
Identification and Mechanistic Analysis of Toxic Degradation Products in the Advanced Oxidation Pathways of Fluoroquinolone Antibiotics


Abstract
:1. Introduction
2. Materials and Methods
2.1. Source of FQ Molecular Structure—PubChem Retrieval Method
2.2. Characterization of the Degradation Capability of FQ Molecules in Advanced Oxidation Systems—DFT Coupled with Negative Index Calculation Method
2.3. Construction of 3D-QSAR Models for the Degradation Capability of FQ Molecules in Advanced Oxidation Systems—SYBYL Software Method
2.4. Construction of Lasso Regression Model for the Degradation Capability of FQ Molecules in Advanced Oxidation Systems—Jupyter Notebook Tool
2.5. Toxicity Risk Assessment of Parent FQs and Their Degradation Products in Advanced Oxidation Systems—VEGA Software Method
3. Results and Discussion
3.1. Evaluation of the Degradation Capability of FQ Molecules in Advanced Oxidation Systems Based on DFT Method
3.2. Cluster-Analysis-Based Characterization of FQ Molecules’ Degradation Features in Advanced Oxidation Systems
3.3. Differential Analysis of the Degradation Capability of FQ Molecules in Advanced Oxidation Systems Based on 3D-QSAR Models
3.4. Toxicity Risk Assessment of Degradation Products of FQ Molecules in Advanced Oxidation Systems Based on Toxicokinetic Models
3.4.1. Human Health Risk Assessment of Degradation Products of FQ Molecules in Advanced Oxidation Systems
3.4.2. Ecological Environmental Risk Assessment of Degradation Products of FQ Molecules in Advanced Oxidation Systems
3.5. Mechanistic Analysis of the Differential Degradation Capability of FQ Molecules in Advanced Oxidation Systems
3.5.1. Differential Analysis of the Degradation Capability of FQ Molecules in Advanced Oxidation Systems Based on 2D-QSAR Model
3.5.2. Mechanistic Analysis of the Differential Degradation Capability of FQ Molecules in Advanced Oxidation Systems Based on Fukui Function
3.5.3. Mechanistic Analysis of the Differential Degradation Capability of FQ Molecules in Advanced Oxidation Systems Based on Fukui Function
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, S.; Wu, Y.; Zheng, H.; Li, H.; Zheng, Y.; Nan, J.; Chang, J.S. Antibiotics degradation by advanced oxidation process (AOPs): Recent advances in ecotoxicity and antibiotic-resistance genes induction of degradation products. Chemosphere 2023, 311, 136977. [Google Scholar] [CrossRef] [PubMed]
- Focazio, M.J.; Kolpin, D.W.; Barnes, K.K.; Furlong, E.T.; Meyer, M.T.; Zaugg, S.D.; Thurman, M.E. A national reconnaissance for pharmaceuticals and other organic wastewater contaminants in the United States—II) Untreated drinking water sources. Sci. Total Environ. 2008, 402, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Tello, A.; Austin, B.; Telfer, T.C. Selective pressure of antibiotic pollution on bacteria of importance to public health. Environ. Health Perspect. 2012, 120, 1100–1106. [Google Scholar] [CrossRef]
- Rosal, R.; Rodríguez, A.; Perdigón-Melón, J.A.; Petre, A.; García-Calvo, E.; Gómez, M.J.; Fernández-Alba, A.R. Occurrence of emerging pollutants in urban wastewater and their removal through biological treatment followed by ozonation. Water Res. 2010, 44, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Larsson, D.J.; de Pedro, C.; Paxeus, N. Effluent from drug manufactures contains extremely high levels of pharmaceuticals. J. Hazard. Mater. 2007, 148, 751–755. [Google Scholar] [CrossRef]
