


Investigation of the Association between Air Pollution and Non-Alcoholic Fatty Liver Disease in the European Population: A Mendelian Randomization Study

Abstract
:1. Introduction
2. Materials and Methods
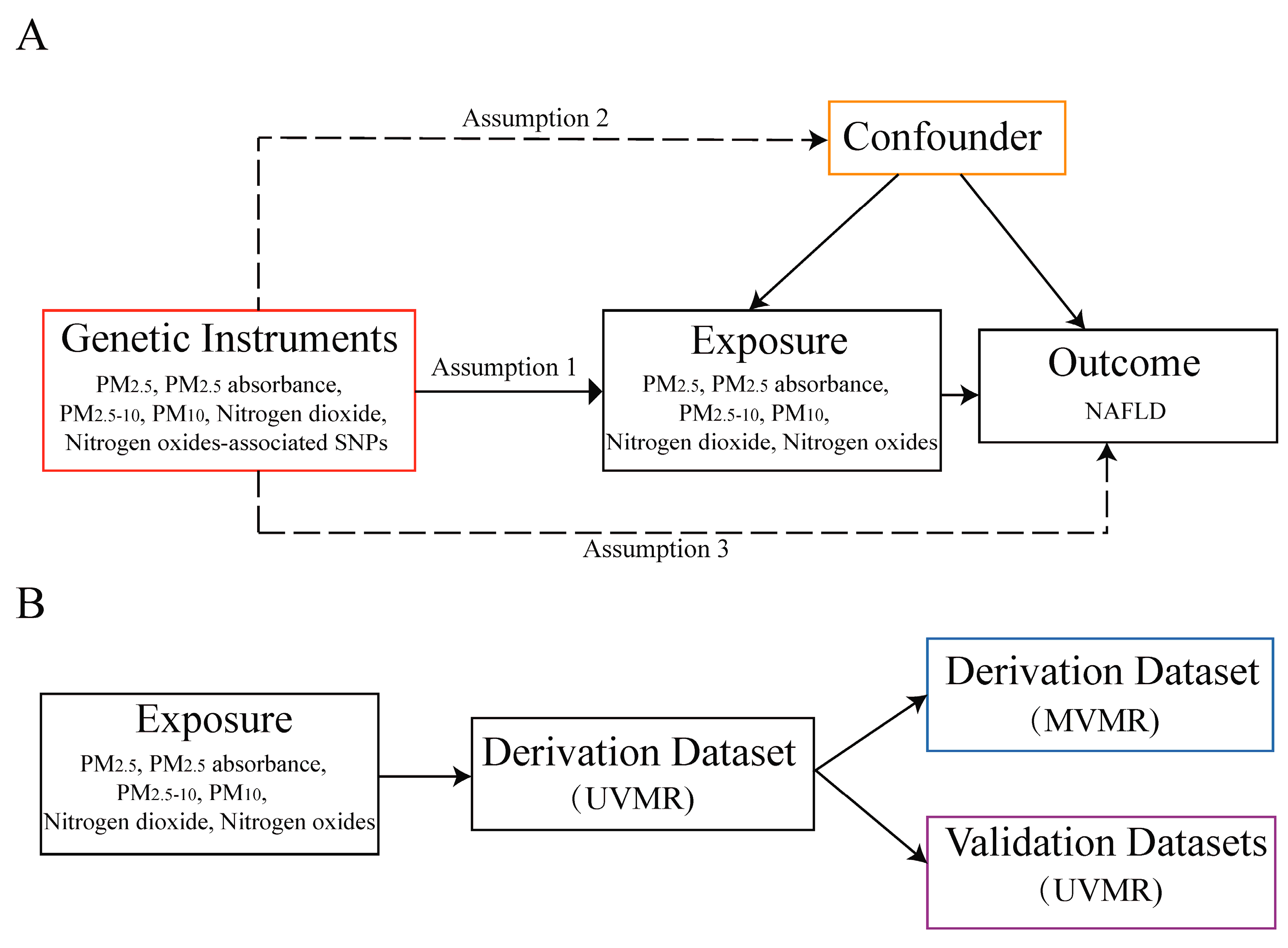
2.1. Study Design
2.2. Data Sources
2.3. Selection of Instrumental Variables
2.4. Mendelian Randomization Analysis
2.5. Sensitivity Analysis
2.6. Statistical Analysis
3. Results
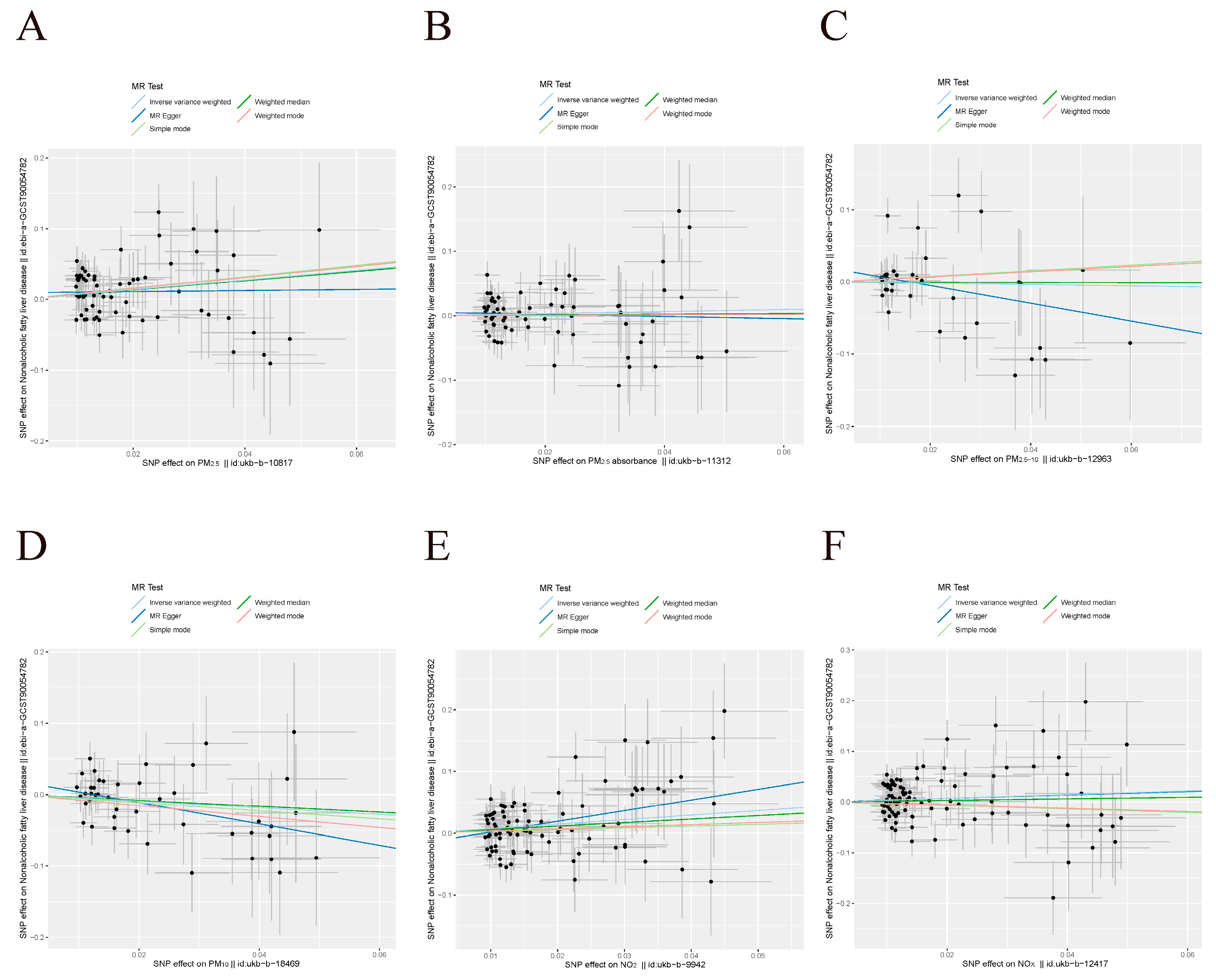
3.1. UVMR Results in the Derivation Dataset
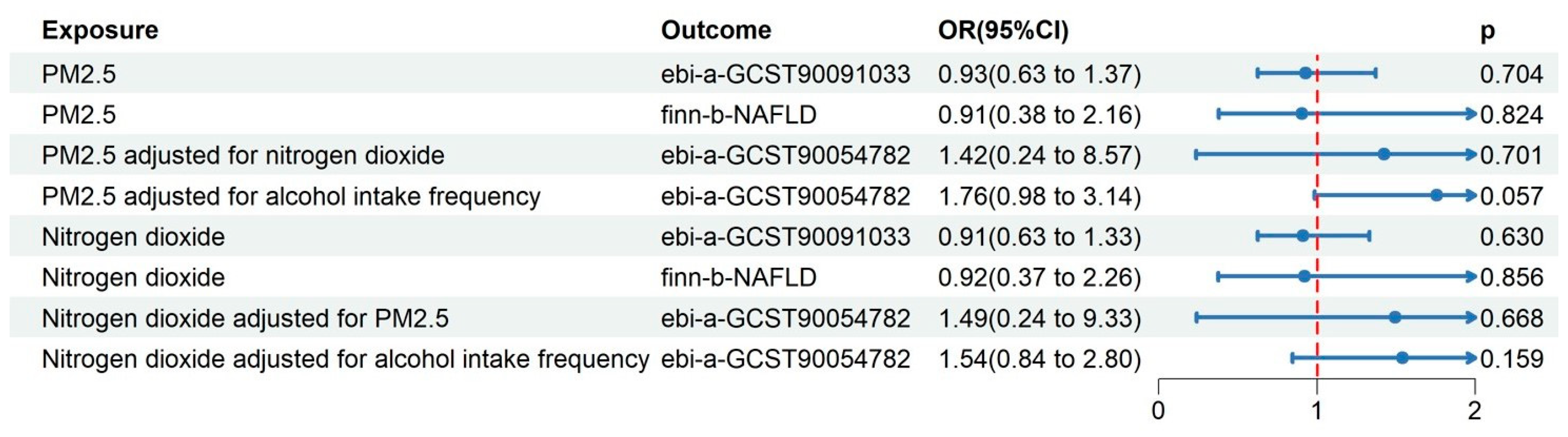
3.2. MVMR Results in the Derivation Dataset
3.3. UVMR Results in the Validation Datasets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PM | particulate matter |
| NO2 | nitrogen dioxide |
| NOx | nitrogen oxides |
| NAFLD | non-alcoholic fatty liver disease |
| MAFLD | metabolic-associated fatty liver disease |
| MR | Mendelian randomization |
| UVMR | univariable Mendelian randomization |
| MVMR | multivariable Mendelian randomization |
| GWAS | Genome-Wide Association Study |
| SNPs | single nucleotide polymorphisms |
| IVs | instrumental variables |
| OR | odds ratio |
| CI | confidence interval |
| UKB | UK Biobank |
| MRC-IEU | UK Medical Research Council Integrative Epidemiology Unit |
| EGCUT | Estonian Genome Center of the University of Tartu |
| STROBE-MR | Strengthening observational studies using Mendelian randomization |
| MR-RAPS | MR-Robust Adjusted Profile Score |
| MR-PRESSO | MR-Pleiotropy RESidual Sum and Outlier |
| mre-IVW/IVW-mre | multiplicative random-effects inverse variance weighting |
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Scorletti, E.; Mosca, A.; Alisi, A.; Byrne, C.D.; Targher, G. Complications, Morbidity and Mortality of Nonalcoholic Fatty Liver Disease. Metab. Clin. Exp. 2020, 111, 154170. [Google Scholar] [CrossRef]
- Targher, G.; Corey, K.E.; Byrne, C.D.; Roden, M. The Complex Link between NAFLD and Type 2 Diabetes Mellitus—Mechanisms and Treatments. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Cazac, G.; Lăcătușu, C.-M.; Mihai, C.; Grigorescu, E.-D.; Onofriescu, A.; Mihai, B. New Insights into Non-Alcoholic Fatty Liver Disease and Coronary Artery Disease: The Liver-Heart Axis. Life 2022, 12, 1189. