Effects of Lead and Cadmium on Brain Endothelial Cell Survival, Monolayer Permeability, and Crucial Oxidative Stress Markers in an in Vitro Model of the Blood-Brain Barrier

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Culture of Human Brain Microvascular Endothelial Cells (hCMEC/D3) and Toxicity Studies
2.3. Determination of Cell Viability
2.4. Intracellular ROS Measurement
2.5. Determination of GSH
2.6. Determination of Catalase Activity
2.7. Dextran Permeability Study
2.8. Trans-Endothelial Electric Resistance (TEER) Measurement
Western Blot Analysis
2.9. Determination of Protein
2.10. Statistical Analysis
3. Results
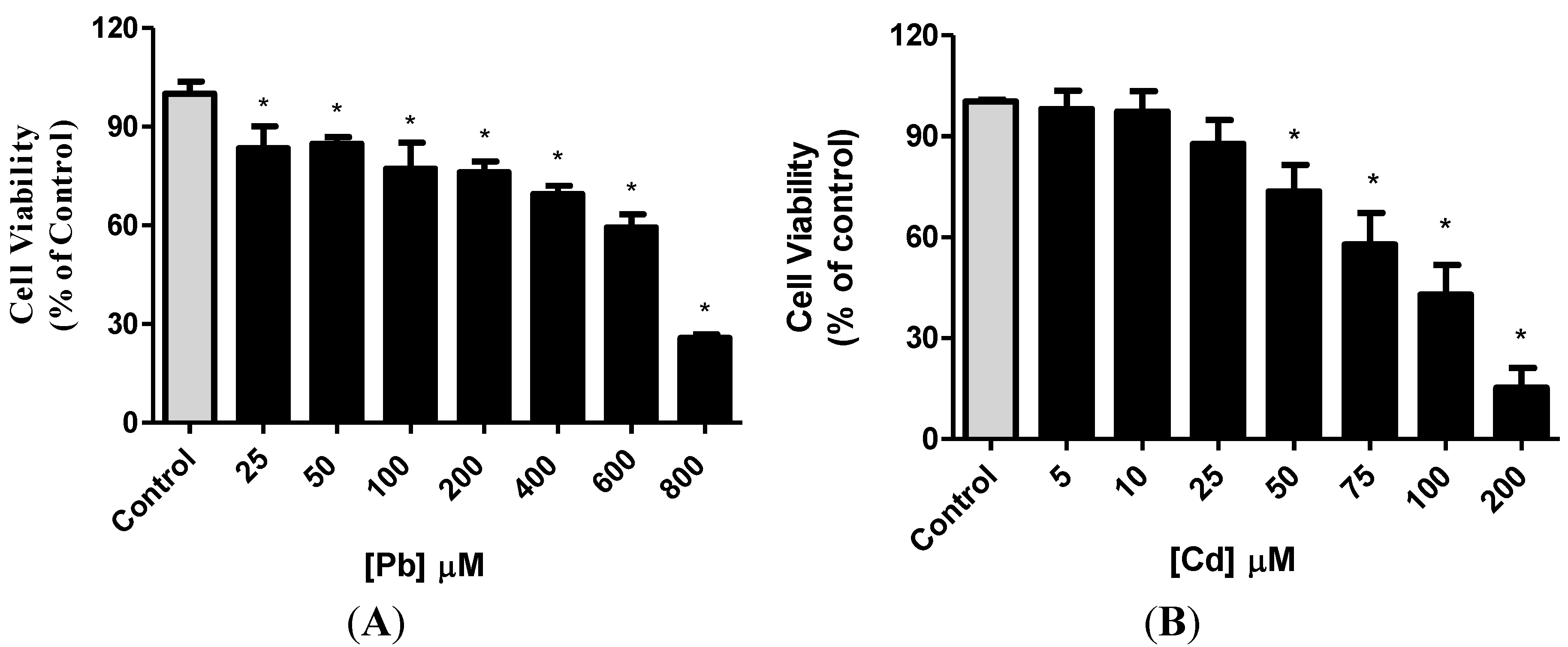
3.1. Effect of Pb or Cd on Cell Viability
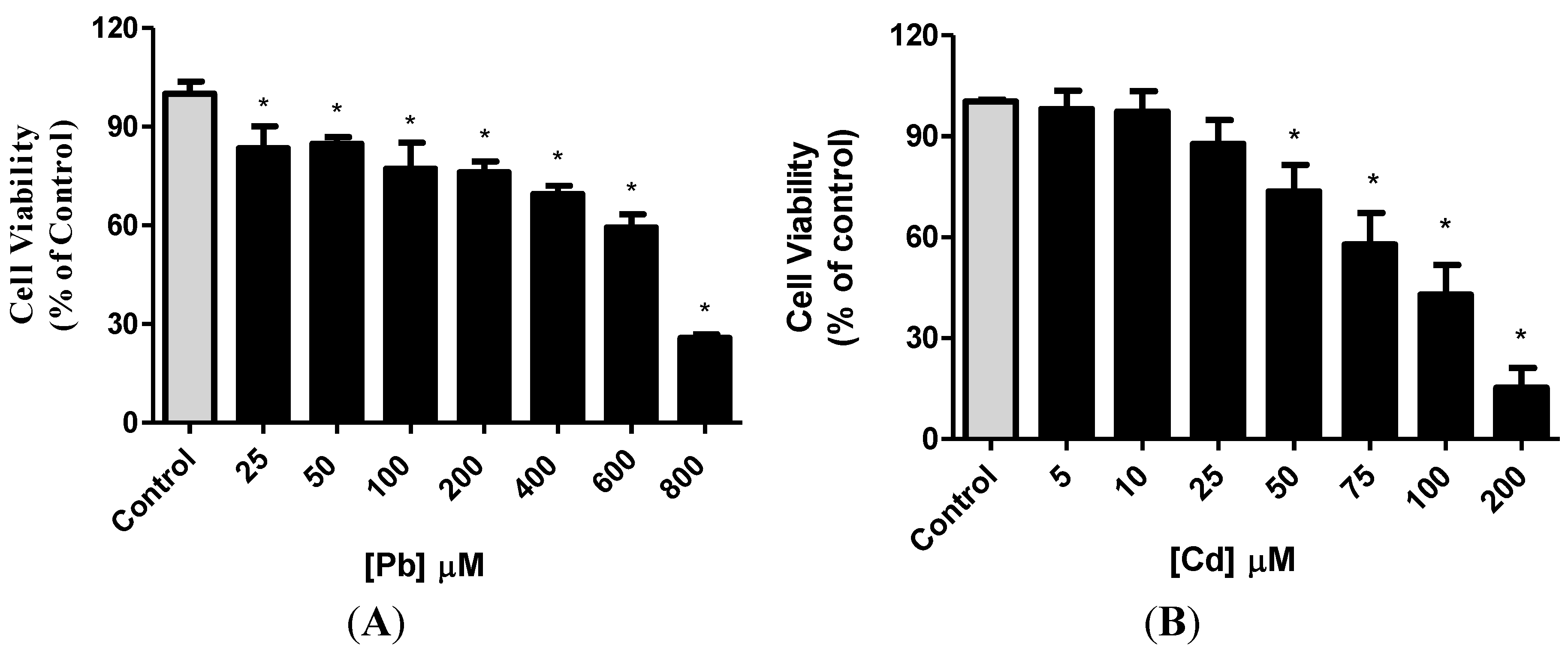
3.2. Effect of Pb or Cd on Intracellular ROS Level
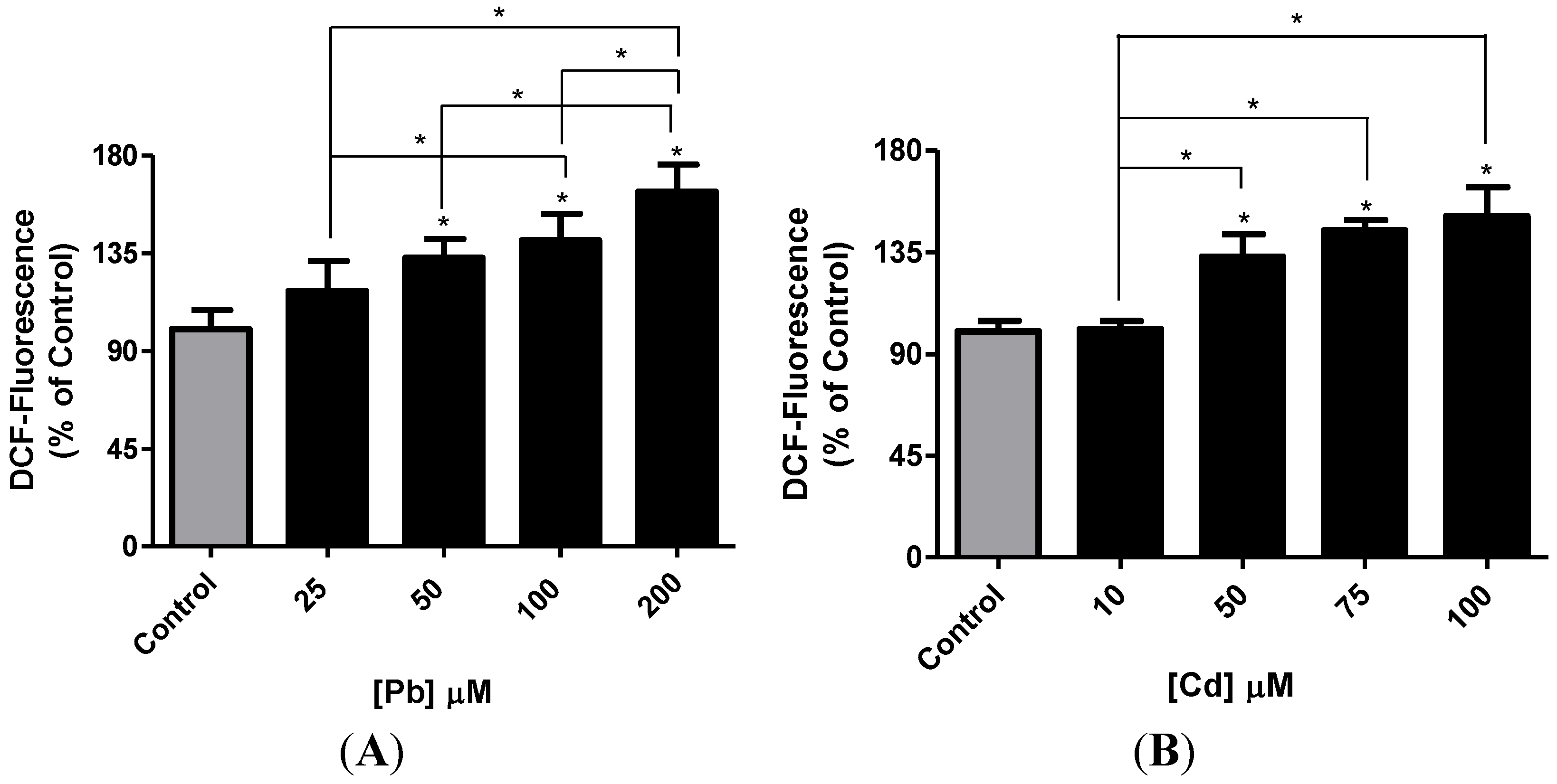
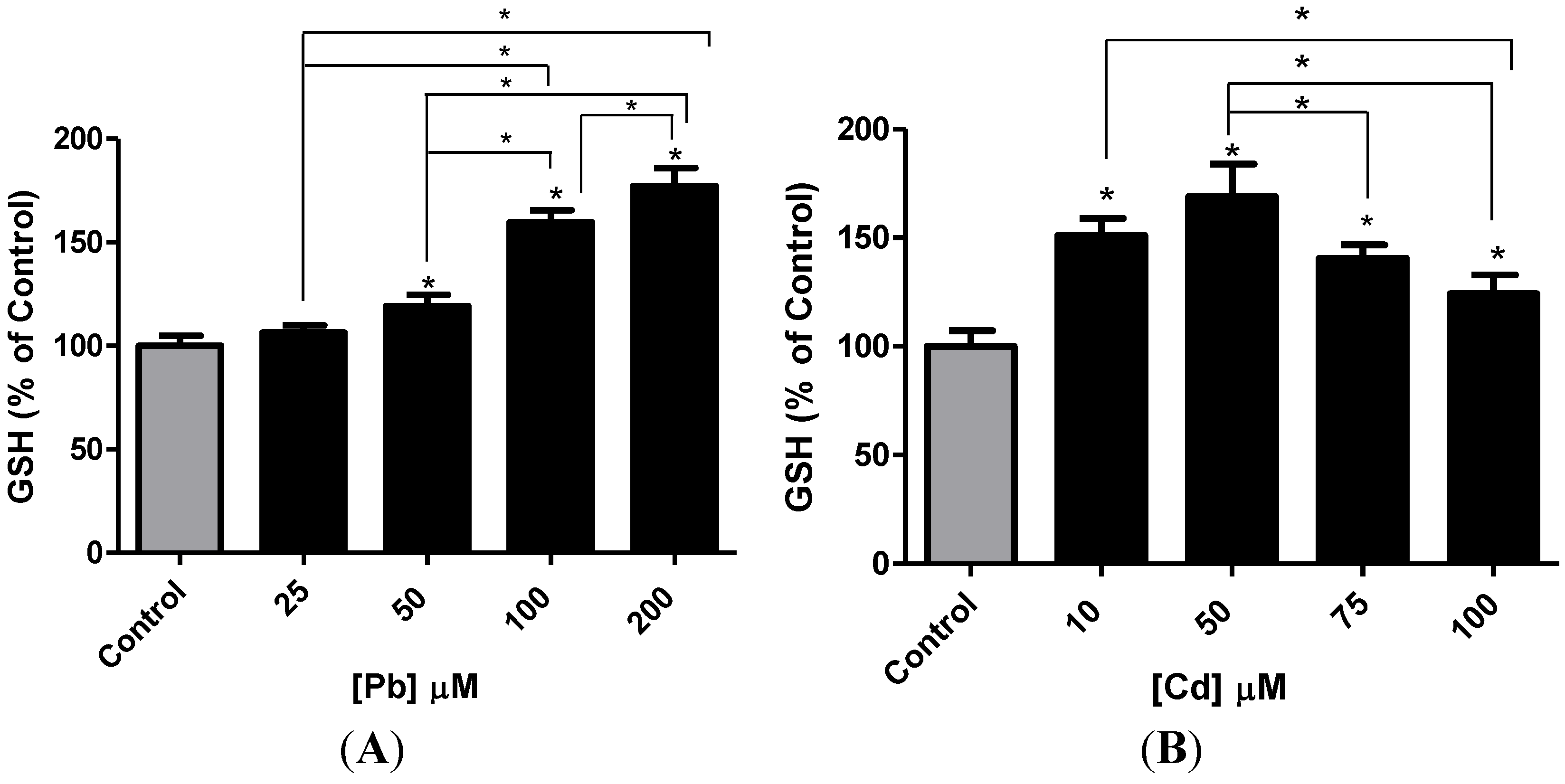
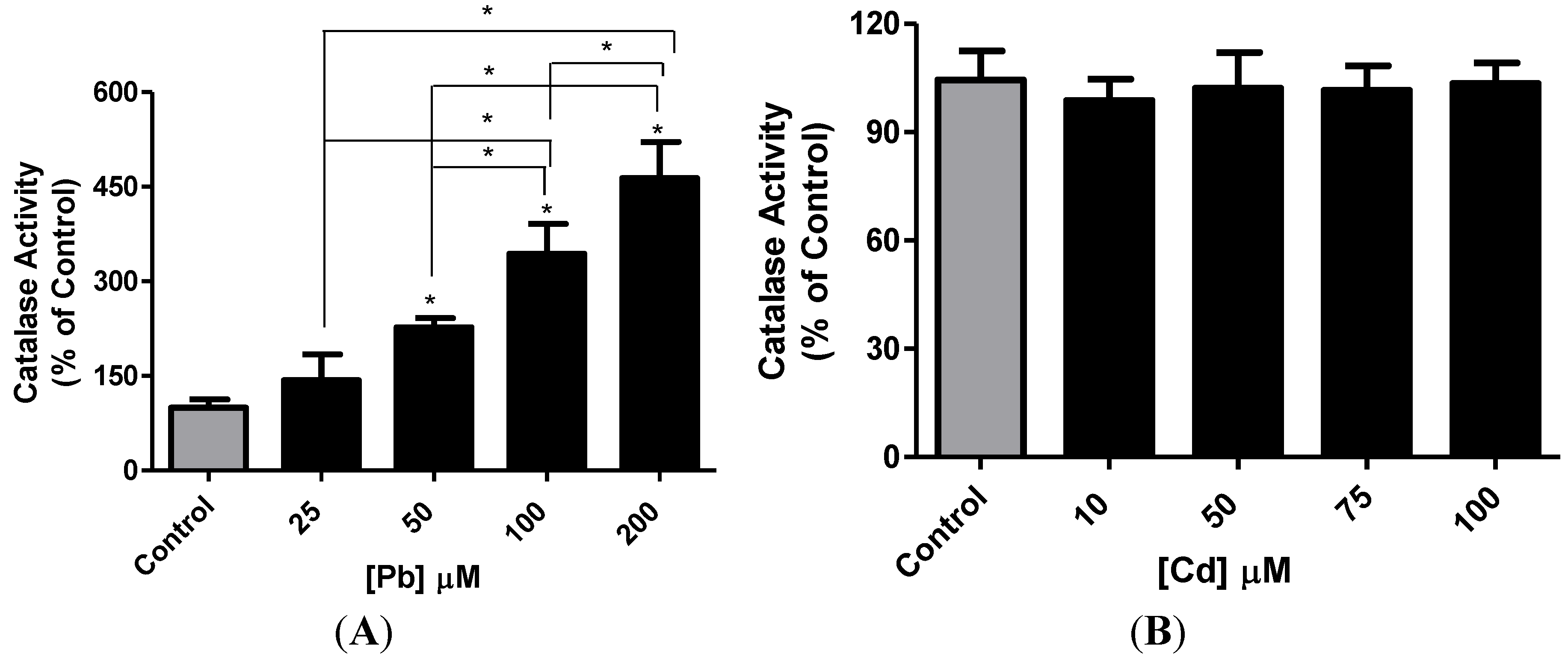
3.3. Effect of Pb or Cd on Intracellular GSH and Catalase Activity
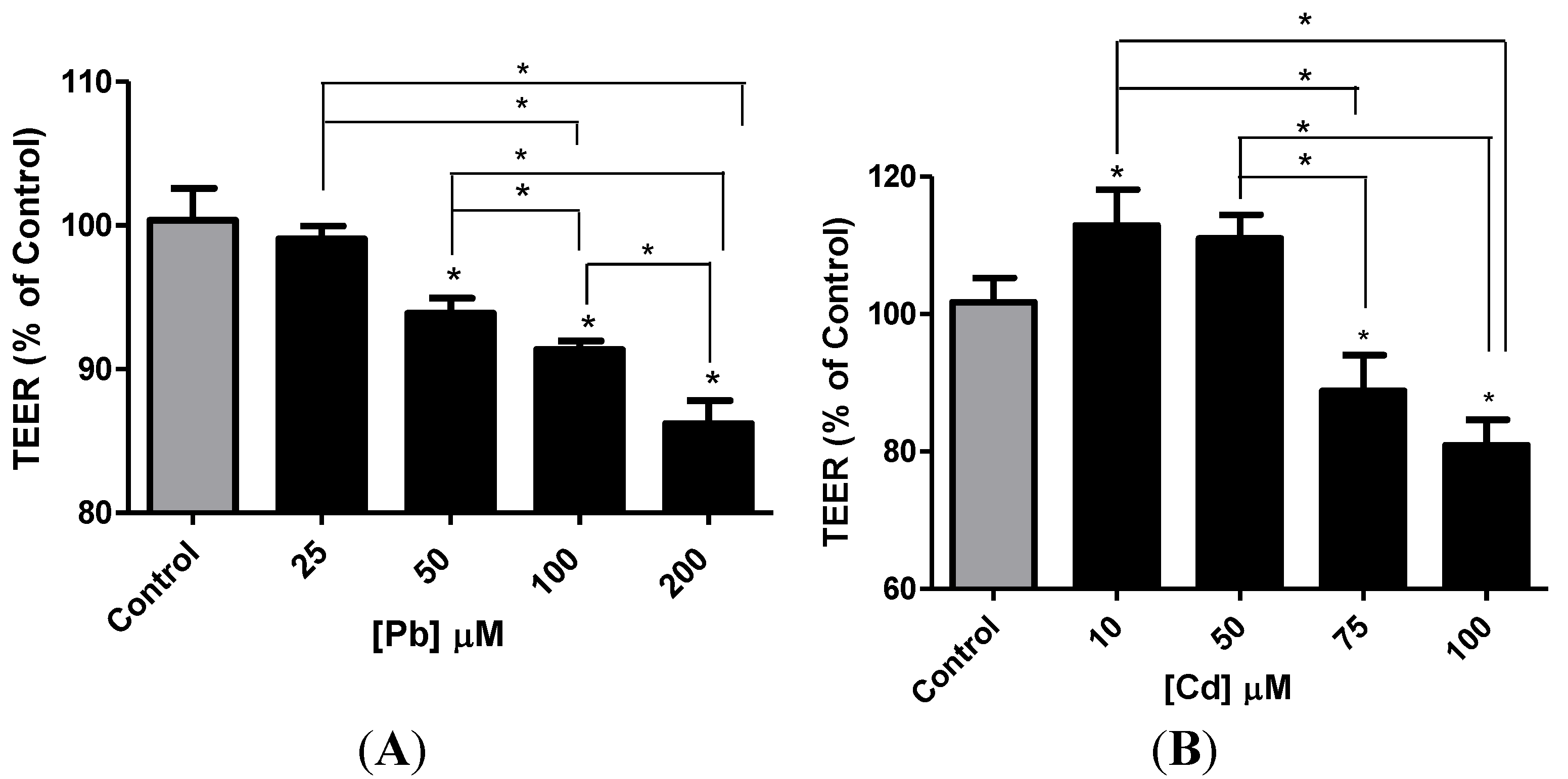
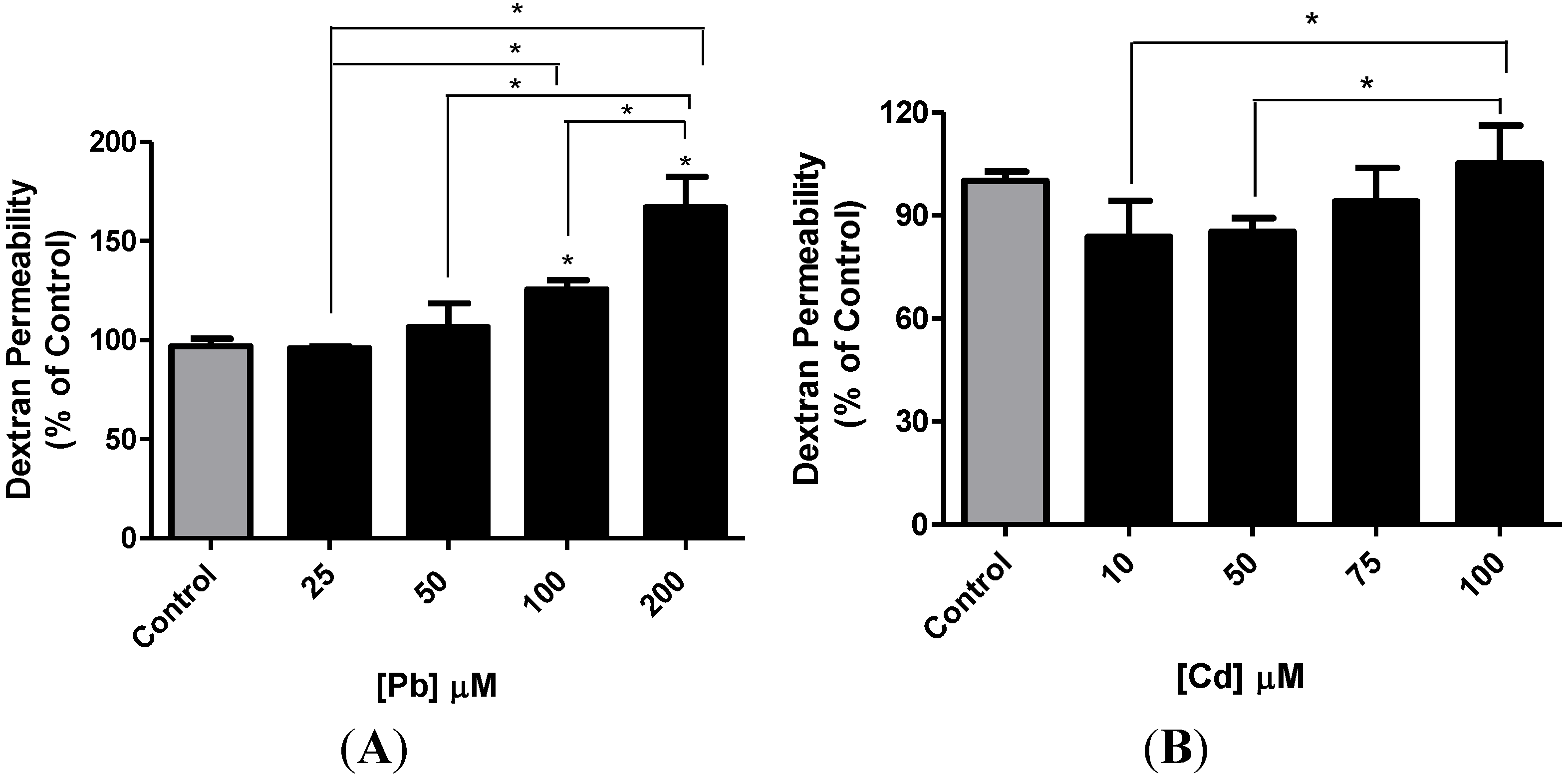
3.4. Effect of Pb or Cd on the Integrity of the hCMEC/D3 Monolayer
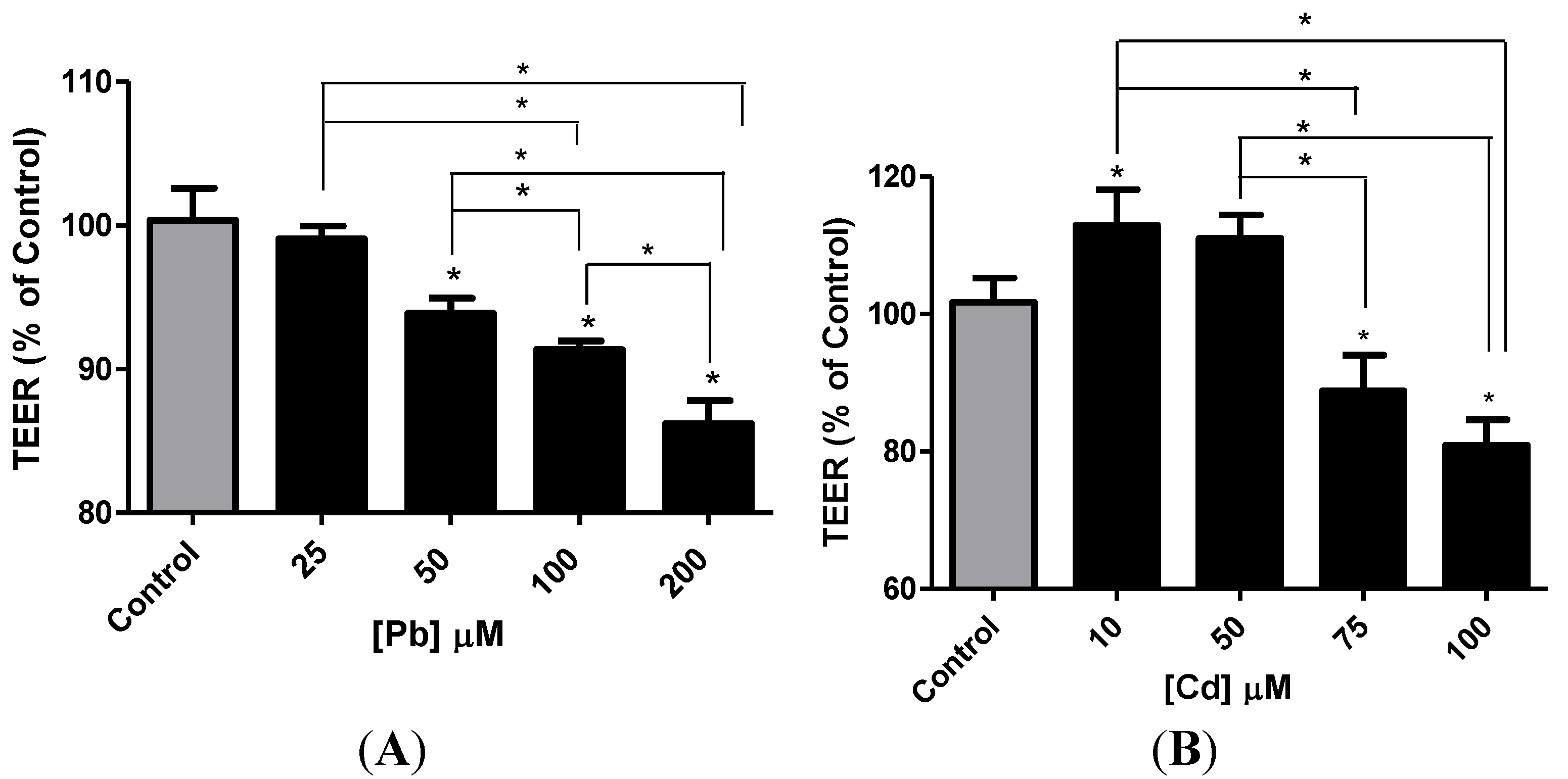
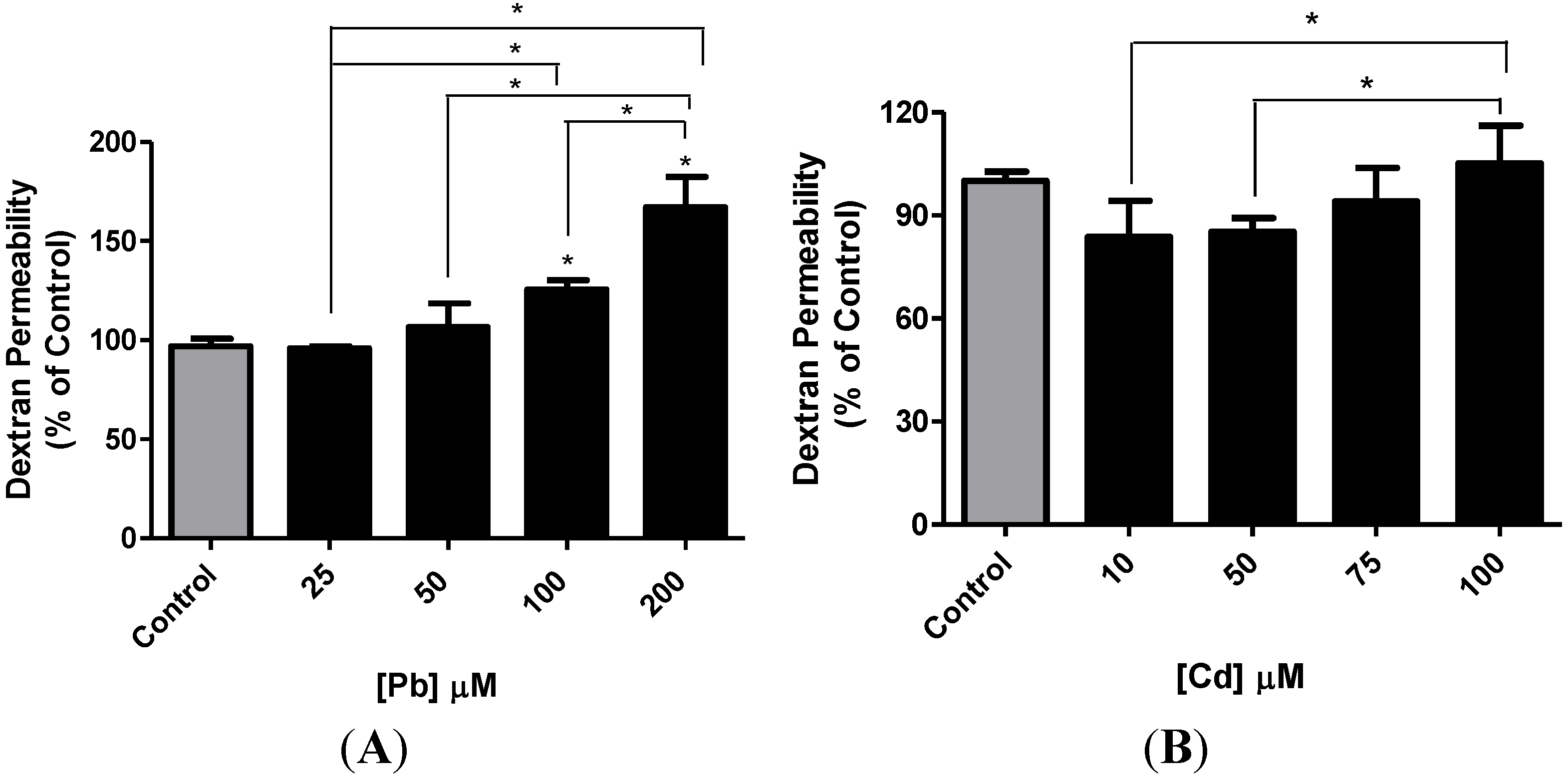
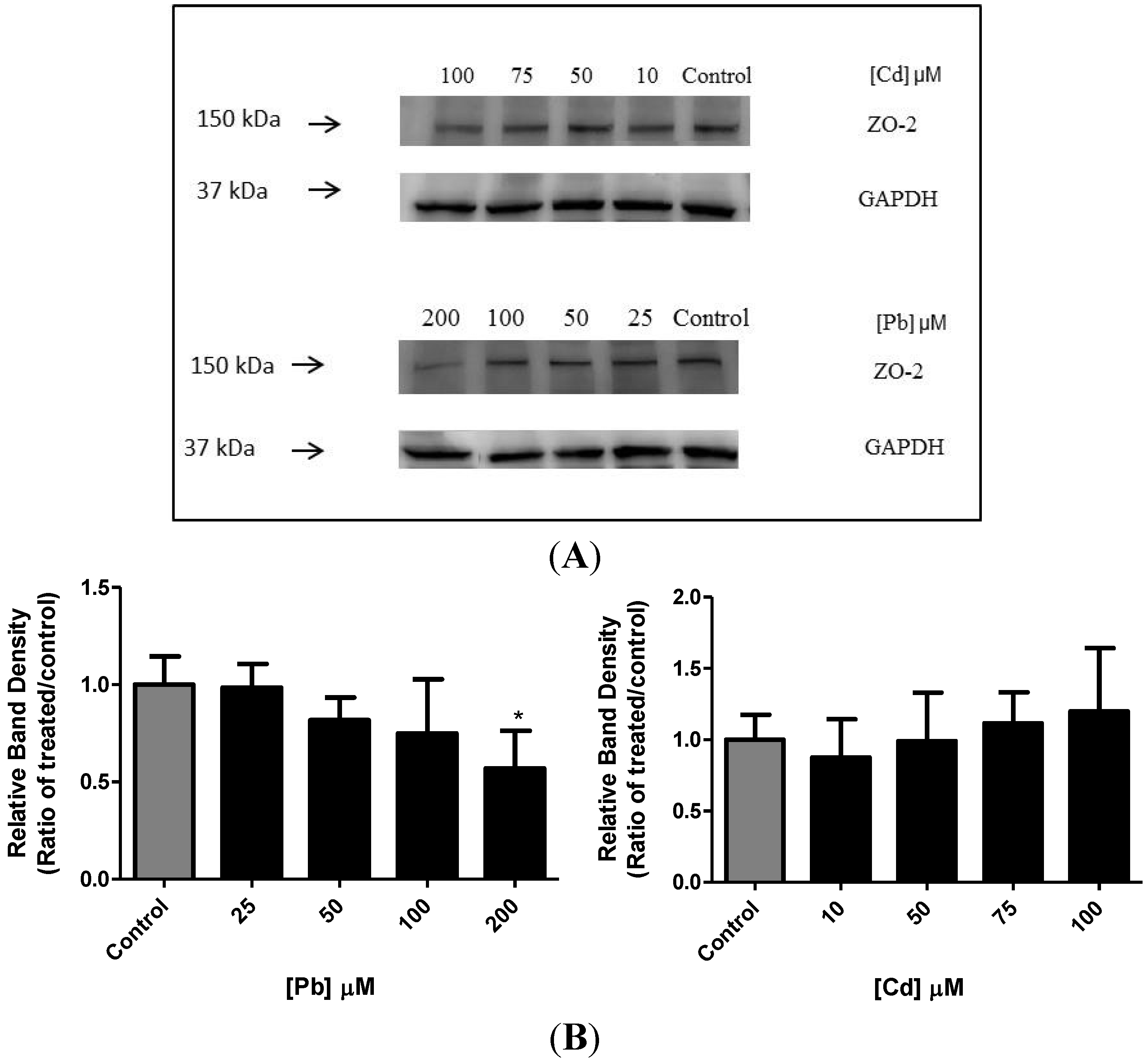
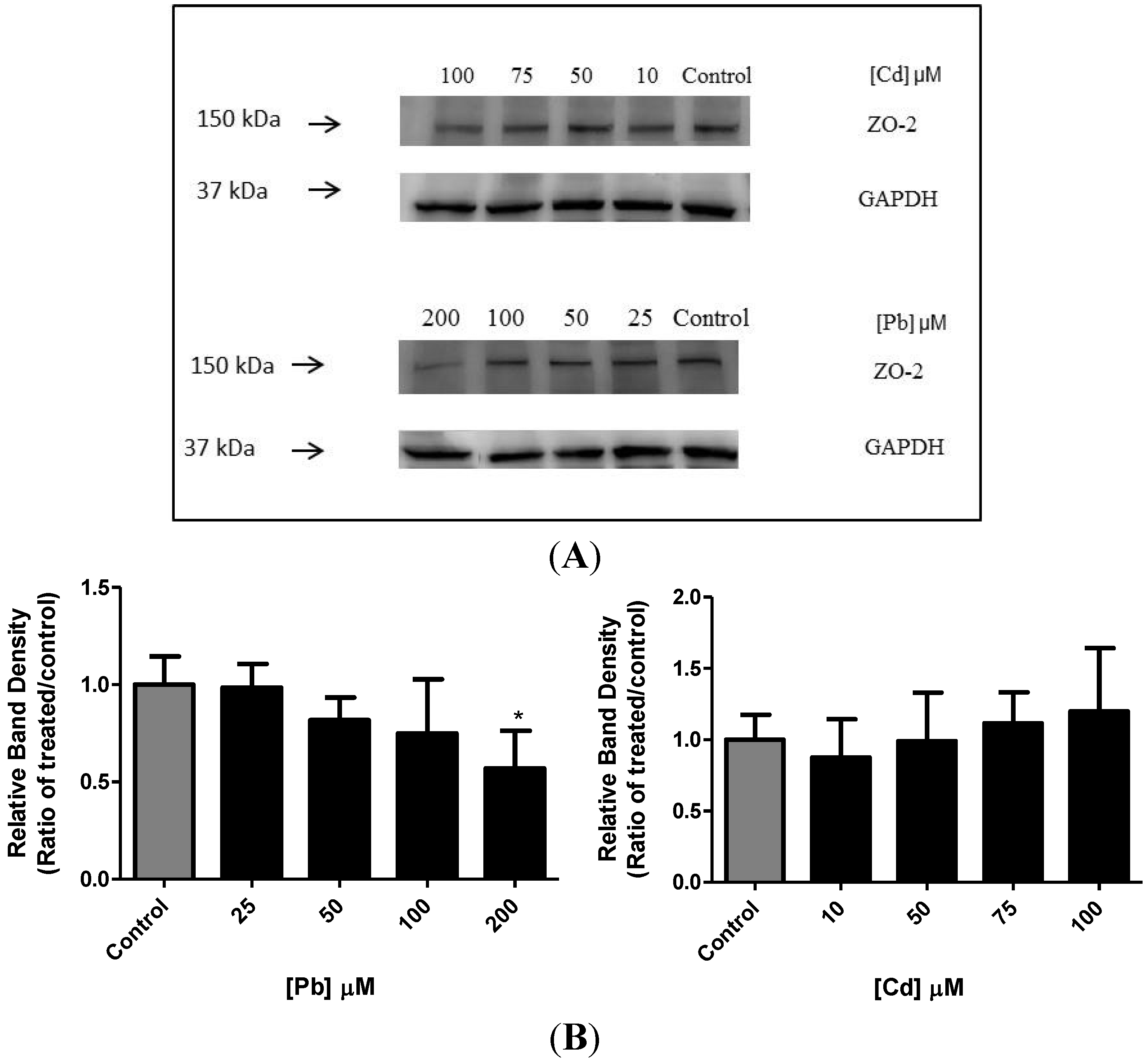
3.5. Effect of Pb or Cd on the Levels of Zona Occluden Protein (ZO-2)
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cory-Slechta, D.A. Relationships between lead-induced learning impairments and changes in dopaminergic, cholinergic, and glutamatergic neurotransmitter system functions. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 391–415. [Google Scholar] [CrossRef]
- Adonaylo, V.N.; Oteiza, P.I. Lead intoxication: Antioxidant defenses and oxidative damage in rat brain. Toxicology 1999, 135, 77–85. [Google Scholar] [CrossRef]
- Bressler, J.; Kim, K.A.; Chakraborti, T.; Goldstein, G. Molecular mechanisms of lead neurotoxicity. Neurochem. Res. 1999, 24, 595–600. [Google Scholar] [CrossRef]
