
iPS Cells—The Triumphs and Tribulations

Department of Craniofacial Development and Stem Cell Biology, Dental Institute, King’s College London, London SE1 9RT, UK
Dent. J. 2016, 4(2), 19; https://doi.org/10.3390/dj4020019
Submission received: 1 March 2016
/
Revised: 18 May 2016
/
Accepted: 27 May 2016
/
Published: 6 June 2016
(This article belongs to the Special Issue Craniofacial Biology for Tooth Repair and Regeneration)
Abstract
:The year 2006 will be remembered monumentally in science, particularly in the stem cell biology field, for the first instance of generation of induced pluripotent stem cells (iPSCs) from mouse embryonic/adult fibroblasts being reported by Takahashi and Yamanaka. A year later, human iPSCs (hiPSCs) were generated from adult human skin fibroblasts by using quartet of genes, Oct4, Sox2, Klf4, and c-Myc. This revolutionary technology won Yamanaka Nobel Prize in Physiology and Medicine in 2012. Like human embryonic stem cells (hESCs), iPSCs are pluripotent and have the capability for self-renewal. Moreover, complications of immune rejection for therapeutic applications would be greatly eliminated by generating iPSCs from individual patients. This has enabled their use for drug screening/discovery and disease modelling in vitro; and for immunotherapy and regenerative cellular therapies in vivo, paving paths for new therapeutics. Although this breakthrough technology has a huge potential, generation of these unusual cells is still slow, ineffectual, fraught with pitfalls, and unsafe for human use. In this review, I describe how iPSCs are being triumphantly used to lay foundation for a fully functional discipline of regenerative dentistry and medicine, alongside discussing the challenges of translating therapies into clinics. I also discuss their future implications in regenerative dentistry field.
1. Introduction
The discovery of Embryonic Stem Cells (ESCs) [1,2,3] incited the search for discovering artificial differentiation techniques to confer the properties of ESCs onto somatic cells by altering epigenomic activity, such that the derived cells are pluripotent and capable of giving rise to embryonic-like stem cells. These techniques are collectively referred to as cellular reprogramming. Yamanaka’s pioneering experiments made it possible in 2006 to create embryonic-like stem cells from mouse fibroblast cultures, without using embryos by engineering his creation in vitro [4].
Human induced pluripotent stem cells (hiPSCs) were generated by using either by retroviral transduction of fibroblasts encoding the original four transcription factors, constitutively expressed the POU domain class 5 transcription factor 1 (Oct3/4), the sex determining region Y-box2 (Sox2), Kruppel-like factor 4 (Klf4), and myelocytomatosis oncogene (c-Myc) also referred as OSKM—“Yamanaka’s cocktail” [5], or by independently determined combination of lentivirally transduced genes Oct3/4, Sox2, NANOG (Nanoghomeobox), and Lin28 [4,6,7]. While these reprogrammed cells have similar developmental potential as authentic hESCs, they come without the baggage of morality and ethics, as they are not derived from human embryos and the possibility of immune rejection from allogeneic transplantation. In addition, these hiPSCs resemble hESCs in their morphology and gene expression and can differentiate into cell types of all the three primary germ layers (ectoderm, endoderm and mesoderm) in vitro and in vivo (Figure 1).
In this review, I present a comprehensive overview of factors playing role in generation of iPSCs and the present day cellular reprogramming alternatives. I will discuss applications and advantages of iPSCs followed by challenges associated with their clinical applications. In the end, I will briefly discuss the future prospects of iPSCs in the field of regenerative dentistry.
2. Factors of Importance in the Generation of iPSCs
The reprogramming factors have their individual role and at the same time, they interact with each other complimentarily. Two methods for delivering the reprogramming transcription factors into the somatic cells are, Integrating Viral Vector Systems and Non-integrating Systems (Figure 2). The viral vector gets integrated into host genome in case of integrating methods. The use of retrovirus and lentivirus falls into this category. However, long-term safety of hiPSCs cannot be assured through mouse studies alone. In addition, even though this method is highly efficient, there is a risk of multiple chromosomal disruptions, any of which may cause genetic dysfunction and/or tumorigenesis. In addition, retroviruses may make iPSCs immunogenic [9]. Thus, we will need to avoid induction methods that involve vector integration into the host genome for the purpose of cell transplantation therapy and hence, altered methodologies have been toiled upon. In non-integrating methods, there is no integration in the host cell genome. The use of Viral vectors like the Adeno virus [10] and Sendai virus [11], plasmid DNA [12,13], synthesized mRNAs [14] and proteins [15] fall under this category. Plasmids such as oriP/EBNA1 (derived from Epstein-bar virus) have been used for reprogramming but they have demonstrated to be of low efficacy [16]. Direct delivery of reprogramming proteins has also been carried out by fusing them with a cell penetrating peptide [15]. A different approach using a single self-replicating RNA replicon, which expressed high levels of Yamanaka factors for transfection into fibroblasts to be reprogrammed into iPSCs, was used and iPSCs displayed all properties of pluripotent stem cells [17]. Finally, small-molecule drugs have been investigated for establishing safe methods of iPSC generation for clinical application because they are non-immunogenic, cost-effective, and easy to handle [18]. Recently, successful reprogramming of mouse somatic cells without transgene introduction was achieved with small-molecule drug combinations [19].
Human skin fibroblasts were used as a starting material for generation of iPSCs and are the major source due to their commercial availability and ease of gene-delivery. However, the process is lowly efficient as typically less than 1% of transfected fibroblasts become iPSCs. In addition, fibroblasts are not amenable to large-scale production due to the technical challenges of establishing stable fibroblast cell lines and the need for invasive skin biopsies to obtain human tissue. iPSCs can also be derived from keratinocytes, mesenchymal cells, adipose stem cells, melanocytes [20] and postmitotic neurons [21].
A more desirable source is human peripheral blood as is easily obtainable through routine, non-invasive clinical procedures. Their use also enables the creation of iPSCs from large number of donor samples stored in biorepositories worldwide. Efforts to develop such methods have yielded iPSCs from human CD34+ blood cells and T-lymphocytes [22,23,24,25]. It was revealed in a particularly key study that hiPSCs can be efficiently derived from T lymphocytes from as little as 1 mL of whole blood [26]. Thus, most, if not all, somatic cells have a potential to become iPSCs, albeit with different efficiencies [27].
3. A Novel Culture System for iPSCs Derivation
In order to use hiPSCs in regenerative medicine, the cells must be prepared using methods compliant with GMP (Good Manufacturing Practice). However, the methods in use up till now use feeder cells to culture iPSCs, which involve complex procedures, and employ culture medium containing serum and numerous other animal-derived constituents, which make it difficult for them to comply with GMP and hence unsuitable for regenerative therapies in humans. Chen et al. [28] recently reported the development of a significantly improved hiPSC culture medium, TeSR™-E8™, which contains only eight completely defined and xeno-free (free of animal-derived constituents) components. TeSR™-E8™ is based on the E8 formulation published by Dr. James Thomson, the lead researcher behind the mTeSR™1 formula [29,30] and contains a minimized set of the components required for maintenance of hiPSCs, providing a simpler medium. This medium is low in protein compared to other conventional feeder-free culture medium such as mTeSR™1 and TeSR™2. Beers et al. [31] described an EDTA-based, enzyme-free passaging protocol for routine maintenance and reprogramming of iPSCs, which achieves maximum cell survival without enzyme neutralization, centrifugation or drug treatment. Wang et al. [32] demonstrated an efficiently scalable culture system using the E8 for the expansion and cryopreservation of hiPSCs under adherent and suspension culture conditions. Another method for generation and maintenance culture of iPSCs that are suitable for use in cell transplantation therapy has been recently developed [33]. They established that recombinant laminin-511 E8 fragments are useful matrices for maintaining hiPSCs when used in combination with a completely xeno-free medium, StemFit™. Using this system, hiPSCs can be easily and stably passaged by dissociating the cells into single cells for long periods, without any chromosomal/karyotype abnormalities. hiPSCs could be generated under feeder-free and xeno-free culture systems from human skin fibroblasts and blood cells, and they possessed differentiation abilities. These results indicate that hiPSCs can be generated and maintained under this novel culture system making them ideal for cell transplantation in humans. In fact, the cell production company Lonza has generated current GMP-qualified xeno-free hiPSC lines from peripheral blood mononuclear cells, which are now available for clinical applications [34].
The microenvironment is also an important factor for consideration when developing a reliable and GMP-compliant cell culture system for regenerative therapies. Matthias Lutolf and colleagues have recently described a 3D culture system that promotes iPSC generation by modulating microenvironmental stiffness, degradability and biochemical composition [35]. They show that exposure to a 3D microenvironment enhances cell reprogramming compared to the traditional 2D environment suggesting that the 3D microenvironment keeps cells in an active proliferation state throughout the reprogramming process and that induction of iPSCs might prove to be faster in 3D conditions. They also show that with respect to a 2D system, the physical cell confinement imposed by the 3D microenvironment can increase the reprogramming efficiency of both mouse and human iPSCs by more than two-fold and, as with a recent 2D culture system, this occurs via an accelerated mesenchymal-to-epithelial transition and increased epigenetic remodelling. 3D models are also able to incorporate the spatial and structural features of tissues and/or organs affected by diseases and thereby could reveal more relevant information. For example, one study recapitulated the pathological phenotypes of Alzheimer’s disease with a 3D model [36]. Researchers have already generated 3D cardiac tissues from hiPSCs that successfully model the human heart [37]. Recently, they also developed organoids with iPSCs that mimic the cerebrum [38].
