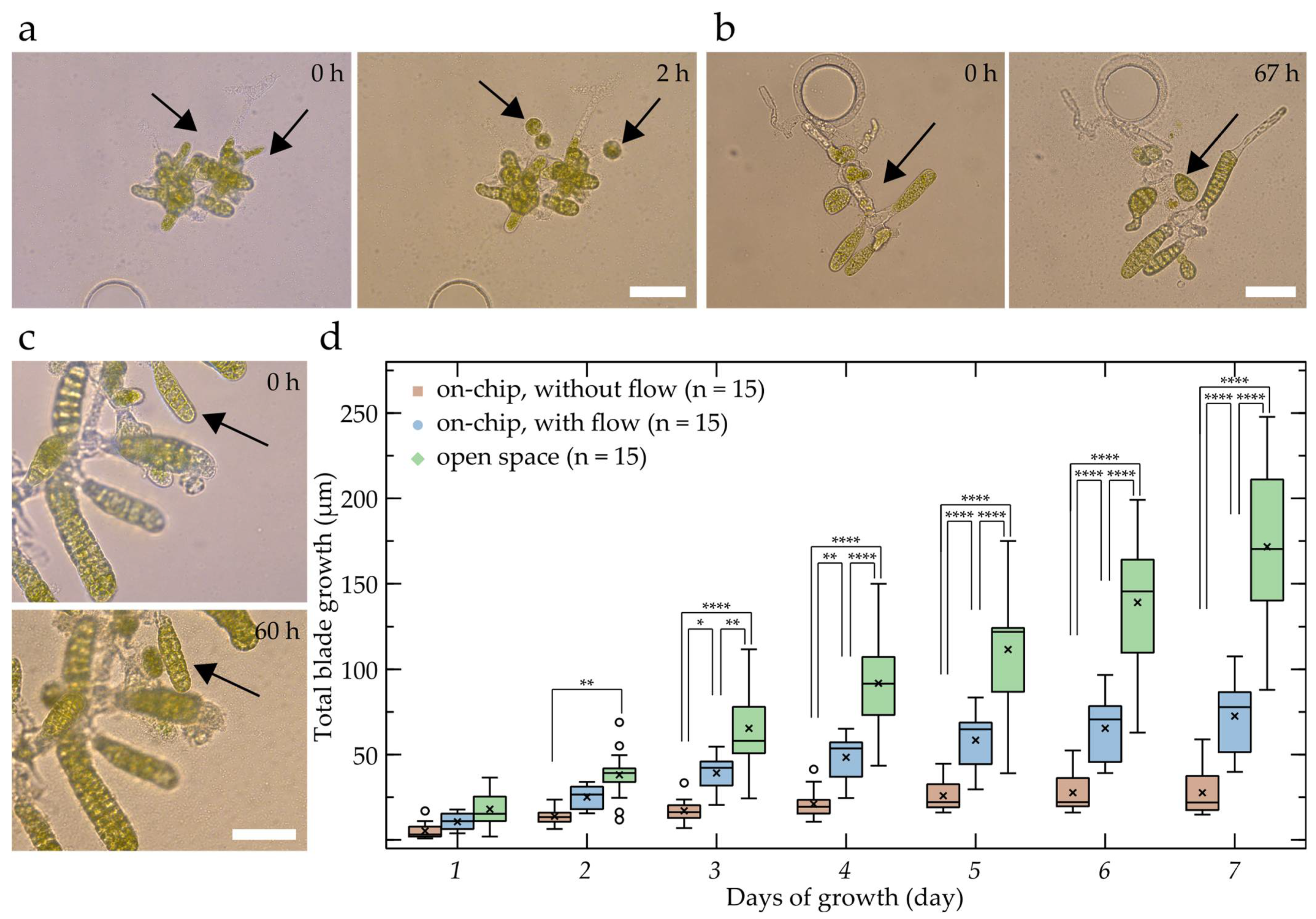
2.2. Microfluidic Cultivation of S. latissima Embryos
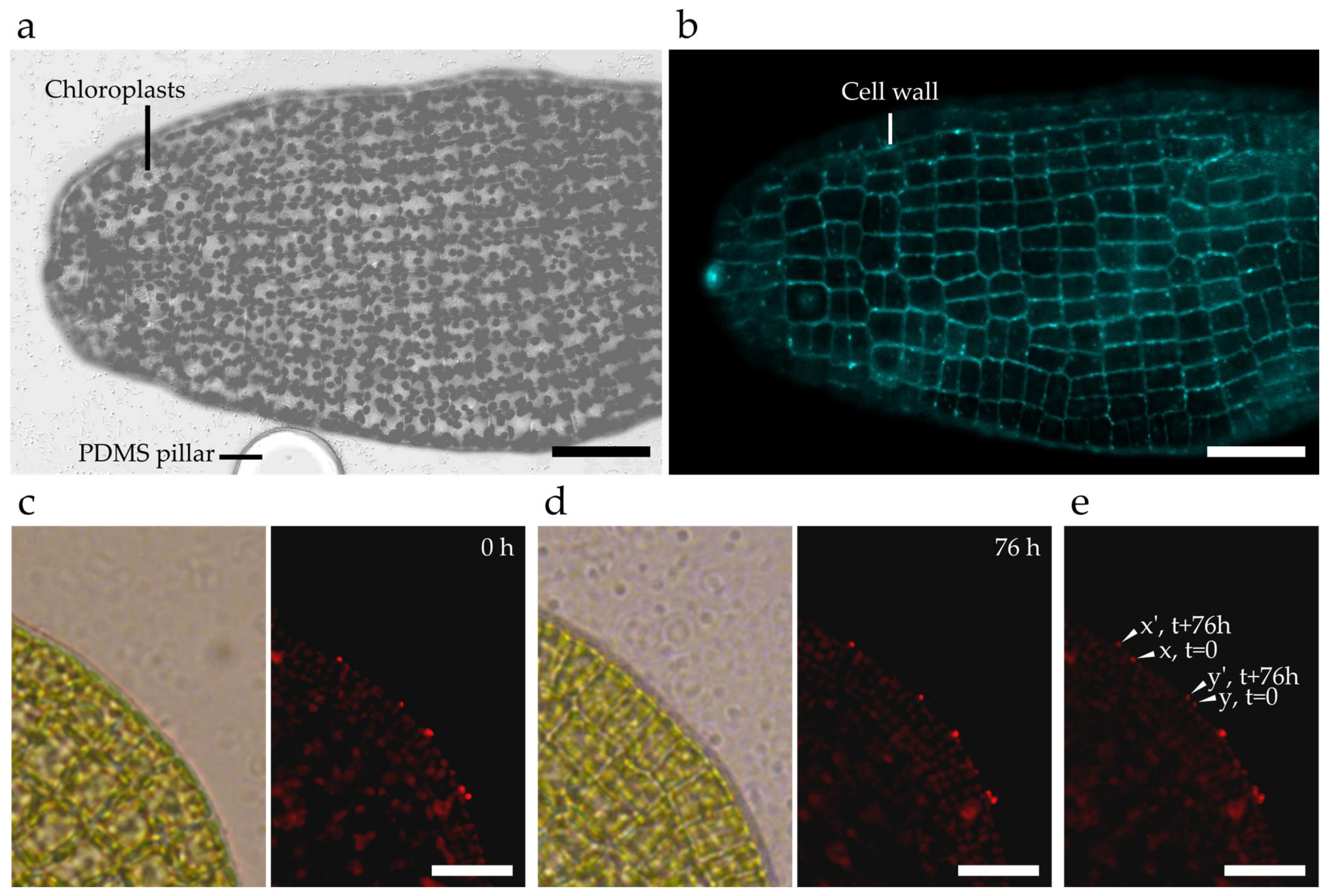
2.3. Assessment of Saccharina Developmental Steps inside Microfluidic Devices
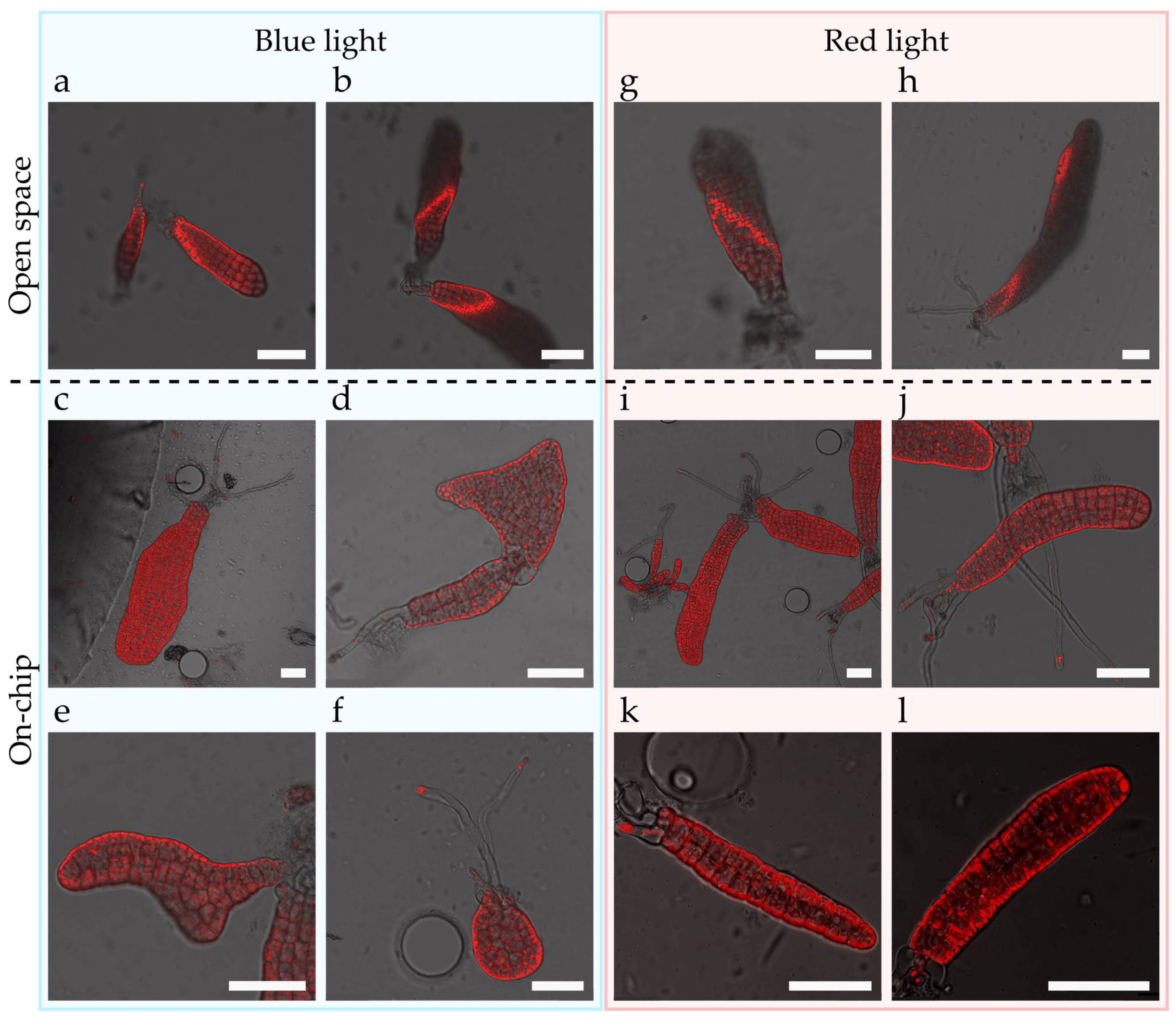
2.4. Cell Biology Protocols to Accurately Image the Cells of the Kelp Embryos
3. Conclusions
4. Materials and Methods
4.1. Production of Saccharina Embryos: General Conditions (Genotype, Storage of the Gametophytes)
4.2. Design and Fabrication of the Microfluidic Device
4.3. Chip Inoculation
4.4. Hydrostatic Pressure-Based Liquid Renewal
4.5. Pump-Based Liquid Renewal
4.6. Setting Up the Protocol for Time-Lapse Monitoring
4.7. Growth Rate Measurements
4.8. Duration of Embryos Viability in Microfluidic Devices
4.9. Cultures and Assessment of the Impact of Culture Density
4.10. Assessment of the Embryonic Developmental Stages
4.11. Impact of Blue and Red Lights in Embryogenesis
4.12. Staining the Embryos with Calcofluor
4.13. Marking the Embryo Surface with Fluorescent Beads
4.14. On-Chip Immunolocalisation
4.15. Statistical Evaluation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Babcock, R.C.; Bustamante, R.H.; Fulton, E.A.; Fulton, D.J.; Haywood, M.D.E.; Hobday, A.J.; Kenyon, R.; Matear, R.J.; Plagányi, E.E.; Richardson, A.J.; et al. Severe Continental-Scale Impacts of Climate Change Are Happening Now: Extreme Climate Events Impact Marine Habitat Forming Communities Along 45% of Australia’s Coast. Front. Mar. Sci. 2019, 6, 411. [Google Scholar] [CrossRef]
- Lotze, H.K.; Tittensor, D.P.; Bryndum-Buchholz, A.; Eddy, T.D.; Cheung, W.W.L.; Galbraith, E.D.; Barange, M.; Barrier, N.; Bianchi, D.; Blanchard, J.L.; et al. Global Ensemble Projections Reveal Trophic Amplification of Ocean Biomass Declines with Climate Change. Proc. Natl. Acad. Sci. USA 2019, 116, 12907–12912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.D.; Ota, Y.; Sumaila, U.R.; Cisneros-Montemayor, A.M.; Cheung, W.W.L. Adaptation Strategies to Climate Change in Marine Systems. Glob. Chang. Biol. 2018, 24, e1–e14. [Google Scholar] [CrossRef] [PubMed]
