
Use of Biomarker Data and Relative Potencies of Mutagenic Metabolites to Support Derivation of Cancer Unit Risk Values for 1,3-Butadiene from Rodent Tumor Data

Abstract
:1. Introduction
2. Background
2.1. Previous Rodent-Based Cancer Risk Assessments for BD
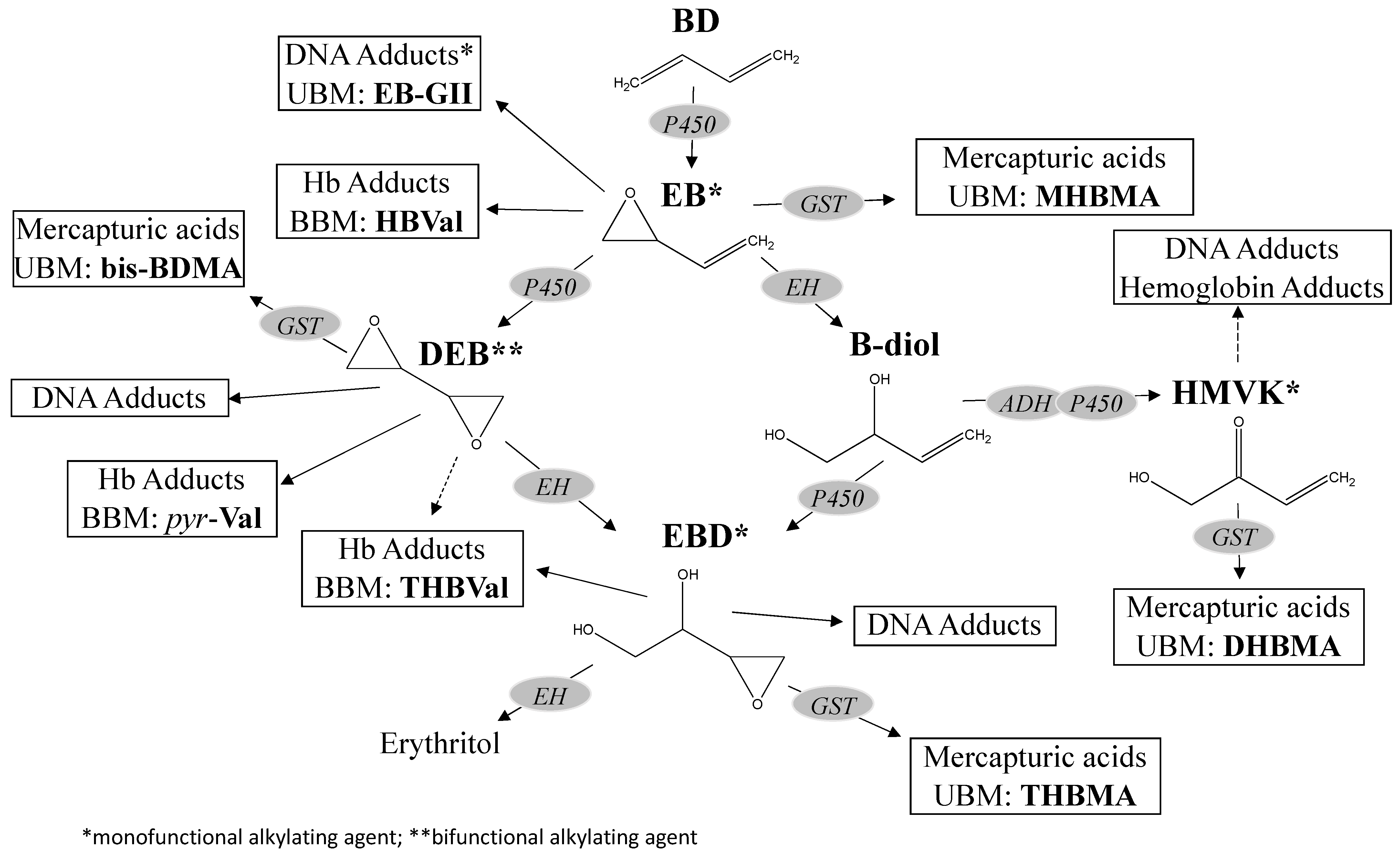
2.2. Metabolism Overview
2.3. Mode of Action Summary
3. Methods
3.1. Exposure Concentration and Duration (CxT) in Tumor Response
3.2. Unit Risk Derivation
3.2.1. Human Equivalent Concentration Calculation
3.2.2. Endpoint/Dataset Selection
3.2.3. Benchmark Dose Modeling
4. Results
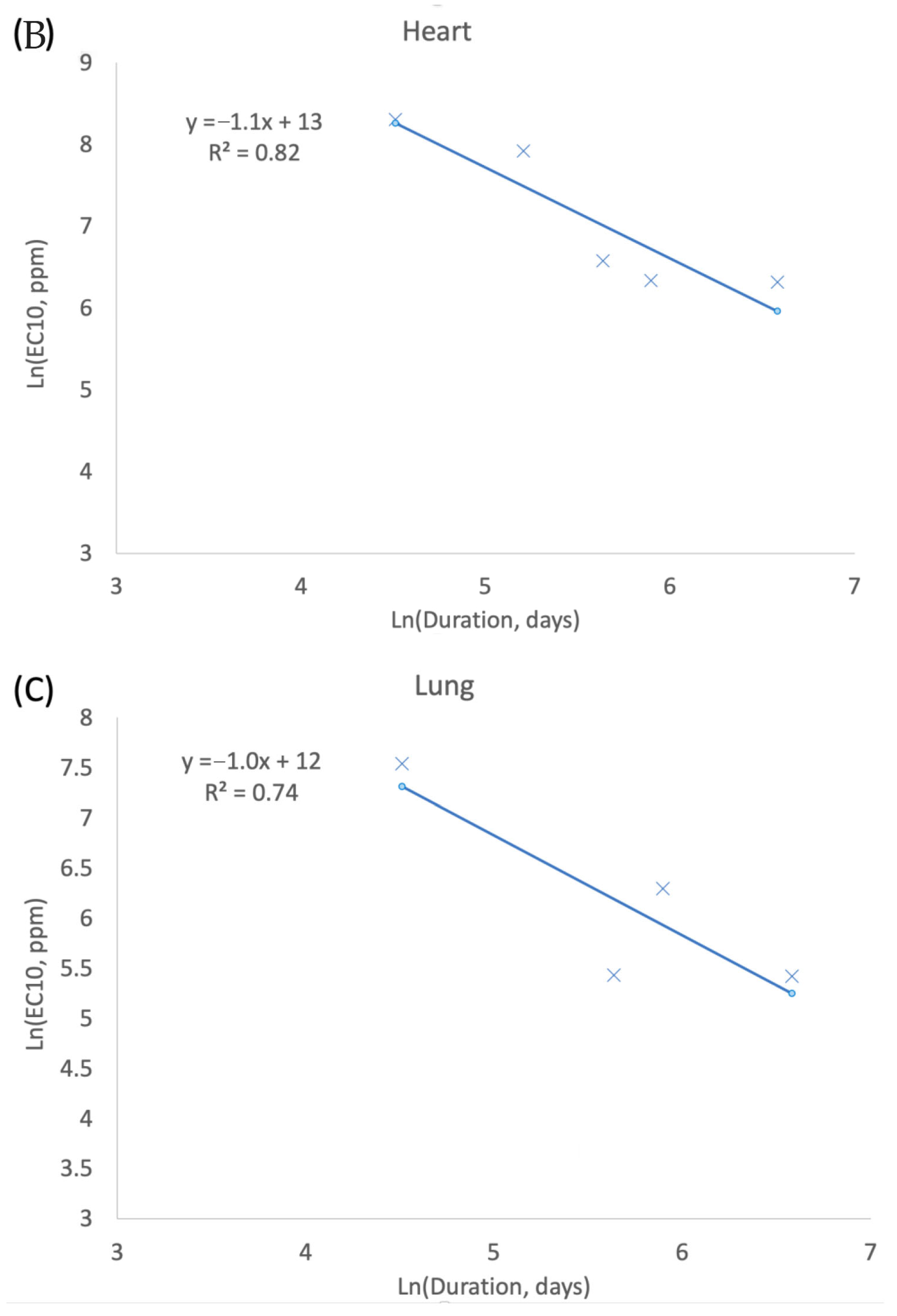
4.1. Exposure Concentration and Duration (CxT)
4.2. Human Equivalent Concentrations
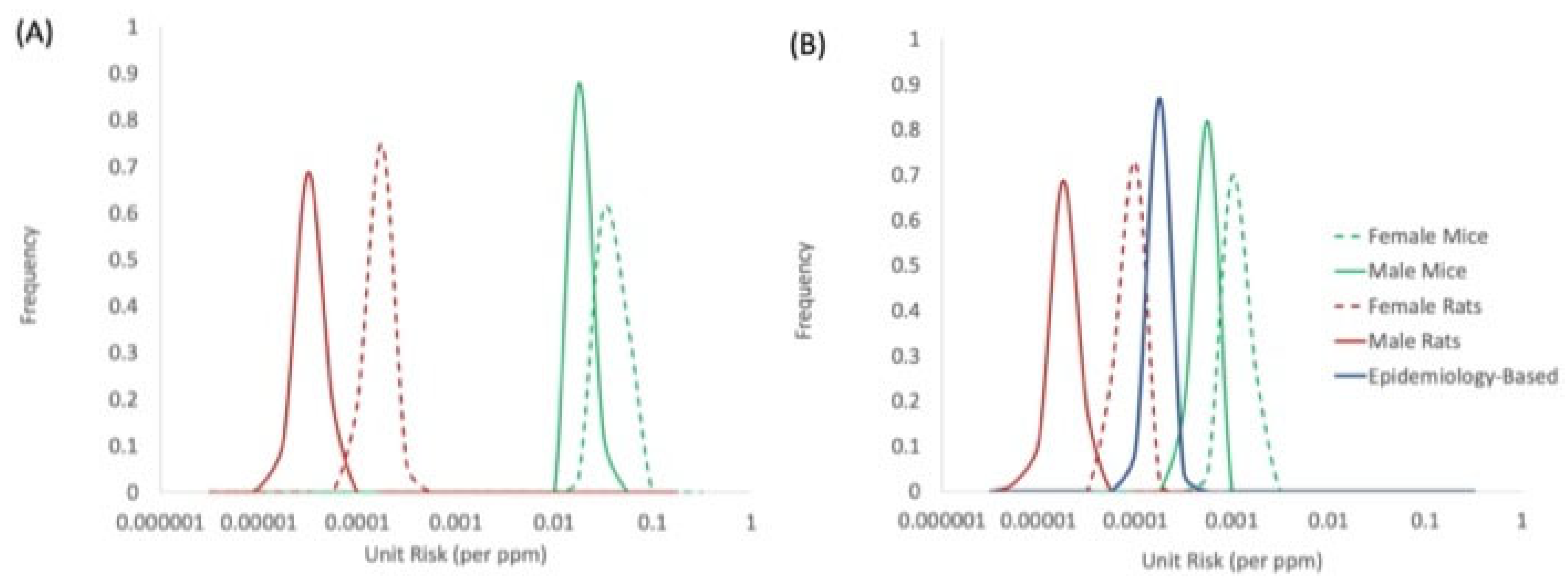
4.3. Unit Risk Values and Species Concordance
4.4. Consideration of Sensitive Subpopulations and Additional Adjustments
5. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sathiakumar, N.; Bolaji, E.B.; Brill, I.; Chen, L.; Tipre, M.; Leader, M.; Arora, T.; Delzell, E. 1,3-Butadiene, styrene and lymphohaematopoietic cancers among North American synthetic rubber polymer workers: Exposure–response analyses. Occup. Environ. Med. 2021, 78, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Sathiakumar, N.; Bolaji, B.; Brill, I.; Chen, L.; Tipre, M.; Leader, M.; Arora, T.; Delzell, E. 1,3-Butadiene, styrene and selected outcomes among synthetic rubber polymer workers: Updated exposure-response analyses. Chem. Interact. 2021, 347, 109600. [Google Scholar] [CrossRef] [PubMed]
- NTP. NTP Toxicology and Carcinogenesis Studies of 1,3-Butadiene (CAS No. 106-99-0) in B6C3F1 Mice (Inhalation Studies). Natl. Toxicol. Program Tech. Rep. Ser. 1993, 434, 1–389. Available online: https://ntp.niehs.nih.gov/ntp/htdocs/lt_rpts/tr434.pdf (accessed on 1 June 2022).