4. Applications of iPSCs
The treatment of multiple diseases is challenging/impossible because of inadequate information is available on the mechanisms involved in the disease progression. To be able to develop the treatment aiming at the root of the disease, diseases need to be modelled. There are already large numbers of disease testing models available but only few of them are adept at simulating human cellular microenvironment and metabolism to some extent. Animal models of rat, mice, monkey, dog, and primates have been used for disease modelling until now. However, their use is limited due to existing variability in the genetic make-up of them, which is mainly accountable for the biological functions, and hence metamorphoses are exhibited compared to humans. Thus, an altered approach is desired which can offer identical environment as human cells and iPSCs seem like an amenable substitute with plethora of advantages. iPSCs do not require multiple proliferation and their derivatives are functional after transplantation in vitro as well as in vivo [39,40,41,42,43]. Hence iPSCs are used in therapeutics for disease modelling, drug screening and regenerative medicine. Another significant use of iPSCs is to generate differentiated cells that are unavailable from adult sources that can integrate into the recipient and replace the damaged or missing cells. Examples of such therapies include retinal pigment epithelial (RPE) cell replacement in macular degeneration, making liver cells to treat liver cirrhosis, the generation of dopaminergic neurons to treat Parkinson disease, or deriving cardiac myocytes or pancreatic islets to treat cardiac disease or diabetes. In regenerative dentistry, iPSCs can be utilised for oral tissue regeneration and development of clinical treatments for the congenital diseases.
4.1. Disease Remodelling
We can group human diseases into three broad categories: genetic, epigenetic and acute environmental [44]. Modelling of all three types is possible in vitro using stem cells, and an excellent way to study the intricate mechanisms and pathways underlying the aetiology and pathophysiology of disease. Stem cells in general are ideal for creating “disease-in-a-dish” models because of their capacity for self-renewal and differentiation, their potential for recapitulating disease pathogenesis, and also their amenability for developing and testing therapeutics [45].
Using patient-specific iPSCs we can recapitulate the suspected effects of the environmental or epigenetic component known to have contributed to the patient’s disease in order to understand its severity or mechanism of action before, during, or after reprogramming. Such models can help us gain better insight into the environmental or epigenetic factors affecting a complex disease in terms of susceptibility, prognosis as well as outcomes. Patient-specific models can also be used as special models, as they can involve known epigenetic changes contributing to the disease. iPSCs can also be used for modelling disease at the organ level as well as understanding systemic diseases.
Multiple diseases have been successfully modelled using iPSCs, for example, Alzheimer’s disease [46,47], Parkinson’s disease [48,49], amyotrophic lateral sclerosis (ALS) or Lou Gehrig’s disease [50,51,52], Huntington disease [53,54], Downs syndrome/trisomy 21 [55], Type 1 diabetes mellitus [56], etc. Blood disorder disease modeling has been focused on sickle cell anaemia and leukemia [57,58,59,60,61]. The cell lines developed for disease modelling were also found to be consistent phenotypically, which is necessary for disease modelling. Fong et al. developed Tauopathy derived iPSCs carrying a TAU-A152T mutation and the phenotypes they observed in the cells from iPSCs were consistent with those in patients with the mutation [62]. There are many human diseases, which have not been recapitulated in small animal models, especially in adult onset diseases [63].
4.2. Drug Screening
iPSCs can be beneficially used for drug discovery and cytotoxic studies in humans. In vitro animal derived cells have been used as testing systems up until now but their incompetence in replicating the “exact” human physiological environment and related phenotypic attributions renders their usage. Sometimes, the benefits proven in the animal models do not turn out to be of value in humans at all. In addition, animal models are inappropriate for testing the drug toxicity as the chemical toxicity differs from animal to animal and the same applies to carcinogenic agents. Finally, a newly discovered drug or therapy must be tested on human cells or test models before being introduced in market so that the results can be concluded for safe administration in humans. These studies include steps such as prediction/identification of a potential drug molecule followed by its synthesis, generation of iPSCs, their differentiation to specific somatic cells, and testing for toxic or non-toxic effects of the synthesized drug on the somatic cells. For toxicity studies, iPSCs from normal and diseased cells are differentiated to specialized cell types.
Only 10% of the drugs that enter clinical trials are able to reach the market approval stage. The cost of developing a drug is increasing with the estimated cost of whole process being US $1.2–1.7 billion per drug compound [64,65,66]. The development of 30% of the medicines was abandoned because of lack of efficacy and 30% due to safety concerns (cardiotoxicity, hepatotoxicity) [67]. The process becomes costlier and extensively time consuming due to early detection failure of drug toxicity in human tissues. Development of toxicity models that predict more precisely before starting the clinical trials may help cut costs and time by demonstrating cardiotoxicity, hepatotoxicity or cytotoxicity caused by the drugs.
iPSCs have been explored by many research groups as their use offer better substitute to conventional tests and better chemical safety assessment as they provide similar environment to human physiological conditions than conventional animal testing models. For example, pluripotent stem cells have been used previously to establish test systems for cytotoxicity [68].
The variability and possibly incomplete reprogramming of iPSCs from somatic cells remain a considerable challenge in the integration of iPSCs into drug discovery. The process of reprogramming involves TET enzymes that mediate the generation of 5-hydroxymethylcytosine (5hmC). A study comparing the activities of TET enzymes in hESCs and hiPSCs suggests that hiPSCs represent an incompletely reprogrammed pluripotent state. This difference in 5hmC detected between hESCs and hiPSCs is also present between clones of hiPSCs, which may account for the variation between hiPSC lines [69]. This variability will likely affect the reproducibility of the results from these studies making it important to develop robust differentiation protocols and reliable assessments of the functionality of a hiPSC-derived disease model before fully integrating these cells into the process of drug development.
As iPSCs can be derived from individual patients, these offer scientists an opportunity for modelling diseases on a patient-by-patient basis. This enables screening the genomic differences between individuals that may help in the progression of disease, and the screening of pharmacological agents to find the ideal one for each individual [70]. During the drug screening procedure, hepatocytes play a central role due to their detoxification capacity. The generation of foetal hepatocyte-like cells from iPSCs resulted in differentiated cells that did not display the functions of fully mature hepatocytes and their viability after cryopreservation is extremely variable [71] which is a major drawback for iPSC-derived drug screening platforms. However, Zhang et al. [72] showed that hiPSC-derived mature hepatocyte-like cells hardly ever proliferated in vitro, and in contrast, hiPSC-derived hepatic endoderm cells exhibited a marked proliferative capability. Due to the lack of standardization of protocols, the amount of available mature hepatocytes have been insufficient for clinical therapy and drug development in the past several decades [73].
4.3. Regenerative Medicine
In regenerative medicine, the injured/degenerated tissues are repaired by generating those tissues with the help of iPSCs in labs and then transplanting them to the site of injury/degeneration. The core concerns with the current transplantation therapy are availability of organ or tissue and immunorejection. There is ever increasing need for organs but quite a shortage of donors, which leads to death of the patients suffering from degenerative disease or accident stricken. In addition, the cell, tissue or organ transplant is only possible from disease-free and physiologically matching donor profile with the patient, hence multiple tests are necessary before accepting/transplanting the donor tissue or organ. The iPSCs are our best bet for the reason being transplanted cells will be differentiated from the repaired iPSCs generated from patient’s own somatic cells. Once the specific cells are formed, they can be directly transplanted to the specific site to cure the degenerative disease. In case of diseased cells possessing mutation, the mutation is first corrected in order to be able to form normal iPSCs, and then these iPSCs are differentiated into specific cell types by providing specific conditions essential for the development of those cells. These repaired cells can then be transplanted into the body of the organism from which the cells for the generation of iPSCs were isolated.
A major difficulty in the application of iPSCs for regenerative medicine is the delivery of reprogramming factors. piggyBac transposon-based approach to generate integration-free iPSCs is highly efficient and most promising [74,75,76] because piggyBac excises without a footprint, leaving the iPSC genome without any genetic alteration and with hallmark pluripotency markers. Though the existence of piggyBac-like elements in the human genome [77] raises the question of safety concerns. Another concern shared by both non-viral and viral vectors, is the integration site specificity and its counterpart insertional mutagenesis. Sequence-specific DNA-binding proteins such as homing nucleases, zinc finger proteins (ZFPs), transcription activator-like effector nucleases (TALENs) and Cas9 of the CRISPR/Cas system have been adapted to introduce gene editing which in turn might cause safety issues, not fully investigated so far [78].
The complete list of conditions, which could be treated with iPSCs in the future, would be beyond the scope of this review but here are a number of conditions that can potentially be treated with this technology. Hematopoietic disorders, liver damage and Spinal cord injury could be treated by the generation of specific cells with the help of iPSCs [79,80,81].
Of late, researchers have been assembling data on the use of iPSC for ex vivo blood expansion of various blood components. They can be used for the generation of Red Blood Cells (RBCs), which could be utilized for generating blood for the purpose of treatment of various damages/diseases prevalent in world. There are several techniques by which we can use iPSCs for the production of RBCs [82].
iPSCs can also be used for the generation of various cells which can help in the repair of many tissues, for example, cardiomyocytes for repairing heart valves, vessels and ischemic tissues, but there are limitations like post treatment side effects, safe delivery and protocol standardization to produce big volumes of pure, good quality cells. These hurdles once overcome, offer great opportunities for iPSC applications for generating cardiovascular cells and studying corresponding diseases [83,84,85].
Another plus is iPSCs can be derived from immune cells as equally as they can redifferentiate into specific immune cell types for clinical immunotherapy. Dedritic cells (DCs) are the most potent antigen-presenting cells. Thus, DC-based cellular vaccination provides a powerful means for immunotherapy, especially against cancer. In addition, antigen-specific negative regulation of immune response by DCs is considered to be a promising approach to treat autoimmune diseases and in transplant medicine. iPSCs may be an ideal source for DCs that can broaden their immunotherapeutic applicability. In 2009, Senju et al. [86] reported that the iPSC-derived DCs are functional in that they effectively processed and presented antigens, stimulated T cells, and produced cytokines.