- Toscano, C.D.; Guilarte, T.R. Lead neurotoxicity: From exposure to molecular effects. Brain Res. Brain Res. Rev. 2005, 49, 529–554. [Google Scholar] [CrossRef]
- Wang, B.; Du, Y. Cadmium and its neurotoxic effects. Oxid. Med. Cell Longev. 2013, 2013, 898034:1–898034:12. [Google Scholar]
- Cao, Y.; Chen, A.; Radcliffe, J.; Dietrich, K.N.; Jones, R.L.; Caldwell, K.; Rogan, W.J. Postnatal cadmium exposure, neurodevelopment, and blood pressure in children at 2, 5, and 7 years of age. Environ. Health Perspect. 2009, 117, 1580–1586. [Google Scholar] [CrossRef]
- Mendez-Armenta, M.; Rios, C. Cadmium neurotoxicity. Environ. Toxicol. Pharmacol. 2007, 23, 350–358. [Google Scholar] [CrossRef]
- Antonio, M.T.; Corredor, L.; Leret, M.L. Study of the activity of several brain enzymes like markers of the neurotoxicity induced by perinatal exposure to lead and/or cadmium. Toxicol. Lett. 2003, 143, 331–340. [Google Scholar] [CrossRef]
- Lafuente, A.; Esquifino, A.I. Cadmium effects on hypothalamic activity and pituitary hormone secretion in the male. Toxicol. Lett. 1999, 110, 209–218. [Google Scholar] [CrossRef]
- Esquifino, A.I.; Marquez, N.; Alvarez-Demanuel, E.; Lafuente, A. Effects of chronic alternating cadmium exposure on the episodic secretion of prolactin in male rats. J. Trace Elem. Med. Biol. 1999, 12, 205–210. [Google Scholar]
- King, L.M.; Banks, W.A.; George, W.J. Differences in cadmium transport to the testis, epididymis, and brain in cadmium-sensitive and -resistant murine strains 129/j and a/j. J. Pharmacol. Exp. Ther. 1999, 289, 825–830. [Google Scholar]
- King, L.M.; Banks, W.A.; George, W.J. Differential zinc transport into testis and brain of cadmium-sensitive and -resistant murine strains. J. Androl. 2000, 21, 656–663. [Google Scholar]
- Shukla, G.S.; Chandra, S.V. Concurrent exposure to lead, manganese, and cadmium and their distribution to various brain regions, liver, kidney, and testis of growing rats. Arch. Environ. Contam. Toxicol. 1987, 16, 303–310. [Google Scholar] [CrossRef]