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, Y.-J.; Park, K.W.; Hwang, Y.S.; Lee, S.H.; Kim, B.J.; Chung, S.J. Association of NO2 and Other Air Pollution Exposures With the Risk of Parkinson Disease. JAMA Neurol. 2021, 78, 800–808. [Google Scholar] [CrossRef]
- Jones, R.R.; Fisher, J.A.; Hasheminassab, S.; Kaufman, J.D.; Freedman, N.D.; Ward, M.H.; Sioutas, C.; Vermeulen, R.; Hoek, G.; Silverman, D.T. Outdoor Ultrafine Particulate Matter and Risk of Lung Cancer in Southern California. Am. J. Respir. Crit. Care Med. 2024, 209, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Dan, L.; Sun, Y.; Yuan, S.; Liu, W.; Chen, X.; Jiang, F.; Fu, T.; Zhang, H.; Deng, M.; et al. Ambient Air Pollution and Risk of Enterotomy, Gastrointestinal Cancer, and All-Cause Mortality among 4,708 Individuals with Inflammatory Bowel Disease: A Prospective Cohort Study. Environ. Health Perspect. 2023, 131, 77010. [Google Scholar] [CrossRef]
- Kentros, P.A.; Huang, Y.; Wylie, B.J.; Khoury-Collado, F.; Hou, J.Y.; de Meritens, A.B.; St Clair, C.M.; Hershman, D.L.; Wright, J.D. Ambient Particulate Matter Air Pollution Exposure and Ovarian Cancer Incidence in the USA: An Ecological Study. BJOG 2023, 131, 690–698. [Google Scholar] [CrossRef]
- Li, Y.; Fan, Z.; Lu, W.; Xu, R.; Liu, T.; Liu, L.; Chen, G.; Lv, Z.; Huang, S.; Zhou, Y.; et al. Long-Term Exposure to Ambient Fine Particulate Matter-Bound Polycyclic Aromatic Hydrocarbons and Cancer Mortality: A Difference-in-Differences Approach. Chemosphere 2023, 340, 139800. [Google Scholar] [CrossRef]
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-Year Trends of the Global Burden of Disease Attributable to Ambient Air Pollution: An Analysis of Data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef]
- Zhao, N.; Wu, W.; Feng, Y.; Yang, F.; Han, T.; Guo, M.; Ren, Q.; Li, W.; Li, J.; Wang, S.; et al. Polymorphisms in Oxidative Stress, Metabolic Detoxification, and Immune Function Genes, Maternal Exposure to Ambient Air Pollution, and Risk of Preterm Birth in Taiyuan, China. Environ. Res. 2021, 194, 110659. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.; Hong, Y.-C. Modification Effect of PARP4 and ERCC1 Gene Polymorphisms on the Relationship between Particulate Matter Exposure and Fasting Glucose Level. Int. J. Environ. Res. Public. Health 2022, 19, 6241. [Google Scholar] [CrossRef]