4.3.1. First ever Clinical Trial
At Riken Centre for Developmental Biology, Japan, the Takahashi team clinical study intended to examine the safety of a human RPE cell product made from each patients’ own iPSCs had been going on. Takahashi’s team conducted safety trials in both monkeys and mice before the first ever iPSC clinical study in humans [87,88]. The animal tests revealed that iPSCs were not rejected and did not cause cancerous growth. Japan health-ministry committee assessed researchers’ safety tests and cleared the team to begin the experimental procedure in September 2014 [89,90]. In an astounding feat of swift clinical translation, Takahashi’s team transplanted its first macular degeneration patient on 12 September 2014 (under GMP conditions) only 7 years after hiPSCs were first ever published [91]. The usual timeline for such translation would be 20 years. Takahashi had reprogrammed some cells from the 70-year old woman patient’s skin to produce iPSCs. She then coaxed those cells to turn on the retinal genes, differentiate into RPE cells and grow into a sheet for implantation. After surgically removing spurious blood vessels and damaged tissue from the affected eye, this cell sheet was transplanted to the damaged area of the retina. The researchers anticipate the cell growth at the transplanted site, which in turn will expectantly repair the pigment epithelium. The procedure has apparently progressed satisfactorily and has been able to stop patient’s vision from further deterioration; however, the experimental procedure was not performed on further individuals when genetic mutations were identified in the iPSCs [92]. In 2015, Zhao et al. [93] demonstrated in humanized mice that an immune response is mounted against iPSC-derived smooth muscle cells but not iPSC-derived RPE, suggesting that autologous iPSC derivatives may have differential immunogenicity.
4.3.2. Ongoing Clinical Trials
Phase I–II clinical trials using hESC-derived insulin-producing progenitor β-cells as a subcutaneous flat encapsulated product, a little smaller than a credit card, is underway by the company ViaCyte (San Diego, CA, USA) to treat type 1 diabetes patients [94].
Recent studies have shown that hESC-derived cardiomyocytes and cardiovascular progenitors are able to halt the deterioration of cardiac function and improve experimentally induced diminished heart function in rodent and monkey models [95,96]. Moreover, in a monkey myocardial infarct model, intramyocardial delivery of 1 billion hESC-derived cardiomyocytes revealed extensive remuscularization; although, potential complications due to ventricular arrhythmias are still a concern.
A 68-year-old patient suffering from severe heart failure was surgically grafted (Paris, France) onto the infarcted area with hESC-derived cardiomyocyte progenitors expressing the early cardiac transcription factor insulin gene enhancer Isl-1 and stage-specific embryonic antigen (SSEA)-1, which are cell markers for cardiac progenitors. The cardiac progenitors were then embedded into a fibrin scaffold to enable the integration of grafted cells from the patch into damaged heart tissue [97]. A coronary artery bypass surgery was performed concomitantly in a non-infarcted area followed by uncomplicated post-operative recovery. The patient symptomatically improved after 3 months, although this may have been owing to revascularization resulting from the coronary bypass surgery. Notably, contractility was echocardiographically observed in the previously akinetic heart patch area, and no arrhythmias, tumour formation, or immunosuppression-related adverse effects were evident [97,98].
Asteria Biotherapeutics (California, USA) with the funding from California Institute for Regenerative Medicine (CIRM) started phase I–II clinical trial on patients suffering from spinal cord injury by injecting them with hESC-derived oligodendrocyte progenitors in 2015. They started the trial with a dose escalation study where the first group of three patients received two million cells. Apparently, the dosage has been safe and one patient has shown encouraging results. At present, they are enrolling patients for the 10 million cell dose trial, which if successful, is to be followed by 20 million cell dose [99].
In Australia, there is a recent announcement by International Stem Cell Corporation (USA) that will begin phase-I clinical trial to treat Parkinson’s disease by injecting 12 sufferers with parthenogenetic (pluripotent human stem cells from unfertilized oocytes) neural stem cells and observe them for 1 year [100].
4.4. Development of iPSC Library
One of the major challenges of the use of ESCs in cell therapies is an immune-mediated rejection after transplantation. Today, this problem can be overcome by direct reprogramming of patients’ somatic cells and by creating an iPSC bank consisting of various human leukocyte antigens (HLA) types thus providing therapeutic tool for the patients needing cell transplantation, free from immune-mediated rejection [101,102]. In addition, each cell line should contain detailed genetic and epigenetic profiles and differentiation potential ‘‘score cards’’ [103,104]. Two works reported that the establishment of 50 unique stem cells lines, having homozygous alleles of the 3 HLA loci (A, B, and DR), would cover 90% of the Japanese population with a faultless match of these loci [105,106]. Considering that derivation and testing iPSCs tailored for individual patients is a time consuming process (about 6 months for each cell line) and costs tens of thousands of dollars, it is quite necessary for cell therapies and regenerative medicine to establish iPSC banks with a sufficient collection of HLA types, thus avoiding additional expenses which are required for iPSC production for each individual patient.
The Center for iPSC Research and Application (CiRA), Kyoto University, Japan under the guidance of Shinya Yamanaka, in collaboration with Kyoto University Hospital, Japan embarked on a project of building the Clinical iPS Cell Bank of Kyoto (CiBK) with lines from approximately top 100 haplotypes donors providing coverage for about 90% of the population in Japan. Yamanaka’s project has an advantage because genetic diversity in Japan is relatively low; elsewhere, therapeutic banks would have to be larger and costlier. Using blood from Japan’s eight cord-blood banks will also make the task easier as the banks hold some 29,000 already HLA-characterized samples. The establishment of iPSCs is to be done at clinical grade, from peripheral blood or umbilical cord blood, in the Cell Processing Center that is part of CiRA [107].
Most iPSC banks outside Japan specialize in cells from people with diseases, for use in research rather than treatment. The California Institute for Regenerative Medicine (CIRM) in San Francisco, for example, plans to bank some 3000 cell lines for distribution to researchers [108].
Recently, the human iPSC initiative (HipSci) reported the establishment of a high-content platform for phenotypic analysis of human iPSC lines [109]. In the described assay, cells are dissociated and seeded as single cells onto 96-well plates coated with fibronectin at three different concentrations. This method allows assessment of cell number, proliferation, morphology and intercellular adhesion. Altogether, this strategy delivers robust quantification of phenotypic diversity within complex cell populations facilitating future identification of the genetic, biological and technical determinants of variance. Approaches such this can be used to benchmark iPSCs from multiple donors and create novel platforms that can readily be tailored for disease modelling and drug discovery. This platform could make the enormous task of developing iPSC library on a world-scale to cater the diverse population less cumbersome.
5. Challenges
5.1. Choosing an Appropriate Somatic Cell Type
Choosing a suitable cell type for reprogramming is of critical concern for future autologous patient-specific iPSC production and clinical therapy. The ideal cell source to be isolated from the patients and used for reprogramming must meet the criteria of easy accessibility with minimal risk procedures, availability in large quantities, relatively high reprogramming efficiency, and fast iPS cell derivation speed. Moad et al. used human prostate and urinary tract cells for the formation of iPSCs and further for studying the mechanisms that regulate the differentiation of prostate and urinary tract cells. With their study, they concluded that iPSCs generated from prostate and urinary tract had better efficiency of differentiation to cells of prostate and urinary tract as compared to iPSCs derived from skin fibroblasts which showed that organ of origin plays an important role in terms of efficiency of differentiation [110]. Table 1 shows a comparison of the different cell origins that have been used for reprogramming.
Another study has shown that persistent donor cell gene expression memory in hiPS cell lines can contribute significantly to the differences among hiPSCs and human ESCs, and adds to the incompleteness of reprogramming [118]. Hence, the optimal cell source for generating patient-specific iPSCs should be carefully selected on a patient-specific basis, when it proves possible to evaluate specific conditions of individual patients in the future [111].
5.2. Variability and Heterogeneity
A vital factor of consideration in iPSCs generation is a concern raised by studies performed on genetic and epigenetic variations in iPSCs and how these variations compromise the utility of iPSCs and undermine their accountability in downstream applications [119]. These variations exist between iPSC lines, between iPSC and ESC lines, between different passages of the same iPSC line, and even between different populations at a specific passage of the same iPSC line. These variations occur due to different sources; some of the variations may be inherited from donor somatic cells, induced or selected by the reprogramming process, or accumulated during culturing; others may simply reflect the innate genetic and epigenetic stability of the pluripotent state of iPSCs. Although each variation is not relevant to the functionality of iPSCs, certain variations may change the properties of iPSCs and their derivatives. For example, the variations may alter the differentiation potential of iPSCs, cause phenotypic changes in iPSC-derived somatic cells, or increase the tumorigenicity or immunogenicity of iPSCs and their derivatives. One of the causes for the varied differentiation capacity is source cell memory, which biases iPSC differentiation into the source cell lineage [120,121,122]. These adverse changes directly affect the utility of iPSCs. Accumulating evidence also indicates that epigenetic mechanisms not only play important roles in the iPSC generation process, but also affect the properties of reprogrammed iPSCs [123]. Extended passage of some iPSC clones in culture did not improve their epigenetic resemblance to ESCs, implying that some human iPSCs retain a residual ‘epigenetic memory’ of their tissue of origin [121]. In a study performed by Deng et al. [124] iPSCs appeared to display more methylation than ESCs. Some studies have identified hotspots or sets of “signature” genes in hiPSCs whose DNA methylation and transcription statuses are clearly different from that of hESCs [124,125,126,127]. However, another comprehensive study concluded that the variations of DNA methylation between hESCs and hiPSC lines are not greater than those between different hESC lines [103]. These studies question the biological consequences of such deviations in pattern for future therapeutic applications of iPSCs and demand more in-depth research to understand the reprogramming process thoroughly.