- Akbari, M.Z.; Xu, Y.; Lu, Z.; Peng, L. Review of antibiotics treatment by advance oxidation processes. Environ. Adv. 2021, 5, 100111. [Google Scholar] [CrossRef]
- Wang, J.; Zhuan, R. Degradation of antibiotics by advanced oxidation processes: An overview. Sci. Total Environ. 2020, 701, 135023. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S. Microbial degradation of sulfamethoxazole in the environment. Appl. Microbiol. Biotechnol. 2018, 102, 3573–3582. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S. Preparation, modification and environmental application of biochar: A review. J. Clean. Prod. 2019, 227, 1002–1022. [Google Scholar] [CrossRef]
- Zhuang, S.; Liu, Y.; Wang, J. Covalent organic frameworks as efficient adsorbent for sulfamerazine removal from aqueous solution. J. Hazard. Mater. 2020, 383, 121126. [Google Scholar] [CrossRef]
- Avramiotis, E.; Frontistis, Z.; Manariotis, I.D.; Vakros, J.; Mantzavinos, D. On the performance of a sustainable rice husk biochar for the activation of persulfate and the degradation of antibiotics. Catalysts 2021, 11, 1303. [Google Scholar] [CrossRef]
- Wang, X.; Brigante, M.; Dong, W.; Wu, Z.; Mailhot, G. Degradation of Acetaminophen via UVA-induced advanced oxidation processes (AOPs). Involvement of different radical species: HO, SO4− and HO2/O2−. Chemosphere 2020, 258, 127268. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez, L.A.; Pascual, J.M.; Martínez, M.D.M.M.; Poyatos Capilla, J.M. Effectiveness of advanced oxidation processes in wastewater treatment: State of the art. Water 2021, 13, 2094. [Google Scholar] [CrossRef]
- Sciscenko, I.; Hắng, H.T.M.; Escudero-Oñate, C.; Oller, I.; Arques, A. Fluorescence spectroscopy and chemometrics: A simple and easy way for the monitoring of fluoroquinolone mixture degradation. ACS Omega 2021, 6, 4663–4671. [Google Scholar] [CrossRef] [PubMed]
- Ao, X.; Zhang, X.; Li, S.; Yang, Y.; Sun, W.; Li, Z. Comprehensive understanding of fluoroquinolone degradation via MPUV/PAA process: Radical chemistry, matrix effects, degradation pathways, and toxicity. J. Hazard. Mater. 2023, 445, 130480. [Google Scholar] [CrossRef]
- Bobu, M.; Yediler, A.; Siminiceanu, I.; Zhang, F.; Schulte-Hostede, S. Comparison of different advanced oxidation processes for the degradation of two fluoroquinolone antibiotics in aqueous solutions. J. Environ. Sci. Health A 2013, 48, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wei, D.; Zhao, H.; Du, Y. Genotoxicity of quinolones: Substituents contribution and transformation products QSAR evaluation using 2D and 3D models. Chemosphere 2014, 95, 220–226. [Google Scholar] [CrossRef]
- Wang, D.; Ning, Q.; Dong, J.; Brooks, B.W.; You, J. Predicting mixture toxicity and antibiotic resistance of fluoroquinolones and their photodegradation products in Escherichia coli. Environ. Pollut. 2020, 262, 114275. [Google Scholar] [CrossRef]
- Wachter, N.; Aquino, J.M.; Denadai, M.; Barreiro, J.C.; Silva, A.J.; Cass, Q.B.; Rocha-Filho, R.C. Electrochemical degradation of the antibiotic ciprofloxacin in a flow reactor using distinct BDD anodes: Reaction kinetics, identification and toxicity of the degradation products. Chemosphere 2019, 234, 461–470. [Google Scholar] [CrossRef]