- Goncalves, J.F.; Fiorenza, A.M.; Spanevello, R.M.; Mazzanti, C.M.; Bochi, G.V.; Antes, F.G.; Stefanello, N.; Rubin, M.A.; Dressler, V.L.; Morsch, V.M.; et al. N-acetylcysteine prevents memory deficits, the decrease in acetylcholinesterase activity and oxidative stress in rats exposed to cadmium. Chem. Biol. Interact. 2010, 186, 53–60. [Google Scholar] [CrossRef]
- Bradbury, M.W.; Deane, R. Permeability of the blood-brain barrier to lead. Neurotoxicology 1993, 14, 131–136. [Google Scholar]
- Savolainen, K.M.; Loikkanen, J.; Naarala, J. Amplification of glutamate-induced oxidative stress. Toxicol. Lett. 1995, 82–83, 399–405. [Google Scholar] [CrossRef]
- Gurer, H.; Ozgunes, H.; Oztezcan, S.; Ercal, N. Antioxidant role of alpha-lipoic acid in lead toxicity. Free Radic. Biol. Med. 1999, 27, 75–81. [Google Scholar] [CrossRef]
- Holtzman, D.; DeVries, C.; Nguyen, H.; Olson, J.; Bensch, K. Maturation of resistance to lead encephalopathy: Cellular and subcellular mechanisms. Neurotoxicology 1984, 5, 97–124. [Google Scholar]
- Qian, Y.; Falahatpisheh, M.H.; Zheng, Y.; Ramos, K.S.; Tiffany-Castiglioni, E. Induction of 78 kd glucose-regulated protein (grp78) expression and redox-regulated transcription factor activity by lead and mercury in c6 rat glioma cells. Neurotox. Res. 2001, 3, 581–589. [Google Scholar] [CrossRef]
- Hermes-Lima, M.; Pereira, B.; Bechara, E.J. Are free radicals involved in lead poisoning? Xenobiotica 1991, 21, 1085–1090. [Google Scholar] [CrossRef]
- Bray, T.M.; Taylor, C.G. Tissue glutathione, nutrition, and oxidative stress. Can. J. Physiol. Pharmacol. 1993, 71, 746–751. [Google Scholar] [CrossRef]
- Patra, R.C.; Rautray, A.K.; Swarup, D. Oxidative stress in lead and cadmium toxicity and its amelioration. Vet. Med. Int. 2011, 2011, 457327. [Google Scholar]
- Casalino, E.; Calzaretti, G.; Sblano, C.; Landriscina, C. Molecular inhibitory mechanisms of antioxidant enzymes in rat liver and kidney by cadmium. Toxicology 2002, 179, 37–50. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Flora, S.J.; Mittal, M.; Mehta, A. Heavy metal induced oxidative stress & its possible reversal by chelation therapy. Indian J. Med. Res. 2008, 128, 501–523. [Google Scholar]