- Henson, S.A.; Cael, B.B.; Allen, S.R.; Dutkiewicz, S. Future Phytoplankton Diversity in a Changing Climate. Nat. Commun. 2021, 12, 5372. [Google Scholar] [CrossRef]
- Pessarrodona, A.; Assis, J.; Filbee-Dexter, K.; Burrows, M.T.; Gattuso, J.-P.; Duarte, C.M.; Krause-Jensen, D.; Moore, P.J.; Smale, D.A.; Wernberg, T. Global Seaweed Productivity. Sci. Adv. 2022, 8, eabn2465. [Google Scholar] [CrossRef]
- Barbier, M.; Charrier, B.; Araujo, R.; Holdt, S.L.; Jacquemin, B.; Rebours, C. Pegasus-Phycomorph European Guidelines for a Sustainable Aquaculture of Seaweeds; COST Action: Brussels, Belgium, 2019. [Google Scholar]
- Charrier, B.; Abreu, M.H.; Araujo, R.; Bruhn, A.; Coates, J.C.; de Clerck, O.; Katsaros, C.; Robaina, R.R.; Wichard, T. Furthering Knowledge of Seaweed Growth and Development to Facilitate Sustainable Aquaculture. New Phytol. 2017, 216, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, F.E. The Structure and Reproduction of the Algae; Cambridge University Press: Cambridge, UK, 1945. [Google Scholar]
- Theodorou, I.; Charrier, B. Brown Algae. In Handbook of Marine Model Organisms in Experimental Biology; CRC Press: Boca Raton, FL, USA, 2021; pp. 27–47. [Google Scholar]
- Lloyd, C.W. How Does the Cytoskeleton Read the Laws of Geometry in Aligning the Division Plane of Plant Cells? Development 1991, 113, 55–65. [Google Scholar] [CrossRef]
- Flanders, D.J.; Rawlins, D.J.; Shaw, P.J.; Lloyd, C.W. Nucleus-Associated Microtubules Help Determine the Division Plane of Plant Epidermal Cells: Avoidance of Four-Way Junctions and the Role of Cell Geometry. J. Cell Biol. 1990, 110, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Bardet, P.