- Owen, P.E.; Glaister, J.R.; Gaunt, I.F.; Pullinger, D.H. Inhalation Toxicity Studies With 1,3-Butadiene 3 Two Year Toxicity/Carcinogenicity Study in Rats. Am. Ind. Hyg. Assoc. J. 1987, 48, 407–413. [Google Scholar] [CrossRef]
- Melnick, R.L.; Huff, J.E. 1,3-Butadiene induces cancer in experimental animals at all concentrations from 6.25 to 8000 parts per million. IARC Sci. Publ. 1993, 127, 309–322. [Google Scholar]
- EPA/630/P-02/002F; A Review of the Reference Dose and Reference Concentration Process. U.S. Environmental Protection Agency|Risk Assessment Forum: Washington, DC, USA, 2002.
- Hazleton. 1,3-Butadiene: Inhalation teratogenicity study in the rat. Hazleton Laboratories Europe. Report 2788-522/3. 1981, p. 318. Available online: https://www.moodys.com/credit-ratings/Hazleton-Laboratories-Corporation-credit-rating-364760/reports?category=&type= (accessed on 1 June 2022).
- Health Canada. Priority Substances List Assessment Report; Cat. no. En40-215/52E; Environment Canada: Edmonton, AB, Canada, 2000; ISBN 0-662-29014-3. [Google Scholar]
- OEHHA. 1,3-Butadiene Reference Exposure Levels; OEHHA: Sacramento, CA, USA, 2013. [Google Scholar]
- Motwani, H.V.; Törnqvist, M. In vivo doses of butadiene epoxides as estimated from in vitro enzyme kinetics by using cob(I)alamin and measured hemoglobin adducts: An inter-species extrapolation approach. Toxicol. Appl. Pharmacol. 2014, 281, 276–284. [Google Scholar] [CrossRef]
- EPA/R-14/002F; Guidance for Applying Quantitative Data to Develop Data-Derived Extrapolation Factors for Interspecies and Intraspecies Extrapolation. U.S. Environmental Protection Agency: Washington, DC, USA, 2014.
- Fred, C.; Tornqvist, M.; Granath, F. Evaluation of Cancer Tests of 1,3-Butadiene Using Internal Dose, Genotoxic Potency, and a Multiplicative Risk Model. Cancer Res. 2008, 68, 8014–8021. [Google Scholar] [CrossRef] [Green Version]
- EPA/630/P-03/001F; Guidelines for Carcinogen Risk Assessment. U.S. Environmental Protection Agency|Risk Assessment Forum: Washington, DC, USA, 2005.
- Bucher, J.R.; Melnick, R.L.; Hildebrandt, P.K. Lack of Carcinogenicity in Mice Exposed Once to High Concentrations of 1,3-Butadiene. J. Nat. Cancer Inst. 1993, 85, 1866–1877. [Google Scholar] [CrossRef]
- EPA-740-R-20-011; Final Scope of the Risk Evaluation for 1,3-Butadiene. CASRN 106-99-0. U.S. Environmental Protection Agency, Office of Chemical Safety and Pollution Prevention: Washington, DC, USA, 2020.
- Himmelstein, M.W.; Acquavella, J.F.; Recio, L.; Medinsky, M.A.; Bond, J.A. Toxicology and Epidemiology of 1,3-Butadiene. Crit. Rev. Toxicol. 1997, 27, 1–108. [Google Scholar] [CrossRef]
- Albertini, R.J.; Srám, R.J.; Vacek, P.M.; Lynch, J.; Nicklas, J.A.; van Sittert, N.J.; Boogaard, P.J.; Henderson, R.F.; Swenberg, J.A.; Tates, A.D.; et al. Biomarkers in Czech workers exposed to 1,3-butadiene: A transitional epidemiologic study. Res. Rep. Health Eff. Inst. 2003, 116, 1–141. [Google Scholar]
- Kirman, C.R.; Albertini, R.J.; Sweeney, L.M.; Gargas, M.L. 1,3-Butadiene: I. Review of metabolism and the implications to human health risk assessment. Crit. Rev. Toxicol. 2010, 40, 1–11. [Google Scholar] [CrossRef]
- Filser, J.G.; Bhowmik, S.; Faller, T.H.; Hutzler, C.; Kessler, W.; Midpanon, S.; Pütz, C.; Schuster, A.; Semder, B.; Veereshwarayya, V.; et al. Quantitative Investigation on the Metabolism of 1,3-Butadiene and of Its Oxidized Metabolites in Once-through Perfused Livers of Mice and Rats. Toxicol. Sci. 2010, 114, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Csanády, G.A.; Guengerich, F.; Bond, J.A. Comparison of the biotransformation of 1,3-butadiene and its metabolite, butadiene monoepoxide, by hepatic and pulmonary tissues from humans, rats and mice. Carcinogenesis 1992, 13, 1143–1153. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, U.; Loeser, E. Species differences in the formation of butadiene monoxide from 1,3-butadiene. Arch. Toxicol. 1985, 57, 222–225. [Google Scholar] [CrossRef]
- Krause, R.J.; Elfarra, A.A. Oxidation of Butadiene Monoxide tomeso-and (±)-Diepoxybutane by cDNA-Expressed Human Cytochrome P450s and by Mouse, Rat, and Human Liver Microsomes: Evidence for Preferential Hydration ofmeso-Diepoxybutane in Rat and Human Liver Microsomes. Arch. Biochem. Biophys. 1997, 337, 176–184. [Google Scholar] [CrossRef]
- Bond, A.J.; Csanády, A.G.; Leavens, T.; Medinsky, A.M. Research strategy for assessing target tissue dosimetry of 1,3-butadiene in laboratory animals and humans. IARC Sci. Publ. 1993, 127, 45–55. [Google Scholar]