Despite the common ability of hiPSCs and hESCs to differentiate into all 3 germ layers, a recent single cell analyses revealed much more heterogeneity in gene expression levels in iPSCs than in ESCs [128]. This suggests exercising caution before assuming that hiPSCs occupy a pluripotent state equivalent to that of hESCs, especially when producing differentiated cells for regenerative therapeutic aims.
5.3. Validation of Pre-Clinical iPSC Therapies
As discussed above, Takahashi’s team successfully transplanted first patient with reprogrammed iPSCs as RPE cells in September 2014 after safety trials on monkeys and mice both. The safety and therapeutic applications of these cells must be meticulously tested in appropriate animal models before advancing to any other clinical trial as well. We need to study minimum number of undifferentiated iPSCs that can cause teratoma or teratocarcinoma in autologous transplantation animal models, as residual undifferentiated cells may still remain after differentiation into specific cell lineages and may lead to tumorigenicity after transplantation.
Another concern is oncogenic transgene integration and insertional mutagenesis may be associated with many of the currently established iPS cell lines, the questions of whether iPSCs generated with different reprogramming technologies as well as their derivatives can induce cancer in the host also need to be strictly evaluated. Even with improvements in the virus-free and transgene-free reprogramming technologies, the cancer-causing possibility of the derived “safe” iPSCs/derivatives still needs to be assessed in animal models before using them clinically for regenerative therapy. In addition, iPS cell therapies need to be validated not only in small animals (mice and rats) but also in large animal models that are anatomically and physiologically more similar to humans. Both monkey [129] and pig [130,131,132] iPS cells have been generated, providing excellent models for transplantation studies. Although the thorough pre-clinical evaluation of iPS cells would be arduous, it is absolutely essential for the future.
5.4. Regulatory and Commercial Hurdles
Given the many probable risks of applying autologous iPS cell treatment to humans, iPSC therapies may encounter strict regulatory restrictions in some parts of the world, including in Europe, United Kingdom and the United States. In addition, clinical GMP-compliant vectors for iPSC reprogramming are currently unavailable. Another issue that may impede the clinical translation is the financial feasibility of producing individualized iPSC therapeutic products. The viability of a business model for patient-specific iPS treatment is still unknown. It may well be the case that few if any pharmaceutical companies will be able to produce cost-effective individualized iPSC products tailored for a single patient at a time. On the other hand, we cannot deny the exceptional potential of iPS therapies. For instance, if researchers are able to solve the immune tolerance problem [133] then one can expect allogeneic transplantation of iPSC products. These cells will need to be made in GMP-compliant, large-scale production for it to be commercially viable [134], and the individual needs or profiles of patients will need to be easily assessed to allow matching and wide distribution. For example, an optimized dose for hematopoietic stem cell transplantation for a 70-kg adult patient was suggested to contain 4.2 × 108 to 5.6 × 108 CD34+ cells [135]. Production of a clinically relevant quantity of hiPSCs and/or their progenies for specific applications, sometimes considered as ~1 to 2 billion [136], in a chemically defined condition by robust, reproducible and economic methods remains a major challenge for advancing hiPSC technology from the bench to the clinic [32]. The cell manufacturing process will need to adhere to additional country-specific guidelines given that iPSC-derived cells may be distributed internationally, even to countries where regulations are yet to be formulated.
6. Future of iPSCs in Regenerative Dentistry
6.1. Gingiva as an iPSC Source
Oral gingiva is often resected during general dental procedures and considered as biomedical waste, is an easily obtainable tissue, and cells can be isolated from patients with minimal discomfort. iPSCs can be easily generated from adult mouse or human gingival fibroblasts by transduction of three Yamanaka factors (Oct3/4, Sox2 and Klf4) without c-Myc and omission of c-Myc oncogen transduction has actually been shown to result in more specific iPSC generation [137] (Figure 3). Furthermore, higher reprogramming efficiency in iPSCs was observed by using gingival fibroblasts compared to conventionally used skin fibroblasts, perhaps because of their high proliferative capability [137].
The gingiva consists of highly vascularized connective tissue under a thin keratinocyte layer, and gingival fibroblasts, which are phenotypically different from other fibroblasts, are the main constituents of this connective tissue [139]. Clinical and experimental findings in patients and animal models have consistently demonstrated that oral mucosa, including gingival tissue, has enhanced wound healing capability compare to skin [140,141], even though both tissue types share similar healing processes and sequences. Another advantage is that gingival fibroblasts contain fibroblastic stem cell population [142]. The multipotent subpopulation and high regenerative capability of gingival fibroblasts may represent a progenitor-like state that could enhance the iPSC reprogramming efficiency of these cells comparatively as less reprogramming would be required to reach the anticipated pluripotent state. Moreover, gingival fibroblasts expand and proliferate well and easily on tissue culture plates [143]; hence primary gingival fibroblast cultures could be established quite effortlessly.
These cells are outstandingly suitable for iPSC-related clinical applications because of their accessibility, ease of culture and reprogramming competency. It may be convenient to apply these cells to achieve the regeneration of complicated tissues/organs, such as the tooth and salivary gland, which are formed through the interaction of epithelial and mesenchymal tissues during organogenesis.
6.2. Tooth Bioengineering
Reciprocal interactions between dental mesenchymal cells derived from Neural Crest (NC) and dental epithelial cells derived from ectodermal epithelium controls tooth development [144,145]. Epithelial-mesenchymal interactions also control the terminal differentiation of odontoblasts and ameloblasts [146,147]. Thus, as a new strategy for tooth regeneration, it is speculated that ectodermal epithelial cells and NC cells induced from iPS cells could be the optimal cell source for the whole tooth regeneration (Figure 4).
6.3. Periodontal Ligament Regeneration
A report by Oshima et al. in 2014 [149] opened the way for generation and regeneration of periodontal tissue by using Dental Follicle (DF) cells from tooth germ at temporally-limited stage of development, while it was restricted to murine specimen. Therefore considering clinical practice in patients for structuring human periodontal tissue with much larger size, researchers need to clarify the characteristics of DF cells at ED 18.5, comparing with those at other stages or periodontal ligament stem cells (PDLSCs), and how they can obtain such DF-like cells from iPSCs because the quantity of PDLSCs localized in periodontal ligament tissue is too small to use in clinical use while PDLSCs are naturally eligible. In addition, which bioactive scaffolds and multifunctional signal molecules support the differentiation of these stem cells also remains to be determined. Sufficient assessment of outcomes from integration of these elements from various aspects is needed for restoration of periodontal function [150].
7. Concluding Remarks and Future Perspectives
During the past decade since its inception, iPSC technology has shown great potential for clinical applications and disease modelling [151,152]. iPSCs are an exceptional source of cells for regenerative medicine as they demonstrate indefinite proliferation, obtainability and plasticity to differentiate into other cell types. There is a minimal risk of immunorejection and no ethical baggage. This potential is further strengthened by combining iPSC technology with genome engineering [153], which allows the correction of mutations in patient-derived iPSCs and use it to our advantage for personalized treatment and disease modelling, as well as modification of reporter lines to facilitate differentiation towards specific cell types. These properties successfully reduce expenditure and risk of clinical trials, and also provide phenotypically consistent cell lines capable of predicting cytotoxic and therapeutic responses of drugs. The field is full of great promises but, as discussed in this review, is still in its infancy and many obstacles remain to be overcome.
Despite the advantages, iPSC technology still faces many pitfalls such as low efficiency and high variability. Donor cell source and vector type still need major contemplation and reprogramming method still needs optimization. Our ultimate goal is to demonstrate therapeutic potential of iPSCs in clinics and it can only be achieved by generating hiPSCs affordably in GMP-compliant manner. The many clinical trials that are underway give us assurance of the coming revolution in near future and the present information available is satisfactorily promising.
Acknowledgments
This review was written as a part of an MSc Regenerative Dentistry coursework at King’s College London. I would like to thank Paul Sharpe and Ana Angelova Volponi at King’s College London for their mentoring.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| iPSCs | induced pluripotent stem cells |
| hiPSCs | Human induced pluripotent stem cells |
| OSKM | Oct4, Sox2, Klf4, and c-Myc |
| hESCs | human embryonic stem cells |
| ESC | Embryonic Stem Cells |
| SOX2 | the sex determining region Y-box2 |
| Klf4 | Kruppel-like factor 4 |
| c-Myc | myelocytomatosis oncogene |
| GMP | Good Manufacturing Practice |
| ECM | Extracellular matrix |
| 5hmc | 5-hydroxymethylcytosine |
| RPE | retinal pigment epithelial |
| RBCs | Red Blood Cells |
| DCs | Dedritic cells |
| SSEA | stage-specific embryonic antigen |
| CIRM | California Institute for Regenerative Medicine |
| HLA | human leukocyte antigens |
| CiRA | Center for iPSC Research and Application |
| CiBK | Clinical iPS Cell Bank of Kyoto |
| NC | Neural Crest |
| DF | Dental Follicle |
| PDLSCs | periodontal ligament stem cells |
References
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.; Waknitz, M.; Swiergiel, J.; Marshall, V.; Jones, J. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.A.; Davis-Dusenbery, B.N.; Eggan, K.C. SnapShot: Directed Differentiation of Pluripotent Stem Cells. Available online: http://www.sciencedirect.com/science/article/pii/S0092867412005946 (accessed on 29 February 2016).