- Yang, S.-I.; Kim, B.-J.; Lee, S.-Y.; Kim, H.-B.; Lee, C.M.; Yu, J.; Kang, M.-J.; Yu, H.-S.; Lee, E.; Jung, Y.-H.; et al. Prenatal Particulate Matter/Tobacco Smoke Increases Infants’ Respiratory Infections: COCOA Study. Allergy Asthma Immunol. Res. 2015, 7, 573–582. [Google Scholar] [CrossRef]
- Emdin, C.A.; Khera, A.V.; Kathiresan, S. Mendelian Randomization. JAMA 2017, 318, 1925–1926. [Google Scholar] [CrossRef]
- Li, F.-R.; Liao, J.; Zhu, B.; Li, X.; Cheng, Z.; Jin, C.; Mo, C.; Wu, X.; Li, Q.; Liang, F. Long-Term Exposure to Air Pollution and Incident Non-Alcoholic Fatty Liver Disease and Cirrhosis: A Cohort Study. Liver Int. 2023, 43, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Timpson, N.J.; Higgins, J.P.T.; Dimou, N.; Langenberg, C.; et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomisation (STROBE-MR): Explanation and Elaboration. BMJ 2021, 375, n2233. [Google Scholar] [CrossRef] [PubMed]
- Eeftens, M.; Beelen, R.; de Hoogh, K.; Bellander, T.; Cesaroni, G.; Cirach, M.; Declercq, C.; Dėdelė, A.; Dons, E.; de Nazelle, A.; et al. Development of Land Use Regression Models for PM(2.5), PM(2.5) Absorbance, PM(10) and PM(Coarse) in 20 European Study Areas; Results of the ESCAPE Project. Environ. Sci. Technol. 2012, 46, 11195–11205. [Google Scholar] [CrossRef]
- Fairfield, C.J.; Drake, T.M.; Pius, R.; Bretherick, A.D.; Campbell, A.; Clark, D.W.; Fallowfield, J.A.; Hayward, C.; Henderson, N.C.; Joshi, P.K.; et al. Genome-Wide Association Study of NAFLD Using Electronic Health Records. Hepatol. Commun. 2022, 6, 297–308. [Google Scholar] [CrossRef]
- Ghodsian, N.; Abner, E.; Emdin, C.A.; Gobeil, É.; Taba, N.; Haas, M.E.; Perrot, N.; Manikpurage, H.D.; Gagnon, É.; Bourgault, J.; et al. Electronic Health Record-Based Genome-Wide Meta-Analysis Provides Insights on the Genetic Architecture of Non-Alcoholic Fatty Liver Disease. Cell Rep. Med. 2021, 2, 100437. [Google