-L.; Guirao, B.; Paoletti, C.; Serman, F.; Léopold, V.; Bosveld, F.; Goya, Y.; Mirouse, V.; Graner, F.; Bellaïche, Y. PTEN Controls Junction Lengthening and Stability during Cell Rearrangement in Epithelial Tissue. Dev. Cell 2013, 25, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Ebbing, A.; Pierik, R.; Bouma, T.; Kromkamp, J.C.; Timmermans, K. How Light and Biomass Density Influence the Reproduction of Delayed Saccharina Latissima Gametophytes (Phaeophyceae). J. Phycol. 2020, 56, 709–718. [Google Scholar] [CrossRef]
- Täuber, S.; Schmitz, J.; Blöbaum, L.; Fante, N.; Steinhoff, H.; Grünberger, A. How to Perform a Microfluidic Cultivation Experiment—A Guideline to Success. Biosensors 2021, 11, 485. [Google Scholar] [CrossRef] [PubMed]
- Sanati Nezhad, A.; Naghavi, M.; Packirisamy, M.; Bhat, R.; Geitmann, A. Quantification of Cellular Penetrative Forces Using Lab-on-a-Chip Technology and Finite Element Modeling. Proc. Natl. Acad. Sci. USA 2013, 110, 8093–8098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamsudhin, N.; Laeubli, N.; Atakan, H.B.; Vogler, H.; Hu, C.; Haeberle, W.; Sebastian, A.; Grossniklaus, U.; Nelson, B.J. Massively Parallelized Pollen Tube Guidance and Mechanical Measurements on a Lab-Ona-Chip Platform. PLoS ONE 2016, 11, e0168138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burri, J.T.; Vogler, H.; Läubli, N.F.; Hu, C.; Grossniklaus, U.; Nelson, B.J. Feeling the Force: How Pollen Tubes Deal with Obstacles. New Phytol. 2018, 220, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Gladkov, A.; Pigareva, Y.; Kutyina, D.; Kolpakov, V.; Bukatin, A.; Mukhina, I.; Kazantsev, V.; Pimashkin, A. Design of Cultured Neuron Networks in Vitro with Predefined Connectivity Using Asymmetric Microfluidic Channels. Sci. Rep. 2017, 7, 15625. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Shinde, A.; Illath, K.; Kar, S.; Nagai, M.; Tseng, F.-G.; Santra, T.S. Microfluidic Platforms for Single Neuron Analysis. Mater. Today Bio. 2022, 13, 100222. [Google Scholar] [CrossRef]