- Zhao, T.; Zhang, Z.-N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced Pluripotent Stem Cells Generated Without Viral Integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of Mouse Induced Pluripotent Stem Cells without Viral Vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly Efficient Reprogramming to Pluripotency and Directed Differentiation of Human Cells with Synthetic Modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human Induced Pluripotent Stem Cells Free of Vector and Transgene Sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, N.; Gros, E.; Li, H.-R.; Kumar, S.; Deacon, D.C.; Maron, C.; Muotri, A.R.; Chi, N.C.; Fu, X.-D.; Yu, B.D.; et al. Efficient Generation of Human iPSCs by a Synthetic Self-Replicative RNA. Cell Stem Cell 2013, 13, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, L.; Xie, X. iPSCs and small molecules: A reciprocal effort towards better approaches for drug discovery. Acta Pharmacol. Sin. 2013, 34, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule Compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Kiskinis, E.; Eggan, K. Progress toward the clinical application of patient-specific pluripotent stem cells. J. Clin. Investig. 2010, 120, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lengner, C.J.; Kirak, O.; Hanna, J.; Cassady, J.P.; Lodato, M.A.; Wu, S.; Faddah, D.A.; Steine, E.J.; Gao, Q.; et al. Reprogramming of Postnatal Neurons into Induced Pluripotent Stem Cells by Defined Factors. Stem Cells 2011, 29, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, Y.-H.; Agarwal, S.; Park, I.-H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.-H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T Cells from Human Peripheral Blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of Induced Pluripotent Stem Cells from Human Terminally Differentiated Circulating T Cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of Human Peripheral Blood Cells to Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.E.; Rondon, E.; Rajesh, D.; Mack, A.; Lewis, R.; Feng, X.; Zitur, L.J.; Learish, R.D.; Nuwaysir, E.F. Derivation of Induced Pluripotent Stem Cells from Human Peripheral Blood T Lymphocytes. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Induced Pluripotent Stem Cells: Past, Present, and Future. Cell Stem Cell 2012, 10, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPS cell derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.E.; Levenstein, M.E.; Jones, J.M.; Berggren, W.T.; Mitchen, E.R.; Frane, J.L.; Crandall, L.J.; Daigh, C.A.; Conard, K.R.; Piekarczyk, M.S.; et al. Derivation of human embryonic stem cells in defined conditions. Nat. Biotechnol. 2006, 24, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.E.; Bergendahl, V.; Levenstein, M.E.; Yu, J.; Probasco, M.D.; Thomson, J.A. Feeder-independent culture of human embryonic stem cells. Nat. Methods 2006, 3, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Beers, J.; Gulbranson, D.R.; George, N.; Siniscalchi, L.I.; Jones, J.; Thomson, J.A.; Chen, G. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 2012, 7, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chou, B.-K.; Dowey, S.; He, C.; Gerecht, S.; Cheng, L. Scalable expansion of human induced pluripotent stem cells in the defined xeno-free E8 medium under adherent and suspension culture conditions. Stem Cell Res. 2013, 11, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Taniguchi, Y.; Senda, S.; Takizawa, N.; Ichisaka, T.; Asano, K.; Morizane, A.; Doi, D.; Takahashi, J.; Nishizawa, M.; et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci. Rep. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghbaderani, B.A.; Tian, X.; Neo, B.H.; Burkall, A.; Dimezzo, T.; Sierra, G.; Rao, M.S. cGMP-Manufactured Human Induced Pluripotent Stem Cells Are Available for Pre-clinical and Clinical Applications. Stem Cell Rep. 2015, 5, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Hubbell, J.A. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 2005, 23, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Loskill, P.; Shao, K.; Huebsch, N.; Hong, S.; Marcus, S.G.; Marks, N.; Mandegar, M.; Conklin, B.R.; Lee, L.P.; et al. Human iPSC-based cardiac microphysiological system for drug screening applications. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Wang, L.; Cai, J.; Yuan, Q.; Yang, X.; Yao, X.; Wong, W.M.; Huang, W.; Li, Z.; Wan, J.B.; et al. Transplanted motoneurons derived from human induced pluripotent stem cells form functional connections with target muscle. Stem Cell Res. 2013, 11, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Chau, M.J.; Deveau, T.C.; Song, M.; Gu, X.; Chen, D.; Wei, L. iPSC Transplantation increases regeneration and functional recovery after ischemic stroke in neonatal rats. Stem Cells 2014, 32, 3075–3087. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cao, J.; Wang, Y.; Lan, T.; Liu, L.; Wang, W.; Jin, N.; Gong, J.; Zhang, C.; Teng, F.; et al. Immunogenicity and functional evaluation of iPSC-derived organs for transplantation. Cell Discov. 2015, 1. [Google Scholar] [CrossRef]
- Korecka, J.A.; Levy, S.; Isacson, O. In vivo modeling of neuronal function, axonal impairment and connectivity in neurodegenerative and neuropsychiatric disorders using induced pluripotent stem cells. Mol. Cell. Neurosci. 2016, 73, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Wang, L.; Xu, L.; Jin, N.; Yan, G.; Xia, J.; Wang, H.; Zhuang, G.; Gao, C.; Meng, L.; et al. Induced Pluripotent Stem Cells Can Effectively Differentiate into Multiple Functional Lymphocyte Lineages in Vivo with Negligible Bias. Stem Cells Dev. 2016, 25, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.B.C.; Daley, G.Q. Reprogramming Cellular Identity for Regenerative Medicine. Cell 2012, 148, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Sterneckert, J.L.; Reinhardt, P.; Schöler, H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014, 15, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Byers, B.; Lee, H.; Reijo Pera, R. Modeling Parkinson’s Disease Using Induced Pluripotent Stem Cells. Curr. Neurol. Neurosci. Rep. 2012, 12, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Chestkov, I.V.; Vasilieva, E.A.; Illarioshkin, S.N.; Lagarkova, M.A.; Kiselev, S.L. Patient-Specific Induced Pluripotent Stem Cells for SOD1-Associated Amyotrophic Lateral Sclerosis Pathogenesis Studies. Acta Nat. 2014, 6, 54–60. [Google Scholar]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways Disrupted in Human ALS Motor Neurons Identified Through Genetic Correction of Mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.-P.; Maragakis, N.J. Induced pluripotent stem cells from ALS patients for disease modeling. Brain Res. 2015, 1607, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.A.; Finkbeiner, S. Modeling Huntington’s disease with induced pluripotent stem cells. Mol. Cell. Neurosci. 2013, 56, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huang, J.S.; Han, C.; Zhang, G.X.; Xu, X.Y.; Shen, Y.; Li, J.; Jiang, H.Y.; Lin, Z.C.; Xiong, N.; et al. Induced Pluripotent Stem Cells in Huntington’s Disease: Disease Modeling and the Potential for Cell-Based Therapy. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Mason, E.A.; Ovchinnikov, D.A.; Wells, C.A.; Wolvetang, E.J. Concise Review: New Paradigms for Down Syndrome Research Using Induced Pluripotent Stem Cells: Tackling Complex Human Genetic Disease. Stem Cells Transl. Med. 2013, 2, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Soejitno, A.; Prayudi, A.K.A. The prospect of induced pluripotent stem cells for diabetes mellitus treatment. Ther. Adv. Endocrinol. Metab. 2011, 2, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Mali, P.; Huang, X.; Dowey, S.N.; Cheng, L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 2011, 118, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Sweeney, C.L.; Chou, B.K.; Choi, U.; Pan, J.; Wang, H.; Dowey, S.N.; Cheng, L.; Malech, H.L. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: Functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood 2011, 117, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Raya, A.; Rodríguez-Pizà, I.; Guenechea, G.; Vassena, R.; Navarro, S.; Barrero, M.J.; Consiglio, A.; Castellà, M.; Río, P.; Sleep, E.; et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 2009, 460, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhan, H.; Mali, P.; Dowey, S.; Williams, D.M.; Jang, Y.Y.; Dang, C.V.; Spivak, J.L.; Moliterno, A.R.; Cheng, L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009, 114, 5473–5480. [Google Scholar] [CrossRef] [PubMed]
- Sebastiano, V.; Maeder, M.L.; Angstman, J.F.; Haddad, B.; Khayter, C.; Yeo, D.T.; Goodwin, M.J.; Hawkins, J.S.; Ramirez, C.L.; Batista, L.F.; et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 2011, 29, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Fong, H.; Wang, C.; Knoferle, J.; Walker, D.; Balestra, M.E.; Tong, L.M.; Leung, L.; Ring, K.L.; Seeley, W.W.; Karydas, A.; et al. Genetic Correction of Tauopathy Phenotypes in Neurons Derived from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2013, 1, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Kaitin, K. Obstacles and Opportunities in New Drug Development. Clin. Pharmacol. Ther. 2008, 83, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Sollano, J.; Kirsch, J.; Bala, M.; Chambers, M.; Harpole, L. The Economics of Drug Discovery and the Ultimate Valuation of Pharmacotherapies in the Marketplace. Clin. Pharmacol. Ther. 2008, 84, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Gunaseeli, I.; Doss, M.; Antzelevitch, C.; Hescheler, J.; Sachinidis, A. Induced Pluripotent Stem Cells as a Model for Accelerated Patient- and Disease-specific Drug Discovery. Curr. Med. Chem. 2010, 17, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Laustriat, D.; Gide, J.; Peschanski, M. Human pluripotent stem cells in drug discovery and predictive toxicology. Biochem. Soc. Trans. 2010, 38. [Google Scholar] [CrossRef] [PubMed]