Scholar] [CrossRef] [PubMed]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen Provides Genetic Insights from a Well-Phenotyped Isolated Population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian Randomization Analysis with Multiple Genetic Variants Using Summarized Data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef]
- Hemani, G.; Tilling, K.; Davey Smith, G. Orienting the Causal Relationship between Imprecisely Measured Traits Using GWAS Summary Data. PLoS Genet. 2017, 13, e1007081. [Google Scholar] [CrossRef]
- Thompson, S.B.; Simon, G. Mendelian Randomization: Methods for Causal Inference Using Genetic Variants, 2nd ed.; Chapman and Hall/CRC: New York, NY, USA, 2021; ISBN 978-0-429-32435-2. [Google Scholar]
- Slob, E.A.W.; Burgess, S. A Comparison of Robust Mendelian Randomization Methods Using Summary Data. Genet. Epidemiol. 2020, 44, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Brion, M.-J.A.; Shakhbazov, K.; Visscher, P.M. Calculating Statistical Power in Mendelian Randomization Studies. Int. J. Epidemiol. 2013, 42, 1497–1501. [Google Scholar] [CrossRef]
- Greco, M.F.D.; Minelli, C.; Sheehan, N.A.; Thompson, J.R. Detecting Pleiotropy in Mendelian Randomisation Studies with Summary Data and a Continuous Outcome. Stat. Med. 2015, 34, 2926–2940. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Chen, C.-Y.; Neale, B.; Do, R. Detection of Widespread Horizontal Pleiotropy in Causal Relationships Inferred from Mendelian Randomization between Complex Traits and Diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Bowden, J.; Fall, T.; Ingelsson, E.; Thompson, S.G. Sensitivity Analyses for Robust Causal Inference from Mendelian Randomization Analyses with Multiple Genetic Variants. Epidemiology 2017, 28, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Thompson, S.G. Interpreting Findings from Mendelian Randomization Using the MR-Egger Method. Eur. J. Epidemiol. 2017, 32, 377–389. [Google Scholar] [CrossRef]
- Gelbard, R.B.; Cripps, M.W. Pitfalls in Study Interpretation. Surg. Infect. (Larchmt) 2021, 22, 646–650. [Google Scholar] [CrossRef]