- Kreuzer, P.E.; Kessler, W.; Welter, H.F.; Baur, C.; Filser, J.G. Enzyme specific kinetics of 1,2-epoxybutene-3 in microsomes and cytosol from livers of mouse, rat, and man. Arch. Toxicol. 1991, 65, 59–67. [Google Scholar] [CrossRef]
- Seaton, M.; Follansbee, M.; Bond, J. Oxidation of 1,2-epoxy-3-butene to 1,2:3,4-diepoxybutane by cDNA-expressed human cytochromes P450 2E1 and 3A4 and human, mouse and rat liver microsomes. Carcinogenesis 1995, 16, 2287–2293. [Google Scholar] [CrossRef]
- Filser, J.G.; Faller, T.H.; Bhowmik, S.; Schuster, A.; Kessler, W.; Pütz, C.; Csanády, G.A. First-pass metabolism of 1,3-butadiene in once-through perfused livers of rats and mice. Chem. Interact. 2001, 135, 249–265. [Google Scholar] [CrossRef]
- Filser, J.G.; Hutzler, C.; Meischner, V.; Veereshwarayya, V.; Csanády, G.A. Metabolism of 1,3-butadiene to toxicologically relevant metabolites in single-exposed mice and rats. Chem. Interact. 2007, 166, 93–103. [Google Scholar] [CrossRef]
- Thornton-Manning, J.R.; Dahl, A.R.; Bechtold, W.E.; Griffith, W.C.; Pei, L.; Henderson, R.F. Gender differences in the metabolism of 1, 3-butadiene in Sprague-Dawley rats following a low level inhalation exposure. Carcinogenesis 1995, 16, 2875–2878. [Google Scholar] [CrossRef]
- Thornton-Manning, J.R.; Dahl, A.R.; Bechtold, W.E.; Griffith, W.C.; Henderson, R.F.; Hederson, R.F. Disposition of butadiene monoepoxide and butadiene diepoxide in various tissues of rats and mice following a low-level inhalation exposure to 1,3-butadiene. Carcinogenesis 1995, 16, 1723–1731. [Google Scholar] [CrossRef]
- Boysen, G.; Georgieva, N.I.; Upton, P.B.; Jayaraj, K.; Li, Y.; Walker, V.E.; Swenberg, J.A. Analysis of Diepoxide-Specific Cyclic N-Terminal Globin Adducts in Mice and Rats after Inhalation Exposure to 1,3-Butadiene. Cancer Res. 2004, 64, 8517–8520. [Google Scholar] [CrossRef] [Green Version]
- Swenberg, J.A.; Boysen, G.; Georgieva, N.; Bird, M.G.; Lewis, R.J. Future directions in butadiene risk assessment and the role of cross-species internal dosimetry. Chem. Interact. 2007, 166, 78–83. [Google Scholar] [CrossRef]
- Georgieva, N.I.; Boysen, G.; Bordeerat, N.; Walker, V.E.; Swenberg, J.A. Exposure-Response of 1,2:3,4-Diepoxybutane–Specific N-Terminal Valine Adducts in Mice and Rats after Inhalation Exposure to 1,3-Butadiene. Toxicol. Sci. 2010, 115, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Albertini, R.J.; Sram, R.J.; Vacek, P.M.; Lynch, J.; Rossner, P.; Nicklas, J.A.; McDonald, J.D.; Boysen, G.; Georgieva, N.; Swenberg, J.A. Molecular epidemiological studies in 1,3-butadiene exposed Czech workers: Female–male comparisons. Chem. Interact. 2007, 166, 63–77. [Google Scholar] [CrossRef]
- Swenberg, J.A.; Bordeerat, N.K.; Boysen, G.; Carro, S.; Georgieva, N.I.; Nakamura, J.; Troutman, J.M.; Upton, P.B.; Albertini, R.J.; Vacek, P.M.; et al. 1,3-Butadiene: Biomarkers and application to risk assessment. Chem. Interact. 2011, 192, 150–154. [Google Scholar] [CrossRef] [Green Version]
- Boysen, G.; Georgieva, N.I.; Bordeerat, N.K.; Šram, R.J.; Vacek, P.; Albertini, R.J.; Swenberg, J.A. Formation of 1,2:3,4-Diepoxybutane-Specific Hemoglobin Adducts in 1,3-Butadiene Exposed Workers. Toxicol. Sci. 2011, 125, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Kotapati, S.; Esades, A.; Matter, B.; Le, C.; Tretyakova, N. High throughput HPLC–ESI−MS/MS methodology for mercapturic acid metabolites of 1,3-butadiene: Biomarkers of exposure and bioactivation. Chem. Interact. 2015, 241, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Albertini, R.J.; Carson, M.L.; Kirman, C.R.; Gargas, M.L. 1,3-Butadiene: II. Genotoxicity profile. Crit. Rev. Toxicol. 2010, 40, 12–73. [Google Scholar] [CrossRef]
- Kirman, C.R.; Albertini, R.A.; Gargas, M.L. 1,3-Butadiene: III. Assessing carcinogenic modes of action. Crit. Rev. Toxicol. 2010, 40 (Suppl. S1), 74–92. [Google Scholar] [CrossRef] [PubMed]
- Walker, V.E.; Degner, A.; Carter, E.W.; Nicklas, J.A.; Walker, D.M.; Tretyakova, N.; Albertini, R.J. 1,3-Butadiene metabolite 1,2,3,4 diepoxybutane induces DNA adducts and micronuclei but not t(9;22) translocations in human cells. Chem. Interact. 2019, 312, 108797. [Google Scholar] [CrossRef] [PubMed]
- Preston, R.J. Chromosomal changes. IARC Sci. Publ. 1999, 146, 395–408. [Google Scholar]
- ten Berge, W.F.; Zwart, A.; Appelman, L.M. Concentration—Time mortality response relationship of irritant and systemically acting vapours and gases. J. Haz. Mat. 1986, 13, 301–309. [Google Scholar] [CrossRef]
- EPA/100/R-12/001; Benchmark Dose Technical Guidance. U.S. Environmental Protection Agency|Risk Assessment Forum: Washington, DC, USA, 2012.