- Seiler, A.; Visan, A.; Buesen, R.; Genschow, E.; Spielmann, H. Improvement of an in vitro stem cell assay for developmental toxicity: The use of molecular endpoints in the embryonic stem cell test. Reprod. Toxicol. 2004, 18, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wu, H.; Li, Y.; Szulwach, K.E.; Lin, L.; Li, X.; Chen, I.P.; Goldlust, I.S.; Chamberlain, S.J.; Dodd, A.; et al. Subtelomeric hotspots of aberrant 5-hydroxymethylcytosine-mediated epigenetic modifications during reprogramming to pluripotency. Nat. Cell Biol. 2013, 15, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Byun, K.; Lee, B. Induced pluripotent stem cells and personalized medicine: Current progress and future perspectives. Anat. Cell Biol. 2011, 44. [Google Scholar] [CrossRef] [PubMed]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly Efficient Generation of Human Hepatocyte–like Cells from Induced Pluripotent Stem Cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Takebe, T.; Sekine, K.; Koike, H.; Zheng, Y.; Taniguchi, H. Identification of proliferating human hepatic cells from human induced pluripotent stem cells. Transplant. Proc. 2014, 46, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, L. Two Effective Routes for Removing Lineage Restriction Roadblocks: From Somatic Cells to Hepatocytes. Int. J. Mol. Sci. 2015, 16, 20873–20895. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBAC transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Norrby, K.; Paca, A.; Mileikovsky, M.; Mohseni, P.; Woltjen, K. Virus free induction of pluripotency and subsequent excision of reprogramming factors. Nature 2009, 458, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Yamazaki, S.; Yamaguchi, T.; Okabe, M.; Masaki, H.; Takaki, S.; Otsu, M.; Nakauchi, H. Generation of Engraftable Hematopoietic Stem Cells From Induced Pluripotent Stem Cells by Way of Teratoma Formation. Mol. Ther. 2013, 21, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kim, Y.; Sharkis, S.; Marchionni, L.; Jang, Y.-Y. In Vivo Liver Regeneration Potential of Human Induced Pluripotent Stem Cells from Diverse Origins. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Nori, S.; Okada, Y.; Yasuda, A.; Tsuji, O.; Takahashi, Y.; Kobayashi, Y.; Fujiyoshi, K.; Koike, M.; Uchiyama, Y.; Ikeda, E.; et al. Grafted human-induced pluripotent stem-cell-derived neurospheres promote motor functional recovery after spinal cord injury in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16825–16830. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.F.; Inoue-Yokoo, T.; Tan, K.S.; Lai, M.I.; Sugiyama, D. Hematopoietic cell differentiation from embryonic and induced pluripotent stem cells. Stem Cell Res. Ther. 2013, 4. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Chen, K.Y.; Naumova, A.V.; Muskheli, V.; Fugate, J.A.; Dupras, S.K.; Reinecke, H.; Xu, C.; Hassanipour, M.; Police, S.; et al. Cardiomyocytes derived from human embryonic stem cells in pro- survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 2007, 25, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Wagner, R.A.; Wilson, K.D.; Xie, X.; Fu, J.-D.; Drukker, M.; Lee, A.; Li, R.A.; Gambhir, S.S.; Weissman, I.L.; et al. Transcriptional and Functional Profiling of Human Embryonic Stem Cell-Derived Cardiomyocytes. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levenberg, S.; Ferreira, L.S.; Chen-Konak, L.; Kraehenbuehl, T.P.; Langer, R. Isolation, differentiation and characterization of vascular cells derived from human embryonic stem cells. Nat. Protoc. 2010, 5, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Senju, S.; Haruta, M.; Matsunaga, Y.; Fukushima, S.; Ikeda, T.; Takahashi, K.; Okita, K.; Yamanaka, S.; Nishimura, Y. Characterization of Dendritic Cells and Macrophages Generated by Directed Differentiation from Mouse Induced Pluripotent Stem Cells. Stem Cells 2009, 27, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Kamao, H.; Mandai, M.; Okamoto, S.; Sakai, N.; Suga, A.; Sugita, S.; Kiryu, J.; Takahashi, M. Characterization of Human Induced Pluripotent Stem Cell-Derived Retinal Pigment Epithelium Cell Sheets Aiming for Clinical Application. Stem Cell Rep. 2014, 2, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Kanemura, H.; Go, M.J.; Shikamura, M.; Nishishita, N.; Sakai, N.; Kamao, H.; Mandai, M.; Morinaga, C.; Takahashi, M.; Kawamata, S. Tumorigenicity Studies of Induced Pluripotent Stem Cell (iPSC)-Derived Retinal Pigment Epithelium (RPE) for the Treatment of Age-Related Macular Degeneration. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Stem Cells Cruise to Clinic. Nature News & Comment. Available online: http://www.nature.com/news/stem-cells-cruise-to-clinic-1.12511 (accessed on 29 February 2016).
- Next-Generation Stem Cells Cleared for Human Trial. Nature News & Comment. Available online: http://www.nature.com/news/next-generation-stem-cells-cleared-for-human-trial-1.15897 (accessed on 29 February 2016).
- Japanese Woman is First Recipient of Next-Generation Stem Cells. Nature News & Comment. Available online: http://www.nature.com/news/japanese-woman-is-first-recipient-of-next-generation-stem-cells-1.15915 (accessed on 29 February 2016).
- First iPS Cell Transplant Patient Makes Progress One Year on. Japan Times. Available online: http://www.japantimes.co.jp/news/2015/10/02/national/science-health/first-ips-cell-transplant-patient-makes-progress-oneyear#.VqeIOvmLSUk (accessed on 29 February 2016).
- Zhao, T.; Zhang, Z.N.; Westenskow, P.D.; Todorova, D.; Hu, Z.; Lin, T.; Rong, Z.; Kim, J.; He, J.; Wang, M.; et al. Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell 2015, 17, 353–359. [Google Scholar] [CrossRef] [PubMed]
- A Crucial Moment in Time for Stem Cell R&D. Biotechnology Focus. Available online: http://biotechnologyfocus.ca/a-crucial-moment-in-time-forstem-cell-rd/ (accessed on 29 February 2016).
- Fernandes, S.; Chong, J.J.H.; Paige, S.L.; Iwata, M.; Torok-Storb, B.; Keller, G.; Murry, C.E. Comparison of Human Embryonic Stem Cell-Derived Cardiomyocytes, Cardiovascular Progenitors, and Bone Marrow Mononuclear Cells for Cardiac Repair. Stem Cell Rep. 2015, 5, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.J.H.; Yang, X.; Don, C.W.; Minami, E.; Liu, Y.-W.; Weyers, J.J.; Murry, C.E. Human Embryonic Stem Cell-Derived Cardiomyocytes Regenerate Non-Human Primate Hearts. Nature 2014, 510, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Cacciapuoti, I.; Parouchev, A.; Benhamouda, N.; Tachdjian, G.; Tosca, L.; et al. Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: First clinical case report. Eur. Heart J. 2015, 36, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Menasché, P.; Vanneaux, V.; Fabreguettes, J.R.; Bel, A.; Tosca, L.; Garcia, S.; Bellamy, V.; Farouz, Y.; Pouly, J.; Damour, O.; et al. Towards a clinical use of human embryonic stem cell-derived cardiac progenitors: A translational experience. Eur. Heart J. 2015, 36, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Spinal Cord Injury and a CIRM-Funded Stem Cell-Based Trial. Available online: http://blog.cirm.ca.gov/2015/10/22/video-spinal-cord-injury-and-a-cirm-funded-stem-cell-based-trial/ (accessed on 29 February 2016).
- Revolutionary Stem Cell Therapy Trial for Parkinson’s Disease to be Held in Australia. ABC News. Available online: http://www.abc.net.au/news/2015-12-15/stem-cell-trial-for-parkinson’s-disease-in-australia/7029722 (accessed on 29 February 2016).
- Turner, M.; Leslie, S.; Martin, N.G.; Peschanski, M.; Rao, M.; Taylor, C.J.; Trounson, A.; Turner, D.; Yamanaka, S.; Wilmut, I. Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell 2013, 13, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Fairchild, P.J. Taming the lion: The challenge of immunity in regenerative medicine. Regen. Med. 2015, 10, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E.M. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, F.; Tokunaga, K.; Nakatsuji, N. Human Leukocyte Antigen Matching Estimations in a Hypothetical Bank of Human Embryonic Stem Cell Lines in the Japanese Population for Use in Cell Transplantation Therapy. Stem Cells 2007, 25, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, N.; Nakajima, F.; Tokunaga, K. HLA-haplotype banking and iPS cells. Nat. Biotechnol. 2008, 26, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.K.; Matsunaga, A.; Takasu, N.; Yamanaka, S. Donor Recruitment and Eligibility Criteria for HLA-Homozygous iPS Cell Bank in Japan. In Stem Cell Banking; Ilic, D., Ed.; Springer: New York, NY, USA, 2014. [Google Scholar]
- Nature. Stem-Cell Pioneer Banks on Future Therapies. Available online: http://www.nature.com/news/stem-cell-pioneer-banks-on-future-therapies-1.11129 (accessed on 29 February 2016).