- Läubli, N.F.; Gerlt, M.S.; Wüthrich, A.; Lewis, R.T.M.; Shamsudhin, N.; Kutay, U.; Ahmed, D.; Dual, J.; Nelson, B.J. Embedded Microbubbles for Acoustic Manipulation of Single Cells and Microfluidic Applications. Anal. Chem. 2021, 93, 9760–9770. [Google Scholar] [CrossRef]
- Deleglise, B.; Lassus, B.; Soubeyre, V.; Alleaume-Butaux, A.; Hjorth, J.J.; Vignes, M.; Schneider, B.; Brugg, B.; Viovy, J.-L.; Peyrin, J.-M. Synapto-Protective Drugs Evaluation in Reconstructed Neuronal Network. PLoS ONE 2013, 8, e71103. [Google Scholar] [CrossRef] [Green Version]
- Läubli, N.F.; Burri, J.T.; Marquard, J.; Vogler, H.; Mosca, G.; Vertti-Quintero, N.; Shamsudhin, N.; DeMello, A.; Grossniklaus, U.; Ahmed, D.; et al. 3D Mechanical Characterization of Single Cells and Small Organisms Using Acoustic Manipulation and Force Microscopy. Nat. Commun. 2021, 12, 2583. [Google Scholar] [CrossRef]
- Charrier, B.; Boscq, S.; Nelson, B.J.; Läubli, N.F. Growth and Labelling of Cell Wall Components of the Brown Alga Ectocarpus in Microfluidic Chips. Front. Mar. Sci. 2021, 8. [Google Scholar] [CrossRef]
- Theodorou, I.; Opsahl-Sorteberg, H.-G.; Charrier, B. Preparation of Zygotes and Embryos of the Kelp Saccharina Latissima for Cell Biology Approaches. Bio. Protoc. 2021, 11. [Google Scholar] [CrossRef]
- Klochkova, T.A.; Motomura, T.; Nagasato, C.; Klimova, A.V.; Kim, G.H. The Role of Egg Flagella in the Settlement and Development of Zygotes in Two Saccharina Species. Phycologia 2019, 58, 145–153. [Google Scholar] [CrossRef]
- Wu, M.-H.; Paul, K.E.; Whitesides, G.M. Patterning Flood Illumination with Microlens Arrays. Appl. Opt. 2002, 41, 2575. [Google Scholar] [CrossRef] [PubMed]
- Davidson, F.F. The Effects of Auxins on the Growth of Marine Algae. Am. J. Bot. 1950, 37, 502. [Google Scholar] [CrossRef]
- Sun, H.; Basu, S.; Brady, S.R.; Luciano, R.L.; Muday, G.K. Interactions between Auxin Transport and the Actin Cytoskeleton in Developmental Polarity of Fucus Distichus Embryos in Response to Light and Gravity. Plant. Physiol. 2004, 135, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Lüning, K.; Dring, M.J. Reproduction Induced by Blue Light in Female Gametophytes of Laminaria Saccharina. Planta 1972, 104, 252–256. [Google Scholar] [CrossRef]