- Xing, Y.; Gao, X.; Li, Q.; Li, X.; Wang, Y.; Yang, Y.; Yang, S.; Lau, P.W.C.; Zeng, Q.; Wang, H. Associations between Exposure to Ambient Particulate Matter and Advanced Liver Fibrosis in Chinese MAFLD Patients. J. Hazard. Mater. 2023, 460, 132501. [Google Scholar] [CrossRef]
- Guo, B.; Guo, Y.; Nima, Q.; Feng, Y.; Wang, Z.; Lu, R.; Baimayangji; Ma, Y.; Zhou, J.; Xu, H.; et al. Exposure to Air Pollution Is Associated with an Increased Risk of Metabolic Dysfunction-Associated Fatty Liver Disease. J. Hepatol. 2022, 76, 518–525. [Google Scholar] [CrossRef]
- Sun, S.; Yang, Q.; Zhou, Q.; Cao, W.; Yu, S.; Zhan, S.; Sun, F. Long-Term Exposure to Air Pollution, Habitual Physical Activity and Risk of Non-Alcoholic Fatty Liver Disease: A Prospective Cohort Study. Ecotoxicol. Environ. Saf. 2022, 235, 113440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, C.; Chang, L.-Y.; Bo, Y.; Lin, C.; Tam, T.; Hoek, G.; Wong, M.C.; Chan, T.-C.; Lau, A.K.; et al. Long-Term Exposure to Ambient Fine Particulate Matter and Liver Enzymes in Adults: A Cross-Sectional Study in Taiwan. Occup. Environ. Med. 2019, 76, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Min, J.-Y.; Seo, Y.-S.; Min, K.-B. Association of Ambient Air Pollution with Increased Liver Enzymes in Korean Adults. Int. J. Environ. Res. Public. Health 2019, 16, 1213. [Google Scholar] [CrossRef]
- Li, W.; Dorans, K.S.; Wilker, E.H.; Rice, M.B.; Long, M.T.; Schwartz, J.; Coull, B.A.; Koutrakis, P.; Gold, D.R.; Fox, C.S.; et al. Residential Proximity to Major Roadways, Fine Particulate Matter, and Hepatic Steatosis. Am. J. Epidemiol. 2017, 186, 857–865. [Google Scholar] [CrossRef]
- Matthiessen, C.; Glaubitz, L.; Lucht, S.; Kälsch, J.; Luedde, T.; Erbel, R.; Stang, A.; Schmidt, B.; Friedman, S.L.; Canbay, A.; et al. Long-Term Exposure to Air Pollution and Prevalent Nonalcoholic Fatty Liver Disease. Environ. Epidemiol. 2023, 7, e268. [Google Scholar] [CrossRef]
- Liang, X.; Liang, L.; Fan, Y. Two-Sample Mendelian Randomization Analysis Investigates Ambient Fine Particulate Matter’s Impact on Cardiovascular Disease Development. Sci. Rep. 2023, 13, 20129. [Google Scholar] [CrossRef]