- Wen, Y.; Zhang, P.P.; An, J.; Yu, Y.X.; Wu, M.H.; Sheng, G.Y.; Fu, J.M.; Zhang, X.Y. Diepoxybutane induces the formation of DNA-DNA rather than DNA-protein cross-links, and single-strand breaks and alkali-labile sites in human hepatocyte L02 cells. Mutat. Res. 2011, 716, 84–91. [Google Scholar] [CrossRef]
- Zhang, P.P.; Wen, Y.; An, J.; Yu, Y.X.; Wu, M.H.; Zhang, X.Y. DNA damage induced by three major metabolites of 1,3-butadiene in human hepatocyte L02 cells. Mutat. Res. 2012, 747, 240–245. [Google Scholar] [CrossRef]
- Meng, R.Q.; Hackfeld, L.C.; Hedge, R.P.; Wisse, A.L.; Redetzke, D.L.; Walker, E.V. Mutagenicity of stereochemical configurations of 1,3-butadiene epoxy metabolites in human cells. Res. Rep. Health Eff. Inst. 2010, 150, 1–34. [Google Scholar]
- Cochrane, E.J.; Skopek, T.R. Mutagenicity of 1,3-butadiene and its epoxide metabolites in human TK6 cells and in splenic T cells isolated from exposed B6C3F1 mice. IARC Sci. Publ. 1993, 127, 195–204. [Google Scholar]
- Erexson, G.L.; Tindall, K.R. Micronuclei and gene mutations in transgenic Big Blue® mouse and rat fibroblasts after exposure to the epoxide metabolites of 1,3-butadiene. Mutat. Res. Toxicol. Environ. Mutagen. 2000, 472, 105–117. [Google Scholar] [CrossRef]
- Adler, I.D.; Kliesch, U.; Nylund, L.; Peltonen, K. In vitro and in vivo mutagenicity of the butadiene metabolites butadiene diolepoxide, butadiene monoepoxide and diepoxybutane. Mutagenesis 1997, 12, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöblom, T.; Lähdetie, J. Micronuclei are induced in rat spermatids in vitro by 1,2,3,4-diepoxybutane but not by 1,2-epoxy-3-butene and 1,2-dihydroxy-3,4-epoxybutane. Mutagenesis 1996, 11, 525–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdez-Flores, C.; Erraguntla, N.; Budinsky, R.A.; Cagen, S.; Kirman, C.R. An Updated Lymphohematopoietic and Bladder Cancers Risk Evaluation for Occupational and Environmental Exposures to 1,3-Butadiene. Chem. -Biol. Interact. 2022; Submitted. [Google Scholar]
- Park, S.L.; Kotapati, S.; Wilkens, L.R.; Tiirikainen, M.; Murphy, S.E.; Tretyakova, N.; Le Marchand, L. 1,3-Butadiene Exposure and Metabolism among Japanese American, Native Hawaiian, and White Smokers. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2240–2249. [Google Scholar] [CrossRef] [Green Version]
- Boldry, E.J.; Patel, Y.M.; Kotapati, S.; Esades, A.; Park, S.L.; Tiirikainen, M.; Stram, D.O.; Le Marchand, L.; Tretyakova, N. Genetic Determinants of 1,3-Butadiene Metabolism and Detoxification in Three Populations of Smokers with Different Risks of Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1034–1042. [Google Scholar] [CrossRef] [Green Version]
- Sangaraju, D.; Boldry, E.J.; Patel, Y.M.; Walker, V.; Stepanov, I.; Stram, D.; Hatsukami, D.; Tretyakova, N. Isotope Dilution nanoLC/ESI+-HRMS3 Quantitation of Urinary N7-(1-Hydroxy-3-buten-2-yl) Guanine Adducts in Humans and Their Use as Biomarkers of Exposure to 1,3-Butadiene. Chem. Res. Toxicol. 2017, 30, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Jokipii Krueger, C.C.; Park, S.L.; Madugundu, G.; Patel, Y.; Le Marchand, L.; Stram, D.O.; Tretyakova, N. Ethnic differences in excretion of butadiene–DNA adducts by current smokers. Carcinogenesis 2021, 42, 694–704. [Google Scholar] [CrossRef]
- Fernandez-Salguero, P.; Hoffman, S.M.; Cholerton, S.; Mohrenweiser, H.; Raunio, H.; Rautio, A.; Pelkonen, O.; Huang, J.D.; Evans, E.W.; Idle, J.R. A genetic polymorphism in coumarin 7-hydroxylation: Sequence of the human CYP2A genes and identification of variant CYP2A6 alleles. Am. J. Hum. Genet. 1995, 57, 651–660. [Google Scholar]