- Anjali, R.; Shanthakumar, S. Insights on the current status of occurrence and removal of antibiotics in wastewater by advanced oxidation processes. J. Environ. Manage. 2019, 246, 51–62. [Google Scholar] [CrossRef]
- Aydogdu, S.; Hatipoglu, A. Aqueous degradation of 6-APA by hydroxyl radical: A theoretical study. J. Mol. Model. 2023, 29, 222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pu, Q.; Li, Y. Bidirectional selection of the functional properties and environmental friendliness of organophosphorus (OP) pesticide derivatives: Design, screening, and mechanism analysis. Sci. Total Environ. 2023, 879, 163043. [Google Scholar] [CrossRef] [PubMed]
- Munkhdalai, L.; Munkhdalai, T.; Park, K.H.; Lee, H.G.; Li, M.; Ryu, K.H. Mixture of activation functions with extended min-max normalization for forex market prediction. IEEE Access 2019, 7, 183680–183691. [Google Scholar] [CrossRef]
- Clark, M.; Cramer, R.D., III; Van Opdenbosch, N. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Pu, Q.; Han, Z.; Li, X.; Li, Q.; Li, Y. Designing and screening of fluoroquinolone substitutes using combined in silico approaches: Biological metabolism–bioconcentration bilateral selection and their mechanism analyses. Green Chem. 2022, 24, 3778–3793. [Google Scholar] [CrossRef]
- Li, Q.; Qiu, Y.; Li, Y. Molecular design of environment-friendly PAE derivatives based on 3D-QSAR assisted with a comprehensive evaluation method combining toxicity and estrogen activities. Water Air Soil Pollut. 2020, 231, 1–13. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Pu, Q.; Fu, R.; Wang, Z.; Li, Y.; Li, X. Innovative screening for functional improved aromatic amine derivatives: Toxicokinetics, free radical oxidation pathway and carcinogenic adverse outcome pathway. J. Hazard. Mater. 2023, 454, 131541. [Google Scholar] [CrossRef]
- Haghighatlari, M.; Hachmann, J. Advances of machine learning in molecular modeling and simulation. Curr. Opin. Chem. Eng. 2019, 23, 51–57. [Google Scholar] [CrossRef]
- Dong, Y.; Georgakis, C.; Santos-Marques, J.; Du, J. Dynamic response surface methodology using Lasso regression for organic pharmaceutical synthesis. Front. Chem. Sci. Eng. 2021, 16, 221–223. [Google Scholar] [CrossRef]
- Patel, P.S.; Pandya, D.M.; Shah, M. A holistic review on the assessment of groundwater quality using multivariate statistical techniques. Environ. Sci. Pollut. Res. 2023, 30, 85046–85070. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Li, Y. Design of environmentally friendly neonicotinoid insecticides with bioconcentration tuning and Bi-directional selective toxic effects. J. Clean. Prod. 2019, 221, 113–121. [Google Scholar] [CrossRef]
- Salahinejad, M.; Ghasemi, J.B. 3D-QSAR studies on the toxicity of substituted benzenes to Tetrahymena pyriformis: CoMFA, CoMSIA and VolSurf approaches. Ecotoxicol. Environ. Saf. 2014, 105, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Segalin, J.; Arsand, J.B.; Jank, L.; Schwalm, C.S.; Streit, L.; Pizzolato, T.M. In silico toxicity evaluation for transformation products of antimicrobials, from aqueous photolysis degradation. Sci. Total Environ. 2022, 828, 154109. [Google Scholar] [CrossRef]
- Drgoňa, J.; Tuor, A.R.; Chandan, V.; Vrabie, D.L. Physics-constrained deep learning of multi-zone building thermal dynamics. Energy Build. 2021, 243, 110992. [Google Scholar] [CrossRef]