- Schneider, L.J.; Santiago, I.; Johnson, B.; Stanley, A.H.; Penaredondo, B.; Lund, A.K. Histological Features of Non-Alcoholic Fatty Liver Disease Revealed in Response to Mixed Vehicle Emission Exposure and Consumption of a High-Fat Diet in Wildtype C57Bl/6 Male Mice. Ecotoxicol. Environ. Saf. 2023, 261, 115094. [Google Scholar] [CrossRef]
- Ding, D.; Ye, G.; Lin, Y.; Lu, Y.; Zhang, H.; Zhang, X.; Hong, Z.; Huang, Q.; Chi, Y.; Chen, J.; et al. MicroRNA-26a-CD36 Signaling Pathway: Pivotal Role in Lipid Accumulation in Hepatocytes Induced by PM2.5 Liposoluble Extracts. Environ. Pollut. 2019, 248, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.-S.; Rinella, M. Non-Alcoholic Fatty Liver Disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Luci, C.; Bourinet, M.; Leclère, P.S.; Anty, R.; Gual, P. Chronic Inflammation in Non-Alcoholic Steatohepatitis: Molecular Mechanisms and Therapeutic Strategies. Front. Endocrinol. 2020, 11, 597648. [Google Scholar] [CrossRef]
- Monserrat-Mesquida, M.; Quetglas-Llabrés, M.; Abbate, M.; Montemayor, S.; Mascaró, C.M.; Casares, M.; Tejada, S.; Abete, I.; Zulet, M.A.; Tur, J.A.; et al. Oxidative Stress and Pro-Inflammatory Status in Patients with Non-Alcoholic Fatty Liver Disease. Antioxidants 2020, 9, 759. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-X.; Ge, C.-X.; Qin, Y.-T.; Gu, T.-T.; Lou, D.-S.; Li, Q.; Hu, L.-F.; Feng, J.; Huang, P.; Tan, J. Prolonged PM2.5 Exposure Elevates Risk of Oxidative Stress-Driven Nonalcoholic Fatty Liver Disease by Triggering Increase of Dyslipidemia. Free Radic. Biol. Med. 2019, 130, 542–556. [Google Scholar] [CrossRef]
- Li, D.; Ji, S.; Guo, Y.; Sang, N. Ambient NO2 Exposure Sex-Specifically Impairs Myelin and Contributes to Anxiety and Depression-like Behaviors of C57BL/6J Mice. J. Hazard. Mater. 2021, 416, 125836. [Google Scholar] [CrossRef]
- Liu, C.-X.; Liu, Y.-B.; Peng, Y.; Peng, J.; Ma, Q.-L. Causal Effect of Air Pollution on the Risk of Cardiovascular and Metabolic Diseases and Potential Mediation by Gut Microbiota. Sci. Total Environ. 2023, 912, 169418. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, W. Causal Effects of Exposure to Ambient Air Pollution on Cancer Risk: Insights from Genetic Evidence. Sci. Total Environ. 2023, 912, 168843. [Google Scholar] [CrossRef]