- Wormhoudt, L.W.; Commandeur, J.N.M.; Vermeulen, N.P.E. Genetic Polymorphisms of HumanN-Acetyltransferase, Cytochrome P450, Glutathione-S-Transferase, and Epoxide Hydrolase Enzymes: Relevance to Xenobiotic Metabolism and Toxicity. Crit. Rev. Toxicol. 1999, 29, 59–124. [Google Scholar] [CrossRef]
- London, S. Lung cancer risk in relation to genetic polymorphisms of microsomal epoxide hydrolase among African-Americans and Caucasians in Los Angeles County. Lung Cancer 2000, 28, 147–155. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Hiyama, K.; Ishioka, S.; Maeda, H.; Maeda, A.; Yamakido, M. Microsomal epoxide hydrolase genotypes and chronic obstructive pulmonary disease in Japanese. Int. J. Mol. Med. 2000, 5, 49–102. [Google Scholar] [CrossRef]
- Degner, A.; Arora, R.; Erber, L.; Chao, C.; Peterson, L.A.; Tretyakova, N.Y. Interindividual Differences in DNA Adduct Formation and Detoxification of 1,3-Butadiene-Derived Epoxide in Human HapMap Cell Lines. Chem. Res. Toxicol. 2020, 33, 1698–1708. [Google Scholar] [CrossRef]
- Fustinoni, S.; Soleo, L.; Warholm, M.; Begemann, P.; Rannug, A.; Neumann, H.-G.; Swenberg, A.J.; Vimercati, L.; Colombi, A. Influence of metabolic genotypes on biomarkers of exposure to 1,3-butadiene in humans. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1082–1090. [Google Scholar]
- Auerbach, A.D. Diagnosis of Fanconi Anemia by Diepoxybutane Analysis. Curr. Protoc. Hum. Genet. 2015, 85, 8.7.1–8.7.17. [Google Scholar] [CrossRef] [Green Version]
- Hines, R.N. Ontogeny of human hepatic cytochromes P450. J. Biochem. Mol. Toxicol. 2007, 21, 169–175. [Google Scholar] [CrossRef]
- Johnsrud, E.K.; Koukouritaki, S.B.; Divakaran, K.; Brunengraber, L.L.; Hines, R.N.; McCarver, D.G. Human hepatic CYP2E1 expression during development. J. Pharmacol. Exp. Ther. 2003, 307, 402–407. [Google Scholar] [CrossRef]
- Matlock, M.K.; Tambe, A.; Elliott-Higgins, J.; Hines, R.N.; Miller, G.P.; Swamidass, S.J. A Time-Embedding Network Models the Ontogeny of 23 Hepatic Drug Metabolizing Enzymes. Chem. Res. Toxicol. 2019, 32, 1707–1721. [Google Scholar] [CrossRef]
- Nieto, A.; Zhang, L.; Bhandari, D.; Zhu, W.; Blount, B.C.; De Jesús, V.R. Exposure to 1,3-Butadiene in the U.S. Population: National Health and Nutrition Examination Survey 2011–2016. Biomarkers 2021, 26, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Kirman, C.R.; North, C.M.; Tretyakova, N.Y.; Erraguntla, N.; Shen, H.; Hays, S.H. Use of Biomarker Data and Metabolite Relative Potencies to Support Derivation of Noncancer Reference Values for 1,3-Butadiene. Regul. Toxicol. Pharmacol. 2022; Submitted. [Google Scholar]
- U.S. EPA. Exposure Factors Handbook 2011 Edition (Final Report); EPA/600/R-09/052F; U.S. Environmental Protection Agency: Washington, DC, USA, 2011. [Google Scholar]
- IRIS Integrated Risk Information System|U.S. Environmental Protection Agency. Record for Acrylamide; IRIS: Washington, DC, USA, 2010. [Google Scholar]
- EPA/635/R-08/012A; Development of a Relative Potency Factor (RPF) Approach for Polycyclic Aromatic Hydrocarbon (PAH) Mixtures. U.S. Environmental Protection Agency: Washington, DC, USA, 2010.
- EPA/100/R-10/005; Recommended Toxicity Equivalence Factors (TEFs) for Human Health Risk Assessments of 2,3,7,8- Tetrachlorodibenzo-p-dioxin and Dioxin-Like Compounds. U.S. Environmental Protection Agency|Risk Assessment Forum: Washington, DC, USA, 2010; Volume 2.