- Lüning, K.; Dring, M.J. Reproduction, Growth and Photosynthesis of Gametophytes of Laminaria Saccharina Grown in Blue and Red Light. Mar. Biol. 1975, 29, 195–200. [Google Scholar] [CrossRef]
- Wang, W.-J.; Sun, X.-T.; Wang, G.-C.; Xu, P.; Wang, X.-Y.; Lin, Z.-L.; Wang, F.-J. Effect of Blue Light on Indoor Seedling Culture of Saccharina Japonica (Phaeophyta). J. Appl. Phycol. 2010, 22, 737–744. [Google Scholar] [CrossRef]
- Deng, Y.; Yao, J.; Fu, G.; Guo, H.; Duan, D. Isolation, Expression, and Characterization of Blue Light Receptor AUREOCHROME Gene From Saccharina Japonica (Laminariales, Phaeophyceae). Mar. Biotechnol. 2014, 16, 135–143. [Google Scholar] [CrossRef]
- Jiang, P.; Qin, S.; Tseng, C.K. Expression of the LacZ Reporter Gene in Sporophytes of the Seaweed Laminaria Japonica (Phaeophyceae) by Gametophyte-Targeted Transformation. Plant Cell Rep. 2003, 21, 1211–1216. [Google Scholar] [CrossRef]
- Badis, Y.; Scornet, D.; Harada, M.; Caillard, C.; Godfroy, O.; Raphalen, M.; Gachon, C.M.M.; Coelho, S.M.; Motomura, T.; Nagasato, C.; et al. Targeted CRISPR-Cas9-based Gene Knockouts in the Model Brown Alga Ectocarpus. New Phytol. 2021, 231, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- le Bail, A.; Billoud, B.; Maisonneuve, C.; Peters, A.F.; Mark Cock, J.; Charrier, B. Early Development Pattern of the Brown Alga Ectocarpus Siliculosus (Ectocarpales, Phaeophyceae) Sporophyte 1. J. Phycol. 2008, 44, 1269–1281. [Google Scholar] [CrossRef] [PubMed]
- Rabillé, H.; Billoud, B.; Rolland, E.; Charrier, B. Dynamic and Microscale Mapping of Cell Growth. In Protocols for Macroalgae Research; CRC Press: Boca Raton, FL, USA; Taylor & Francis: Oxfordshire, UK, 2018; pp. 349–364. [Google Scholar]