- Monteiro, H.P.; Abdalla, D.S.; Faljoni-Alario, A.; Bechara, E.J. Generation of active oxygen species during coupled autoxidation of oxyhemoglobin and delta-aminolevulinic acid. Biochim. Biophys. Acta 1986, 881, 100–106. [Google Scholar] [CrossRef]
- Monteiro, H.P.; Abdalla, D.S.; Arcuri, A.S.; Bechara, E.J. Oxygen toxicity related to exposure to lead. Clin. Chem. 1985, 31, 1673–1676. [Google Scholar]
- Wang, H.; Joseph, J.A. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 1999, 27, 612–616. [Google Scholar] [CrossRef]
- Winters, R.A.; Zukowski, J.; Ercal, N.; Matthews, R.H.; Spitz, D.R. Analysis of glutathione, glutathione disulfide, cysteine, homocysteine, and other biological thiols by high-performance liquid chromatography following derivatization by n-(1-pyrenyl)maleimide. Anal.Biochem. 1995, 227, 14–21. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Monnet-Tschudi, F.; Zurich, M.G.; Boschat, C.; Corbaz, A.; Honegger, P. Involvement of environmental mercury and lead in the etiology of neurodegenerative diseases. Rev. Environ. Health 2006, 21, 105–117. [Google Scholar]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef]
- Jiang, L.F.; Yao, T.M.; Zhu, Z.L.; Wang, C.; Ji, L.N. Impacts of cd(ii) on the conformation and self-aggregation of Alzheimer’s tau fragment corresponding to the third repeat of microtubule-binding domain. Biochim. Biophys. Acta 2007, 1774, 1414–1421. [Google Scholar] [CrossRef]
- Okuda, B.; Iwamoto, Y.; Tachibana, H.; Sugita, M. Parkinsonism after acute cadmium poisoning. Clin. Neurol. Neurosurg. 1997, 99, 263–265. [Google Scholar] [CrossRef]
- Ercal, N.; Treeratphan, P.; Hammond, T.C.; Matthews, R.H.; Grannemann, N.H.; Spitz, D.R. In vivo indices of oxidative stress in lead-exposed c57bl/6 mice are reduced by treatment with meso-2,3-dimercaptosuccinic acid or n-acetylcysteine. Free Radic. Biol. Med. 1996, 21, 157–161. [Google Scholar] [CrossRef]
- Gurer, H.; Ozgunes, H.; Neal, R.; Spitz, D.R.; Ercal, N. Antioxidant effects of n-acetylcysteine and succimer in red blood cells from lead-exposed rats. Toxicology 1998, 128, 181–189. [Google Scholar] [CrossRef]