- Nakamura, J.; Carro, S.; Gold, A.; Zhang, Z. An unexpected butadiene diolepoxide-mediated genotoxicity implies alternative mechanism for 1,3-butadiene carcinogenicity. Chemosphere 2020, 266, 129149. [Google Scholar] [CrossRef]
- Elfarra, A.A.; Zhang, X.-Y. Alcohol Dehydrogenase- and Rat Liver Cytosol-Dependent Bioactivation of 1-Chloro-2-hydroxy-3-butene to 1-Chloro-3-buten-2-one, a Bifunctional Alkylating Agent. Chem. Res. Toxicol. 2012, 25, 2600–2607. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yu, Y.-X.; Luan, Y.; An, J.; Yin, D.-G.; Zhang, X.-Y. Bioactivation of 1-chloro-2-hydroxy-3-butene, an in vitro metabolite of 1,3-butadiene, by rat liver microsomes. Chem. Interact. 2018, 282, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-J.; Tang, W.-F.; Xiang, M.-H.; Yan, J.; Cao, X.; Zhou, C.-H.; Chang, Y.; Xi, J.; Cao, Y.-Y.; Luan, Y.; et al. Isotope Dilution LC/ESI(-)-MS-MS Quantitation of Urinary 1,4-Bis(N-Acetyl-S-Cysteinyl)-2-Butanone in Mice and Rats as the Biomarker Of. Chem. Interact. 2019, 311, 108760. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assessor (Year) | Endpoint | Dataset | DR Model | POD Type | POD Value | Species Extrapolation Assumption | Low-Dose Extrapolation Assumption | Unit Risk (ppm−1) |
|---|---|---|---|---|---|---|---|---|
| USEPA [6] | Leydig cell, pancreatic exocrine cell, Zymbal gland, mammary gland, thyroid follicular cell | Male and Female Rats [7] | Multistage | LEC10 | NS | Air concentration equivalence | Linear | 0.0042–0.056 |
| Lymphocytic lymphomas, histiocytic sarcomas, heart hemangiosarcomas, lung, forestomach, Harderian gland, liver, preputial gland, ovary, mammary gland | Male and Female Mice [3] | Multistage-Weibull time-to-tumor | LEC10 | 0.7–13.3 ppm | Air concentration equivalence | Linear | 0.0064–0.29 | |
| Health Canada [8] | Multiple | Male and Female Rats [7] | Multistage | TC05 | 4.7–905 mg/m3 | Air concentration equivalence | NA | 0.00012–0.024 * |
| Multiple | Male & Female Mice [3] | Multistage | TC05 | 1.4–23 mg/m3 | Air concentration equivalence | NA | 0.0048–0.079 * | |
| OEHHA [9] | Multiple | Female MouseMale and Female Mice [3]; Male and Female Rats [7] | Multistage | NS | NS | Surface area scaling | Linear | 0.077 0.002–0.16 |
| Gender | Duration (Reference) | Exposure | Concentration (ppm) | Lymphoma | Histiocytic Sarcoma | Heart | Alveolar–Bronchiolar | Forestomach | Mammary Gland | Liver | Harderian | Preputial, Ovary |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Male | Acute [14] | 2 h (1×) | 0 | 7/59 | NR | NR | 8/59 | 0/59 | 0/59 | 17/59 | NR | NR |
| 1000 | 8/58 | NR | NR | 9/58 | 1/58 | 0/58 | 21/58 | NR | NR | |||
| 5000 | 8/58 | NR | NR | 12/57 | 1/58 | 0/58 | 21/58 | NR | NR | |||
| 10,000 | 10/58 | NR | NR | 8/58 | 3/58 | 1/58 | 18/58 | NR | NR | |||
| Long-term [3] | 6 h/d, 5 d/wk, 40 wk | 200 | 8/50 | 5/50 | 15/50 | 36/50 | 3/50 | NR | 33/49 | 27/50 | 1/50 | |
| 6 h/d, 5 d/wk, 52 wk | 312 | 8/50 | 7/50 | 33/50 | 32/50 | 9/50 | NR | 25/50 | 30/50 | 4/50 | ||
| 6 h/d, 5 d/wk, 13 wk | 625 | 22/50 | 2/50 | 7/50 | 28/50 | 7/50 | NR | 24/49 | 23/50 | 5/50 | ||
| 6 h/d, 5 d/wk, 26 wk | 625 | 33/50 | 2/50 | 13/50 | 17/50 | 10/50 | NR | 13/50 | 13/50 | 3/50 | ||
| Lifetime [3] | 6 h/d, 5 d/wk, 103 wk | 0 | 4/50 | 0/50 | 0/50 | 21/50 | 1/50 | NR | 21/50 | 6/50 | 0/50 | |
| 6.25 | 2/50 | 0/50 | 0/49 | 23/50 | 0/50 | NR | 23/50 | 7/50 | 0/50 | |||
| 20 | 4/50 | 4/50 | 1/50 | 19/50 | 0/50 | NR | 30/50 | 9/50 | 0/50 | |||
| 62.5 | 6/50 | 5/50 | 5/48 | 31/49 | 1/50 | NR | 25/48 | 20/50 | 0/50 | |||
| 200 | 2/50 | 7/50 | 20/48 | 35/50 | 8/50 | NR | 33/48 | 31/50 | 5/50 | |||
| 625 | 51/73 | 4/73 + | 4/73 + | 3/73 + | 4/73 + | NR | 5/72 + | 6/73 + | 0/73 + | |||
| Female | Acute [14] | 2 h (1×) | 0 | 13/57 | NR | NR | 3/56 | 0/57 | 2/57 | 5/56 | NR | 0/53 |
| 1000 | 19/56 | NR | NR | 4/56 | 1/56 | 1/56 | 6/55 | NR | 0/52 | |||
| 5000 | 18/57 | NR | NR | 0/57 | 0/57 | 3/57 | 8/57 | NR | 1/53 | |||
| 10,000 | 13/58 | NR | NR | 3/58 | 0/58 | 4/58 | 3/58 | NR | 0/56 | |||
| Lifetime [3] | 6 h/d, 5 d/wk, 103 wk | 0 | 6/50 | 3/50 | 0/50 | 4/50 | 0/50 | 0/50 | 15/49 | 8/50 | 1/49 | |
| 6.25 | 12/50 | 2/50 | 0/50 | 15/50 | 0/50 | 2/50 | 14/49 | 10/50 | 0/49 | |||
| 20 | 11/50 | 7/50 | 0/50 | 19/50 | 3/50 | 4/50 | 15/50 | 7/50 | 1/48 | |||
| 62.5 | 7/50 | 4/50 | 1/49 | 24/50 | 2/50 | 12/50 | 19/50 | 15/50 | 9/50 | |||