- Khalifa, N.M.; Srour, A.M.; Abd El-Karim, S.S.; Saleh, D.O.; Al-Omar, M.A. Synthesis and 2D-QSAR study of active benzofuran-based vasodilators. Molecules 2017, 22, 1820. [Google Scholar] [CrossRef]
- Ghose, A.K.; Pritchett, A.; Crippen, G.M. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships III: Modeling hydrophobic interactions. J. Comput. Chem. 1988, 9, 80–90. [Google Scholar] [CrossRef]
- Liu, P.; Wu, Z.; Lee, J.; Cravotto, G. Sonocatalytic degrading antibiotics over activated carbon in cow milk. Food Chem. 2024, 432, 137168. [Google Scholar] [CrossRef]
- Sharma, V.; Goswami, R.; Madan, A.K. Eccentric connectivity index: A novel highly discriminating topological descriptor for structure− property and structure− activity studies. J. Chem. Inf. Comput. Sci. 1997, 37, 273–282. [Google Scholar] [CrossRef]
- Tian, Z.; Li, G.; Bai, M.; Hou, X.; Li, X.; Zhao, C.; Zhu, Q.; Du, C.; Li, M.; Liu, W.; et al. Microbial mechanisms of refractory organics degradation in old landfill leachate by a combined process of UASB-A/O-USSB. Sci. Total Environ. 2022, 848, 157737. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhou, X.; Chen, D.; Li, F.; Xue, T.; Farag, A.S. Ternary Fe 3 O 4@ PANI@ Au nanocomposites as a magnetic catalyst for degradation of organic dyes. Sci. China Technol. Sci. 2017, 60, 749–757. [Google Scholar] [CrossRef]
- Platts, J.A.; Butina, D.; Abraham, M.H.; Hersey, A. Estimation of molecular linear free energy relation descriptors using a group contribution approach. J. Chem. Inf. Comput. Sci. 1999, 39, 835–845. [Google Scholar] [CrossRef]
- Pucci, R.; Angilella, G.G.N. Density functional theory, chemical reactivity, and the Fukui functions. Found. Chem. 2022, 24, 59–71. [Google Scholar] [CrossRef]
- Bao, C.; Zhao, J.; Sun, Y.; Zhao, X.; Zhang, X.; Zhu, Y.; Xing, B. Enhanced degradation of norfloxacin by Ce-mediated Fe-MIL-101: Catalytic mechanism, degradation pathways, and potential applications in wastewater treatment. Environ. Sci. Nano 2021, 8, 2347–2359. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Comparison of computational methods for atomic charges. Acta Phys.-Chim. Sin. 2012, 28, 1–18. [Google Scholar] [CrossRef]
- Asghar, A.; Bello, M.M.; Raman, A.A.A.; Daud, W.M.A.W.; Ramalingam, A.; Zain, S.B.M. Predicting the degradation potential of Acid blue 113 by different oxidants using quantum chemical analysis. Heliyon 2019, 5, e02396. [Google Scholar] [CrossRef]








{kind=link}
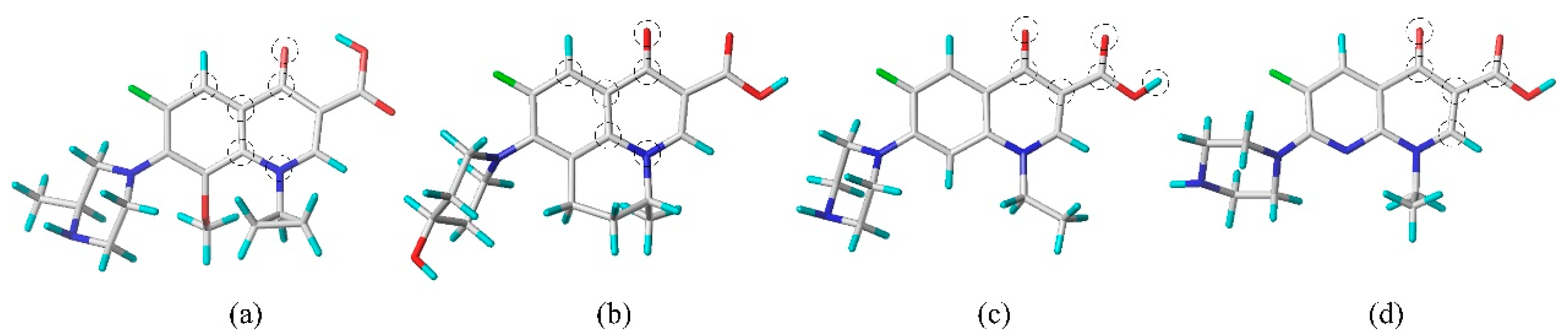{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FQ Molecule | Bond Dissociation Energy (kJ/mol) | ||||||
|---|---|---|---|---|---|---|---|
| Bond Dissociation Energy 1 | Bond Dissociation Energy 2 | Bond Dissociation Energy 3 | Bond Dissociation Energy 4 | Bond Dissociation Energy 5 | Average Bond Dissociation Energy 2, 3, 5 | Average Bond Dissociation Energy 1–5 | |
| CIP | 315.215 | 475.796 | 428.048 | 345.621 | 420.067 | 441.304 | 396.949 |
| CLI | - | 476.809 | 427.754 | - | 417.685 | 440.750 | - |
| ENO | 332.520 | 469.497 | 435.749 | 354.007 | 419.707 | 441.651 | 402.296 |
| FLE | 323.916 | 478.663 | 432.779 | 356.372 | 419.095 | 443.513 | 402.165 |
| GAT | 345.581 | 478.644 | 436.300 | 355.487 | 419.518 | 444.821 | 407.106 |