- Charrier, B.; Rabillé, H.; Billoud, B. Gazing at Cell Wall Expansion under a Golden Light. Trends Plant Sci. 2019, 24, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Bidhendi, A.J.; Chebli, Y.; Geitmann, A. Fluorescence Visualization of Cellulose and Pectin in the Primary Plant Cell Wall. J. Microsc. 2020, 278, 164–181. [Google Scholar] [CrossRef]
- Shaw, S.L.; Dumais, J.; Long, S.R. Cell Surface Expansion in Polarly Growing Root Hairs of Medicago Truncatula. Plant Physiol. 2000, 124, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabillé, H.; Billoud, B.; Tesson, B.; le Panse, S.; Rolland, É.; Charrier, B. The Brown Algal Mode of Tip Growth: Keeping Stress under Control. PLoS Biol. 2019, 17, e2005258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabillé, H.; Torode, T.A.; Tesson, B.; le Bail, A.; Billoud, B.; Rolland, E.; le Panse, S.; Jam, M.; Charrier, B. Alginates along the Filament of the Brown Alga Ectocarpus Help Cells Cope with Stress. Sci. Rep. 2019, 9, 12956. [Google Scholar] [CrossRef] [Green Version]
- le Bail, A.; Charrier, B. Culture Methods and Mutant Generation in the Filamentous Brown Algae Ectocarpus Siliculosus. Methods Mol. Biol. 2013, 959, 323–332. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clerc, T.; Boscq, S.; Attia, R.; Kaminski Schierle, G.S.; Charrier, B.; Läubli, N.F. Cultivation and Imaging of S. latissima Embryo Monolayered Cell Sheets Inside Microfluidic Devices. Bioengineering 2022, 9, 718. https://doi.org/10.3390/bioengineering9110718
Clerc T, Boscq S, Attia R, Kaminski Schierle GS, Charrier B, Läubli NF. Cultivation and Imaging of S. latissima Embryo Monolayered Cell Sheets Inside Microfluidic Devices. Bioengineering. 2022; 9(11):718. https://doi.org/10.3390/bioengineering9110718
Chicago/Turabian StyleClerc, Thomas, Samuel Boscq, Rafaele Attia, Gabriele S. Kaminski Schierle, Bénédicte Charrier, and Nino F. Läubli. 2022. "Cultivation and Imaging of S. latissima Embryo Monolayered Cell Sheets Inside Microfluidic Devices" Bioengineering 9, no. 11: 718. https://doi.org/10.3390/bioengineering9110718