- Ribarov, S.R.; Bochev, P.G. Lead-hemoglobin interaction as a possible source of reactive oxygen species—A chemiluminescent study. Arch. Biochem. Biophys. 1982, 213, 288–292. [Google Scholar] [CrossRef]
- Sandhir, R.; Julka, D.; Gill, K.D. Lipoperoxidative damage on lead exposure in rat brain and its implications on membrane bound enzymes. Pharmacol.Toxicol. 1994, 74, 66–71. [Google Scholar] [CrossRef]
- Solliway, B.M.; Schaffer, A.; Pratt, H.; Yannai, S. Effects of exposure to lead on selected biochemical and haematological variables. Pharmacol. Toxicol. 1996, 78, 18–22. [Google Scholar] [CrossRef]
- Chen, L.; Liu, L.; Huang, S. Cadmium activates the mitogen-activated protein kinase (mapk) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2a and 5. Free Radic. Biol. Med. 2008, 45, 1035–1044. [Google Scholar] [CrossRef]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Kontos, C.D.; Huang, S. Cadmium induction of reactive oxygen species activates the mtor pathway, leading to neuronal cell death. Free Radic. Biol. Med. 2011, 50, 624–632. [Google Scholar] [CrossRef]
- Hermes-Lima, M.; Valle, V.G.; Vercesi, A.E.; Bechara, E.J. Damage to rat liver mitochondria promoted by delta-aminolevulinic acid-generated reactive oxygen species: Connections with acute intermittent porphyria and lead-poisoning. Biochim. Biophys. Acta 1991, 1056, 57–63. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Fuhr, B.J.; Rabenstein, D.L. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. Ix. The binding of cadmium, zinc, lead, and mercury by glutathione. J. Am. Chem. Soc. 1973, 95, 6944–6950. [Google Scholar] [CrossRef]
- Kamiyama, T.; Miyakawa, H.; Li, J.P.; Akiba, T.; Liu, J.H.; Liu, J.; Marumo, F.; Sato, C. Effects of one-year cadmium exposure on livers and kidneys and their relation to glutathione levels. Res. Commun. Mol. Pathol. Pharmacol. 1995, 88, 177–186. [Google Scholar]