- Leha, A.; Moens, N.; Meleckyte, R.; Culley, O.J.; Gervasio, M.K.; Kerz, M.; Reimer, A.; Cain, S.; Streeter, I.; Folarin, A.; et al. A high-content platform to characterise human induced pluripotent stem cell lines. Methods 2016, 96, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Moad, M.; Pal, D.; Hepburn, A.C.; Williamson, S.C.; Wilson, L.; Lako, M.; Armstrong, L.; Hayward, S.W.; Franco, O.E.; Cates, J.M.; et al. A Novel Model of Urinary Tract Differentiation, Tissue Regeneration, and Disease: Reprogramming Human Prostate and Bladder Cells into Induced Pluripotent Stem Cells. Eur. Urol. 2013, 64, 753–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Longaker, M.T.; Wu, J.C. Human iPS cell-based therapy: Considerations before clinical applications. Cell Cycle 2010, 9, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Panetta, N.J.; Gupta, D.M.; Wilson, K.D.; Lee, A.; Jia, F.; Hu, S.; Cherry, A.M.; Robbins, R.C.; Longaker, M.T.; et al. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 15720–15725. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Maherali, N.; Kulalert, W.; Hochedlinger, K. Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J. Cell Sci. 2009, 122, 3502–3510. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Montserrat, N.; Aasen, T.; Gonzalez, F.; Rodríguez-Pizà, I.; Vassena, R.; Raya, A.; Boué, S.; Barrero, M.J.; Corbella, B.A.; et al. Generation of Induced Pluripotent Stem Cells from Human Cord Blood Using OCT4 and SOX2. Cell Stem Cell 2009, 5, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.; Olmer, R.; Schwanke, K.; Wunderlich, S.; Merkert, S.; Hess, C.; Zweigerdt, R.; Gruh, I.; Meyer, J.; Wagner, S.; et al. Generation of Induced Pluripotent Stem Cells from Human Cord Blood. Cell Stem Cell 2009, 5, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Greber, B.; Araúzo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Schöler, H.R. Direct reprogramming of human neural stem cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, Z.; Wilson, K.D.; Wu, Y.; Hu, S.; Quertermous, T.; Wu, J.C. Persistent Donor Cell Gene Expression among Human Induced Pluripotent Stem Cells Contributes to Differences with Human Embryonic Stem Cells. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhang, Y. Genetic and epigenetic variations in iPSCs: Potential causes and implications for application. Cell Stem Cell 2013, 13, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Huo, H.; Loh, Y.H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhang, Y. Embryonic stem cell and induced pluripotent stem cell: An epigenetic perspective. Cell Res. 2013, 23, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; Antosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.; Yu, J.; et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of Aberrant Epigenomic Reprogramming in Human Induced Pluripotent Stem Cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Toyoda, M.; Yamazaki-Inoue, M.; Fukawatase, Y.; Chikazawa, E.; Sakaguchi, H.; Akutsu, H.; Umezawa, A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Diep, D.; Gore, A.; Panopoulos, A.D.; Montserrat, N.; Plongthongkum, N.; Kumar, S.; Fung, H.L.; Giorgetti, A.; Bilic, J.; et al. Identification of a specific reprogramming-associated epigenetic signature in human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16196–16201. [Google Scholar] [CrossRef] [PubMed]
- Narsinh, K.H.; Sun, N.; Sanchez-Freire, V.; Lee, A.S.; Almeida, P.; Hu, S.; Jan, T.; Wilson, K.D.; Leong, D.; Rosenberg, J.; et al. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J. Clin. Investig. 2011, 121, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, F.; Yong, J.; Zhang, P.; Hou, P.; Li, H.; Jiang, W.; Cai, J.; Liu, M.; Cui, K.; et al. Generation of Induced Pluripotent Stem Cells from Adult Rhesus Monkey Fibroblasts. Cell Stem Cell 2008, 3, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.A.; Xu, J.; Yang, J.; Peng, M.; Qin, D.; Li, W.; Jiang, Z.; Chen, J.; Deng, K.; Zhong, M.; et al. Generation of Induced Pluripotent Stem Cell Lines from Tibetan Miniature Pig. J. Biol. Chem. 2009, 284, 17634–17640. [Google Scholar] [CrossRef] [PubMed]
- Alexenko, A.P.; Sachdev, S.; Sinha, S.; Roberts, R.M.; Ezashi, T.B.P.; Telugu, V.L. Derivation of induced pluripotent stem cells from pig somatic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10993–10998. [Google Scholar]
- Wu, Z.; Chen, J.; Ren, J.; Bao, L.; Liao, J.; Cui, C.; Rao, L.; Li, H.; Gu, Y.; Dai, H.; et al. Generation of Pig Induced Pluripotent Stem Cells with a Drug-Inducible System. J. Mol. Cell Biol. 2009, 1, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Swijnenburg, R.-J.; Schrepfer, S.; Govaert, J.A.; Cao, F.; Ransohoff, K.; Sheikh, A.Y.; Haddad, M.; Connolly, A.J.; Davis, M.M.; Robbins, R.C.; et al. Immunosuppressive therapy mitigates immunological rejection of human embryonic stem cell xenografts. Proc. Natl. Acad. Sci. USA 2008, 105, 12991–12996. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W.; Cavanagro, J.; Deans, R.; Feigel, E.; Horowitz, E.; Keating, A.; Rao, M.; Turner, M.; Wilmut, I.; Yamanaka, S. Harmonizing standards for producing clinical-grade therapies from pluripotent stem cells. Nat. Biotechnol. 2014, 32, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.; Frankfurt, O.; Altman, J.; Evens, A.; Tallman, M.; Gordon, L.; Williams, S.; Winter, J.; Krishnamurthy, J.; Duffey, S.; et al. Optimizing the CD34+cell dose for reduced-intensity allogeneic hematopoietic stem cell transplantation. Leuk. Lymphoma 2009, 50, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, D.E.; Jing, D.; Lock, L.T.; Tzanakakis, E.S. Scalable Stirred-Suspension Bioreactor Culture of Human Pluripotent Stem Cells. Tissue Eng. Part A 2010, 16, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Egusa, H.; Okita, K.; Kayashima, H.; Yu, G.; Fukuyasu, S.; Saeki, M.; Matsumoto, T.; Yamanaka, S.; Yatani, H. Gingival Fibroblasts as a Promising Source of Induced Pluripotent Stem Cells. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, M.; Egusa, H.; Otsu, K.; Saitoh, I.; Miura, T.; Harada, H. Frontier dental research on iPS cells. J. Oral Biosci. 2013, 55, 191–199. [Google Scholar] [CrossRef]
- Stephens, P.; Davies, K.J.; Occleston, N.; Pleass, R.D.; Kon, C.; Daniels, J.; Khaw, P.T.; Thomas, D.W. Skin and oral fibroblasts exhibit phenotypic differences in extracellular matrix reorganization and matrix metalloproteinase activity. Bri. J. Dermatol. 2001, 144, 229–237. [Google Scholar] [CrossRef]
- Sciubba, J.J.; Waterhouse, J.P.; Meyer, J. A fine structural comparison of the healing of incisional wounds of mucosa and skin. J. Oral Pathol. Med. 1978, 7, 214–227. [Google Scholar] [CrossRef]
- Walsh, L.J.; L’Estrange, P.R.; Seymour, G.J. High magnification in situ viewing of wound healing in oral mucosa. Aust. Dent. J. 1996, 41, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Egusa, H.; Sonoyama, W.; Nishimura, M.; Atsuta, I.; Akiyama, K. Stem cells in dentistry Part I: Stem cell sources. J. Prosthodont. Res. 2012, 56, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, C.; Cimasoni, G. Functional Characteristics of Gingival and Periodontal Ligament Fibroblasts. J. Dent. Res. 1996, 75, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Thesleff, I.; Sharpe, P. Signalling networks regulating dental development. Mech. Dev. 1997, 67, 111–123. [Google Scholar] [CrossRef]
- Jernvall, J.; Thesleff, I. Reiterative signaling and patterning during mammalian tooth morphogenesis. Mech. Dev. 2000, 92, 19–29. [Google Scholar] [CrossRef]
- Ruch, J.; Lesot, H.; Begue-Kirn, C. Odontoblast differentiation. Int. J. Dev. Biol. 1995, 39, 51–68. [Google Scholar] [PubMed]
- Imai, H.; Osumi-Yamashita, N.; Ninomiya, Y.; Eto, K. Contribution of Early-Emigrating Midbrain Crest Cells to the Dental Mesenchyme of Mandibular Molar Teeth in Rat Embryos. Dev. Biol. 1996, 176, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Otsu, K.; Kumakami-Sakano, M.; Fujiwara, N.; Kikuchi, K.; Keller, L.; Lesot, H.; Harada, H. Stem cell sources for tooth regeneration: Current status and future prospects. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Inoue, K.; Nakajima, K.; Tachikawa, T.; Yamazaki, H.; Isobe, T.; Sugawara, A.; Ogawa, M.; Tanaka, C.; Saito, M.; et al. Functional tooth restoration by next-generation bio-hybrid implant as a bio-hybrid artificial organ replacement therapy. Sci. Rep. 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Akamine, A. Quest for the development of tooth root/periodontal ligament complex by tissue engineering. Integr. Mol. Med. 2014, 1. [Google Scholar] [CrossRef]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug testing. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Trounson, A.; DeWitt, N.D. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol. 2016, 17, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Jaenisch, R. Highly efficient gene targeting of expressed and silent genes in human ESCs and iPSCs using zinc finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Directed Differentiation of Pluripotent Stem Cells [8]. Highlighted here are some of strategies for directing the differentiation of Embryonic Stem Cells (ESCs) and induced pluripotent stem cells (iPSCs) into defined cell types. Most cell types and pathways depicted correspond to published work on human cells, expect for the production of spermatozoa, oocyte-like cells, otic hair cells, cortical layers, and optic cup, which were generated with mouse ESCs or iPSCs. This figure is reproduced from Williams, Davis-Dusenbery and Eggan [8]; published by Elsevier under open-access license policies.
Figure 1.
Directed Differentiation of Pluripotent Stem Cells [8]. Highlighted here are some of strategies for directing the differentiation of Embryonic Stem Cells (ESCs) and induced pluripotent stem cells (iPSCs) into defined cell types. Most cell types and pathways depicted correspond to published work on human cells, expect for the production of spermatozoa, oocyte-like cells, otic hair cells, cortical layers, and optic cup, which were generated with mouse ESCs or iPSCs. This figure is reproduced from Williams, Davis-Dusenbery and Eggan [8]; published by Elsevier under open-access license policies.