| 200 | 9/50 | 7/50 | 21/50 | 25/50 | 4/50 | 15/50 | 16/50 | 20/50 | 8/50 | |||
| 625 | 32/80 | 4/80 + | 23/80 + | 22/78 + | 22/80 + | 16/80 + | 2/80 + | 9/80 + | 6/79 + |
| Target Tissues | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gender | Duration | Exposure | Concentration (ppm) | Pancreas | Zymbal | Mammary | Thyroid | Glial Cell | Testis, Uterus |
| Male | Lifetime | 6 h/d, 5 d/wk, 103 wk | 0 | 3/100 | 1/100 | 1/100 | 3/100 | 1/100 | 0/100 |
| 1000 | 1/100 | 1/100 | 2/100 | 5/100 | 4/100 | 3/100 | |||
| 8000 | 11/100 | 2/100 | 0/100 | 1/100 | 5/100 | 8/100 | |||
| Female | Lifetime | 6 h/d, 5 d/wk, 103 wk | 0 | 2/100 | 0/100 | 50/100 | 0/100 | NR | 1/100 |
| 1000 | 0/100 | 0/100 | 79/100 | 4/100 | NR | 4/100 | |||
| 8000 | 0/100 | 4/100 | 81/100 | 11/100 | NR | 5/100 | |||
| Metabolite-Specific Unit Internal Dose (nM*h per ppm*h BD) 1 | |||||
|---|---|---|---|---|---|
| Mouse | Rat | Human 1 | |||
| Metabolite | Female | Male | Female | Male | Male |
| EB | 13 ± 2 | 15 ± 2 | 0.77 ± 0.1 | 0.72 ± 0.1 | 0.11 ± 0.076 |
| DEB | 27 ± 7 | 38 ± 8 | 1.45 ± 0.2 | 1.37 ± 0.3 | 0.024 ± 0.020 |
| EBD | 266 ± 71 | 210 ± 30 | 19 ± 0.9 | 19 ± 2 | 52 ± 36 |
| Metabolite | |||||
|---|---|---|---|---|---|
| Endpoint | EB | DEB | EDB | In Vitro Cell System | Reference |
| DNA Damage | 1.00 | 11.21 | 0.961 | Human hepatocytes, pH 11.9 | [43,44] |
| 1.00 | 4.22 | 0.955 | Human hepatocytes, pH 9 | ||
| DNA Damage Mean ± SD | 1.00 | 7.72 ± 4.94 | 0.96 ± 0.004 | ||
| Mutations | 1.00 | 81.66 | 2.10 | Human TK6 (HPRT) | [45] |
| 1.00 | 277.12 | 4.46 | Human TK6 (TK) | ||
| 1.00 | 58.10 | 0.45 | Human TK6 (HPRT) | [46] | |
| 1.00 | 114.83 | 0.71 | Human TK6 (TK) | ||
| 1.00 | 49.08 | 0.35 | BB Mouse Fibroblasts | [47] | |
| — 2 | — 2 | — 2 | BB Rat Fibroblasts | ||
| 1.00 | 4.20 | 3.87 | SA T100 | [48] | |
| Mutations Mean ± SD | 1.00 | 97.5 ± 95.3 | 1.99 ± 1.81 | ||
| Micronuclei | 1.00 | 128.28 | 0.58 | BB Mouse Fibroblasts | [47] |
| 1.00 | 124.08 | 0.74 | BB Rat Fibroblasts | ||
| — 2 | — 2 | — 2 | Rat spermatids | [49] | |
| Micronuclei Mean ± SD | 1.00 | 126.18 ± 2.97 | 0.66 ± 0.12 | ||
| Overall Mean ± SD 3 | 1.00 | 85.28 ± 82.81 | 1.52 ± 1.48 | ||
| Individual Metabolites | |||||
|---|---|---|---|---|---|
| Parameter (units) | Species/Extrapolation | EB | DEB | EBD | Metabolites Combined 3 |
| Genotoxicity Index (nM*h per ppm*h BD) 1 | Female Mouse | 13.0 | 2303 | 404 | 2719 |
| Male Mouse | 15.0 | 3241 | 319 | 3574 | |
| Female Rat | 0.77 | 124 | 28.8 | 153 | |
| Male Rat | 0.72 | 117 | 28.5 | 146 | |
| Human | 0.109 | 2.04 | 79.2 | 81.4 | |
| EFAK (Unitless) 2 | Human: Female Mouse | 0.00842 | 0.000886 | 0.196 | 0.0300 4 |
| Human: Male Mouse | 0.00730 | 0.000630 | 0.249 | 0.0228 4 | |
| Human: Female Rat | 0.142 | 0.0165 | 2.75 | 0.531 4 | |
| Human: Male Rat | 0.152 | 0.0175 | 2.75 | 0.556 4 | |
| Dataset | Range of Model Fit Statistics for Individual Tumor Types | Unit Risk for Combined Tumor Types (ppm−1 HEC) * | |||
|---|---|---|---|---|---|
| Dataset | N | Range of Observation, (HEC, ppm Continuous) | p-Values | AICs | |
| Female Mouse (Table 2) | 558 | 52–27,800 | 0.103–0.867 | 81.6–349.1 | 8.8 × 10−4 (5.7 × 10−4–1.2 × 10−3) |
| Male Mouse (Table 2) | 756 | 49–36,550 | 0.052–0.966 | 35.6–337.3 | 3.5 × 10−4 (2.8 × 10−4–4.3 × 10−4) |
| Female Rat (Table 3) | 300 | 336–2690 | 0.00016–0.969 | 35.7–357 | 6.7 × 10−5 (4.2 × 10−5–9.6 × 10−5) |
| Male Rat (Table 3) | 300 | 321–2570 | 0.131–0.163 | 88.7–109 | 1.4 × 10−5 (7.5 × 10−6–2.1 × 10−5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirman, C.R.; Hays, S.M. Use of Biomarker Data and Relative Potencies of Mutagenic Metabolites to Support Derivation of Cancer Unit Risk Values for 1,3-Butadiene from Rodent Tumor Data. Toxics 2022, 10, 394. https://doi.org/10.3390/toxics10070394
Kirman CR, Hays SM. Use of Biomarker Data and Relative Potencies of Mutagenic Metabolites to Support Derivation of Cancer Unit Risk Values for 1,3-Butadiene from Rodent Tumor Data. Toxics. 2022; 10(7):394. https://doi.org/10.3390/toxics10070394
Chicago/Turabian StyleKirman, Christopher R., and Sean M. Hays. 2022. "Use of Biomarker Data and Relative Potencies of Mutagenic Metabolites to Support Derivation of Cancer Unit Risk Values for 1,3-Butadiene from Rodent Tumor Data" Toxics 10, no. 7: 394. https://doi.org/10.3390/toxics10070394