- Karmakar, R.; Banik, S.; Bandyopadhyay, S.; Chatterjee, M. Cadmium-induced alterations of hepatic lipid peroxidation, glutathione S-transferase activity and reduced glutathione level and their possible correlation with chromosomal aberration in mice: A time course study. Mutat. Res. 1998, 397, 183–190. [Google Scholar]
- Hsu, J.M. Lead toxicity as related to glutathione metabolism. J. Nutr. 1981, 111, 26–33. [Google Scholar]
- Christie, N.T.; Costa, M. In vitro assessment of the toxicity of metal compounds: IV. Disposition of metals in cells: Interactions with membranes, glutathione, metallothionein, and DNA. Biol. Trace. Elem. Res. 1984, 6, 139–158. [Google Scholar] [CrossRef]
- Patra, R.C.; Swarup, D.; Dwivedi, S.K. Antioxidant effects of alpha tocopherol, ascorbic acid and L-methionine on lead induced oxidative stress to the liver, kidney and brain in rats. Toxicology 2001, 162, 81–88. [Google Scholar] [CrossRef]
- Gong, P.; Chen, F.X.; Ma, G.F.; Feng, Y.; Zhao, Q.; Wang, R. Endomorphin 1 effectively protects cadmium chloride-induced hepatic damage in mice. Toxicology 2008, 251, 35–44. [Google Scholar] [CrossRef]
- Chaurasia, S.S.; Kar, A. Protective effects of vitamin e against lead-induced deterioration of membrane associated type-i iodothyronine 5'-monodeiodinase (5'd-i) activity in male mice. Toxicology 1997, 124, 203–209. [Google Scholar] [CrossRef]
- Tandon, S.K.; Singh, S.; Prasad, S.; Srivastava, S.; Siddiqui, M.K. Reversal of lead-induced oxidative stress by chelating agent, antioxidant, or their combination in the rat. Environ. Res. 2002, 90, 61–66. [Google Scholar] [CrossRef]
- Waisberg, M.; Joseph, P.; Hale, B.; Beyersmann, D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 2003, 192, 95–117. [Google Scholar] [CrossRef]
- Plateel, M.; Dehouck, M.P.; Torpier, G.; Cecchelli, R.; Teissier, E. Hypoxia increases the susceptibility to oxidant stress and the permeability of the blood-brain barrier endothelial cell monolayer. J. Neurochem. 1995, 65, 2138–2145. [Google Scholar]
- Bar-Or, A.; Nuttall, R.K.; Duddy, M.; Alter, A.; Kim, H.J.; Ifergan, I.; Pennington, C.J.; Bourgoin, P.; Edwards, D.R.; Yong, V.W. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain 2003, 126, 2738–2749. [Google Scholar] [CrossRef]