- Ning, P.; Guo, X.; Qu, Q.; Li, R. Exploring the Association between Air Pollution and Parkinson’s Disease or Alzheimer’s Disease: A Mendelian Randomization Study. Environ. Sci. Pollut. Res. Int. 2023, 30, 123939–123947. [Google Scholar] [CrossRef]
- Qiu, S.; Hu, Y.; Liu, G. Mendelian Randomization Study Supports the Causal Effects of Air Pollution on Longevity via Multiple Age-Related Diseases. npj Aging 2023, 9, 29. [Google Scholar] [CrossRef]
- Au Yeung, S.L.; Gill, D. Concerns over Using the Mendelian Randomization Design to Investigate the Effect of Air Pollution. Sci. Total Environ. 2024, 917, 170474. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exposure/Outcome | Dataset | Sample Size or Case/Control | NSNP | Unit | Population | Consortium/Cohort | Year | |
|---|---|---|---|---|---|---|---|---|
| GWAS ID | PMID | |||||||
| Exposure | ||||||||
| PM2.5 | ukb-b-10817 | —— | 423,796 | 9,851,867 | SD | European | MRC-IEU | 2018 |
| PM2.5 absorbance | ukb-b-11312 | —— | 423,796 | 9,851,867 | SD | European | MRC-IEU | 2018 |
| PM2.5–10 | ukb-b-12963 | —— | 423,796 | 9,851,867 | SD | European | MRC-IEU | 2018 |
| PM10 | ukb-b-18469 | —— | 423,796 | 9,851,867 | SD | European | MRC-IEU | 2018 |
| NO2 | ukb-b-9942 | —— | 456,380 | 9,851,867 | SD | European | MRC-IEU | 2018 |
| NOx | ukb-b-12417 | —— | 456,380 | 9,851,867 | SD | European | MRC-IEU | 2018 |
| Alcohol intake frequency | ukb-b-5779 | —— | 462,346 | 9,851,867 | SD | European | MRC-IEU | 2018 |
| Outcome | ||||||||
| NAFLD | ebi-a-GCST90054782 | 34535985 | 4,761/373,227 | 9,097,254 | Event | European | UK Biobank | 2021 |
| NAFLD | ebi-a-GCST90091033 | 34841290 | 8,434/770,180 | 6,784,388 | Event | European | eMERGE, UK Biobank, FinnGen and Estonian Biobank | 2021 |
| NAFLD | finn-b-NAFLD | —— | 894/217,898 | 16,380,466 | Event | European | FinnGen | 2021 |
| Exposure | Method | OR (95% CI) | p | NSNP | F Statistic Median (Min, Max) | p (Cochran’s Q Heterogeneity Test) | p (MR-Egger Intercept Test) | p (MR-PRESSO Global Test) |
|---|---|---|---|---|---|---|---|---|
| PM2.5 | IVW | 2.88 (0.73–11.33) | 0.129 | 13 | 26.0 (24.1, 30.1) | 0.027 | 0.504 | 0.030 |