| LEV | 323.651 | 480.971 | 424.094 | 362.482 | 419.069 | 441.378 | 402.053 |
| LOM | 302.263 | 468.812 | 421.597 | 335.379 | 420.174 | 436.861 | 389.645 |
| MOX | - | 474.677 | 430.099 | - | 420.721 | 441.832 | - |
| NAD | - | 485.717 | 434.137 | - | 422.396 | 447.417 | - |
| NOR | 334.738 | 473.383 | 420.770 | 343.804 | 421.537 | 438.563 | 398.846 |
| OFL | 285.316 | 472.396 | 424.136 | 338.739 | 418.284 | 438.272 | 387.774 |
| PEF | 332.362 | 478.256 | 433.633 | 355.115 | 420.883 | 444.257 | 404.050 |
| RUF | 322.860 | 483.039 | 435.182 | 354.429 | 420.621 | 446.281 | 403.226 |
| SPA | 329.582 | 479.695 | 435.563 | 359.746 | 424.528 | 446.595 | 405.822 |
| TOS | - | 481.703 | 439.522 | - | 418.397 | 446.541 | - |
| GEM | - | 477.578 | 434.822 | - | 417.869 | 443.423 | - |
| CoMSIA | q2 | N | R2 | SEE | F | r2pred | SEP |
|---|---|---|---|---|---|---|---|
| Piperazine ring cleavage | 0.516 | 5 | 1.000 | 0.001 | 227534.936 | 0.972 | 0.043 |
| Defluorination | 0.595 | 6 | 1.000 | 0.002 | 26018.716 | 0.992 | 0.062 |
| Hydroxylation | 0.628 | 7 | 1.000 | 0.008 | 1926.276 | 0.878 | 0.018 |
| Piperazine ring hydroxylation | 0.622 | 2 | 0.991 | 0.037 | 268.732 | 0.999 | 0.006 |
| CoMSIA | Hydrogen Bond Acceptor Field (%) | Hydrophobic Field (%) | Electrostatic Field (%) | Hydrogen Bond Donor Field (%) | Steric Field (%) |
|---|---|---|---|---|---|
| Piperazine ring cleavage | 49.30 | 21.00 | 15.40 | 7.30 | 7.00 |
| Defluorination | 43.20 | 22.30 | 18.20 | 8.60 | 7.80 |
| Hydroxylation | 25.30 | 24.70 | 24.50 | 6.20 | 19.20 |
| Piperazine ring hydroxylation | 40.60 | 16.90 | 28.50 | 7.70 | 6.30 |
| Implicit Variable | Feature Parameter | Feature Importance |
|---|---|---|
| Bond dissociation energy 1 | (AlogP)2 | 0.713 |
| MATS2m | 0.187 | |
| maxsssCH | 0.069 | |
| GATS5c | 0.022 | |
| ATS6s | 0.005 | |
| MATS1m | 0.002 | |
| SP-2 | 0.002 | |
| SM1_Dze | <0.001 | |
| AVP-1 | <0.001 | |
| Bond dissociation energy 2 | ECCEN | 0.737 |
| MATS8i | 0.132 | |
| MATS3c | 0.065 | |
| nHaaCH | 0.006 | |
| SpMin3_Bhm | 0.003 | |
| SpMin8_Bhi | 0.001 | |
| EE_Dzp | <0.001 | |
| MAXDP | <0.001 | |
| Bond dissociation energy 3 | ALogp2 | 0.847 |
| GATS6e | 0.105 | |
| SP-2 | 0.023 | |
| GATS3i | 0.001 | |
| SpMAD_Dt | <0.001 | |
| GATS5i | <0.001 | |
| C1SP2 | <0.001 | |
| Bond dissociation energy 4 | MLFER_BH | 0.749 |
| SP-2 | 0.119 | |
| GATS6i | 0.085 | |
| Sare | 0.037 | |
| SpMax8_Bhe | 0.005 | |
| SP-5 | 0.003 | |
| GATS6e | 0.002 | |
| ATS0m | <0.001 | |
| ATS3i | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, S.; Wang, Z.; Pu, Q.; Li, X.; Cui, Y.; Yang, H.; Li, Y. Identification and Mechanistic Analysis of Toxic Degradation Products in the Advanced Oxidation Pathways of Fluoroquinolone Antibiotics. Toxics 2024, 12, 203. https://doi.org/10.3390/toxics12030203
Sun S, Wang Z, Pu Q, Li X, Cui Y, Yang H, Li Y. Identification and Mechanistic Analysis of Toxic Degradation Products in the Advanced Oxidation Pathways of Fluoroquinolone Antibiotics. Toxics. 2024; 12(3):203. https://doi.org/10.3390/toxics12030203
Chicago/Turabian StyleSun, Shuhai, Zhonghe Wang, Qikun Pu, Xinao Li, Yuhan Cui, Hao Yang, and Yu Li. 2024. "Identification and Mechanistic Analysis of Toxic Degradation Products in the Advanced Oxidation Pathways of Fluoroquinolone Antibiotics" Toxics 12, no. 3: 203. https://doi.org/10.3390/toxics12030203