- Balbuena, P.; Li, W.; Magnin-Bissel, G.; Meldrum, J.B.; Ehrich, M. Comparison of two blood-brain barrier in vitro systems: Cytotoxicity and transfer assessments of malathion/oxon and lead acetate. Toxicol. Sci. 2010, 114, 260–271. [Google Scholar] [CrossRef]
- Bressler, J.P.; Goldstein, G.W. Mechanisms of lead neurotoxicity. Biochem. Pharmacol. 1991, 41, 479–484. [Google Scholar] [CrossRef]
- Hossain, M.A.; Russell, J.C.; Miknyoczki, S.; Ruggeri, B.; Lal, B.; Laterra, J. Vascular endothelial growth factor mediates vasogenic edema in acute lead encephalopathy. Ann. Neurol. 2004, 55, 660–667. [Google Scholar] [CrossRef]
- Rohrer, S.R.; Shaw, S.M.; Lamar, C.H. Cadmium induced endothelial cell alterations in the fetal brain from prenatal exposure. Acta Neuropathol. 1978, 44, 147–149. [Google Scholar] [CrossRef]
- Shukla, A.; Shukla, G.S.; Srimal, R.C. Cadmium-induced alterations in blood-brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum. Exp. Toxicol. 1996, 15, 400–405. [Google Scholar] [CrossRef]
- Shi, L.Z.; Zheng, W. Early lead exposure increases the leakage of the blood-cerebrospinal fluid barrier, in vitro. Hum. Exp. Toxicol. 2007, 26, 159–167. [Google Scholar] [CrossRef]
- Dyatlov, V.A.; Platoshin, A.V.; Lawrence, D.A.; Carpenter, D.O. Lead potentiates cytokine- and glutamate-mediated increases in permeability of the blood-brain barrier. Neurotoxicology 1998, 19, 283–291. [Google Scholar]
- Balda, M.S.; Flores-Maldonado, C.; Cereijido, M.; Matter, K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J. Cell. Biochem. 2000, 78, 85–96. [Google Scholar] [CrossRef]
- Persidsky, Y.; Heilman, D.; Haorah, J.; Zelivyanskaya, M.; Persidsky, R.; Weber, G.A.; Shimokawa, H.; Kaibuchi, K.; Ikezu, T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in hiv-1 encephalitis (hive). Blood 2006, 107, 4770–4780. [Google Scholar] [CrossRef]