| MR Egger | 9.33 (0.25–347.19) | 0.251 | ||||||
| Weighted median | 4.83 (1.03–22.65) | 0.046 | ||||||
| IVW-mre | 2.88 (0.73–11.33) | 0.129 | ||||||
| MR-RAPS | 3.11 (1.12–8.60) | 0.029 | ||||||
| PM2.5 (Outlier-corrected) | IVW | 4.26 (1.24–14.64) | 0.021 | 12 | 25.9 (24.1, 30.1) | 0.147 | 0.783 | 0.173 |
| MR Egger | 6.53 (0.26–163.21) | 0.280 | ||||||
| Weighted median | 5.59 (1.27–24.63) | 0.023 | ||||||
| IVW-mre | 4.26 (1.24–14.64) | 0.021 | ||||||
| MR-RAPS | 4.59 (1.56–13.44) | 0.006 |
| Exposure | Method | OR (95% CI) | p | NSNP | F Statistic Median (Min, Max) | p (Cochran’s Q Heterogeneity Test) | p (MR-Egger Intercept Test) | p (MR-PRESSO Global Test) |
|---|---|---|---|---|---|---|---|---|
| PM2.5 | IVW | 1.99 (1.22–3.22) | 0.005 | 72 | 21.1 (19.6, 30.1) | 0.321 | 0.317 | 0.313 |
| MR Egger | 1.08 (0.30–3.88) | 0.908 | ||||||
| Weighted median | 1.94 (0.98–3.80) | 0.055 | ||||||
| IVW-mre | 1.99 (1.22–3.22) | 0.005 | ||||||
| MR-RAPS | 2.06 (1.26–3.35) | 0.004 | ||||||
| NO2 | IVW | 2.08 (1.27–3.40) | 0.004 | 89 | 22.1 (19.5, 37.8) | 0.028 | 0.099 | 0.031 |
| MR Egger | 5.63 (1.58–20.07) | 0.009 | ||||||
| Weighted median | 1.76 (0.92–3.36) | 0.085 | ||||||
| IVW-mre | 2.08 (1.27–3.40) | 0.004 | ||||||
| MR-RAPS | 2.17 (1.39–3.40) | 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Zhang, Y.; Yuan, Y.; Xie, Z.; Li, L. Investigation of the Association between Air Pollution and Non-Alcoholic Fatty Liver Disease in the European Population: A Mendelian Randomization Study. Toxics 2024, 12, 228. https://doi.org/10.3390/toxics12030228
Yang J, Zhang Y, Yuan Y, Xie Z, Li L. Investigation of the Association between Air Pollution and Non-Alcoholic Fatty Liver Disease in the European Population: A Mendelian Randomization Study. Toxics. 2024; 12(3):228. https://doi.org/10.3390/toxics12030228
Chicago/Turabian StyleYang, Jing, Yaqi Zhang, Yin Yuan, Zhongyang Xie, and Lanjuan Li. 2024. "Investigation of the Association between Air Pollution and Non-Alcoholic Fatty Liver Disease in the European Population: A Mendelian Randomization Study" Toxics 12, no. 3: 228. https://doi.org/10.3390/toxics12030228