- Collins, N.T.; Cummins, P.M.; Colgan, O.C.; Ferguson, G.; Birney, Y.A.; Murphy, R.P.; Meade, G.; Cahill, P.A. Cyclic strain-mediated regulation of vascular endothelial occludin and zo-1: Influence on intercellular tight junction assembly and function. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 62–68. [Google Scholar] [CrossRef]
- Balbuena, P.; Li, W.; Ehrich, M. Assessments of tight junction proteins occludin, claudin 5 and scaffold proteins zo1 and zo2 in endothelial cells of the rat blood-brain barrier: Cellular responses to neurotoxicants malathion and lead acetate. Neurotoxicology 2011, 32, 58–67. [Google Scholar] [CrossRef]
- Wang, W.; Duan, B.; Xu, H.; Xu, L.; Xu, T.L. Calcium-permeable acid-sensing ion channel is a molecular target of the neurotoxic metal ion lead. J. Biol. Chem. 2006, 281, 2497–2505. [Google Scholar] [CrossRef]
- Prozialeck, W.C. Evidence that e-cadherin may be a target for cadmium toxicity in epithelial cells. Toxicol. Appl. Pharmacol. 2000, 164, 231–249. [Google Scholar] [CrossRef]
- Prozialeck, W.C.; Grunwald, G.B.; Dey, P.M.; Reuhl, K.R.; Parrish, A.R. Cadherins and ncam as potential targets in metal toxicity. Toxicol. Appl. Pharmacol. 2002, 182, 255–265. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tobwala, S.; Wang, H.-J.; Carey, J.W.; Banks, W.A.; Ercal, N. Effects of Lead and Cadmium on Brain Endothelial Cell Survival, Monolayer Permeability, and Crucial Oxidative Stress Markers in an in Vitro Model of the Blood-Brain Barrier. Toxics 2014, 2, 258-275. https://doi.org/10.3390/toxics2020258
Tobwala S, Wang H-J, Carey JW, Banks WA, Ercal N. Effects of Lead and Cadmium on Brain Endothelial Cell Survival, Monolayer Permeability, and Crucial Oxidative Stress Markers in an in Vitro Model of the Blood-Brain Barrier. Toxics. 2014; 2(2):258-275. https://doi.org/10.3390/toxics2020258
Chicago/Turabian StyleTobwala, Shakila, Hsiu-Jen Wang, Joshua Warren Carey, William A. Banks, and Nuran Ercal. 2014. "Effects of Lead and Cadmium on Brain Endothelial Cell Survival, Monolayer Permeability, and Crucial Oxidative Stress Markers in an in Vitro Model of the Blood-Brain Barrier" Toxics 2, no. 2: 258-275. https://doi.org/10.3390/toxics2020258