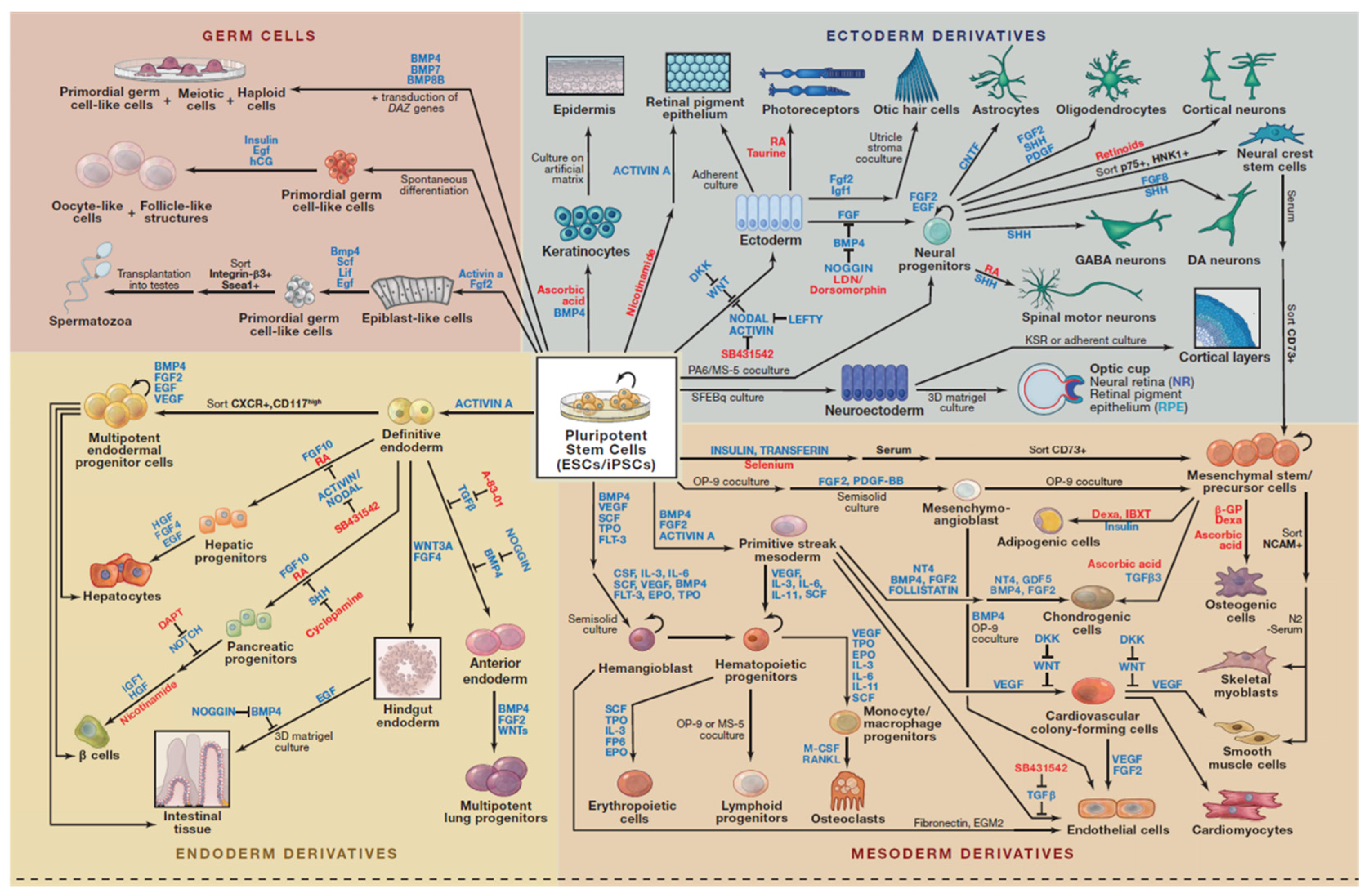
Figure 2.
An overview of key reprogramming methods available for the generation of iPSCs from various somatic cell sources and their possible applications. Adult stem cells or iPSCs can be expanded in culture and differentiated into the disease-affected cells that can be used to recapitulated disease pathogenesis in vitro. Patient-specific disease models can be used to identify new biomarkers for improved diagnostic procedures, such as earlier detection of disease onset. These disease models can also be used to identify compounds that alleviate disease pathology in vitro, which can be further developed into novel drugs. Such compounds can be identified by carrying out a phenotypic screen using the disease model to identify compounds that may act through novel mechanisms. Stem-cell-derived cells can also form the basis for cell replacement therapies.
Figure 2.
An overview of key reprogramming methods available for the generation of iPSCs from various somatic cell sources and their possible applications. Adult stem cells or iPSCs can be expanded in culture and differentiated into the disease-affected cells that can be used to recapitulated disease pathogenesis in vitro. Patient-specific disease models can be used to identify new biomarkers for improved diagnostic procedures, such as earlier detection of disease onset. These disease models can also be used to identify compounds that alleviate disease pathology in vitro, which can be further developed into novel drugs. Such compounds can be identified by carrying out a phenotypic screen using the disease model to identify compounds that may act through novel mechanisms. Stem-cell-derived cells can also form the basis for cell replacement therapies.
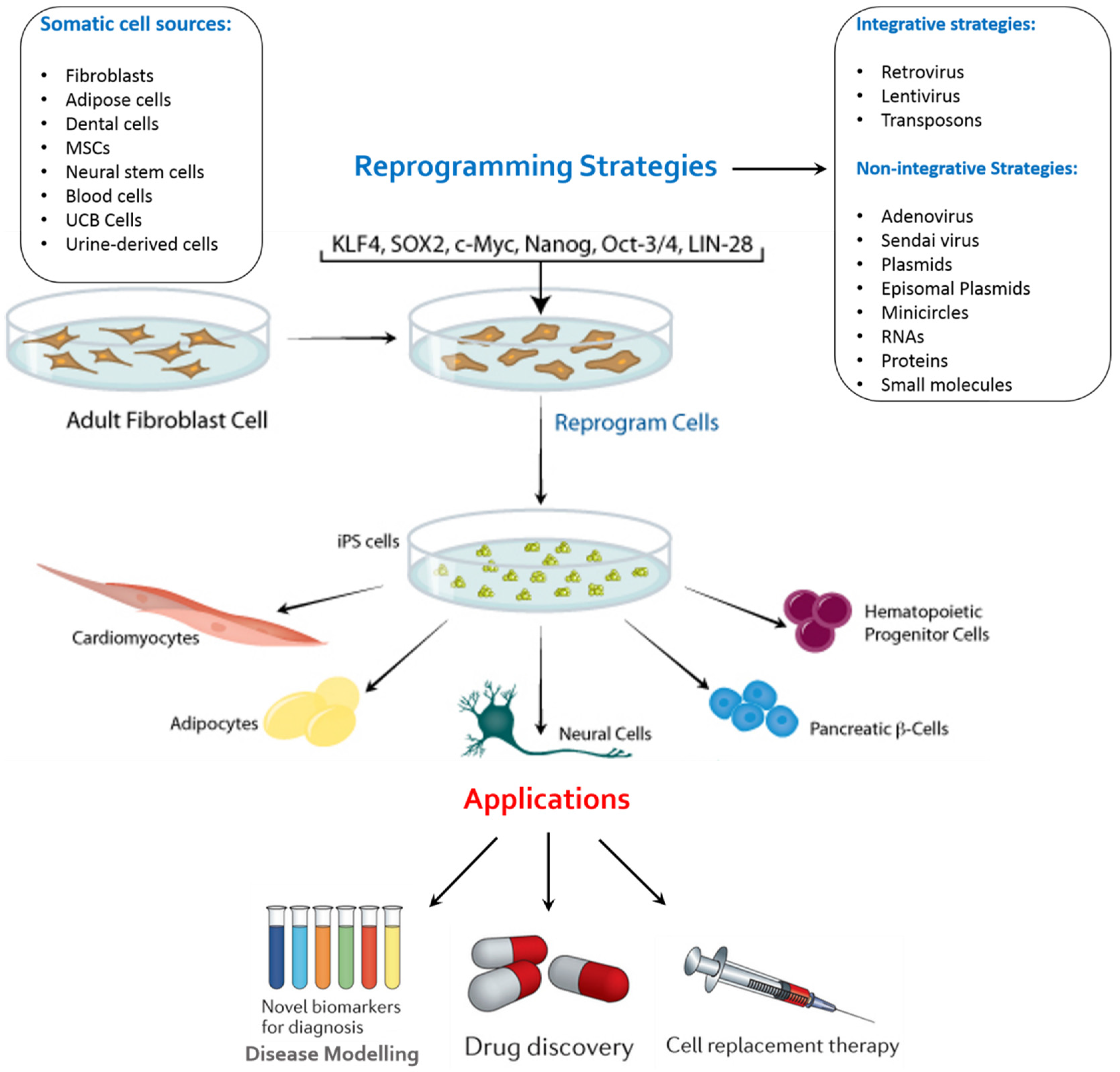
Figure 3.
Generation of iPS cells from gingiva [138]. Gingival tissues from adult mice and patients (resected during dental implant surgery) can be used for iPS cell generation. Isolated gingival fibroblasts were easily reprogrammed by the transduction of three factors (Oct3/4, Sox2, and Klf4) without the c-Myc oncogene. Red iPS colonies in the figure show robust staining for alkaline phosphatase (ALP), which is associated with undifferentiated pluripotent stem cells. This figure is reproduced from the study by Egusa et al. [137] under open-access license policies.
Figure 3.
Generation of iPS cells from gingiva [138]. Gingival tissues from adult mice and patients (resected during dental implant surgery) can be used for iPS cell generation. Isolated gingival fibroblasts were easily reprogrammed by the transduction of three factors (Oct3/4, Sox2, and Klf4) without the c-Myc oncogene. Red iPS colonies in the figure show robust staining for alkaline phosphatase (ALP), which is associated with undifferentiated pluripotent stem cells. This figure is reproduced from the study by Egusa et al. [137] under open-access license policies.
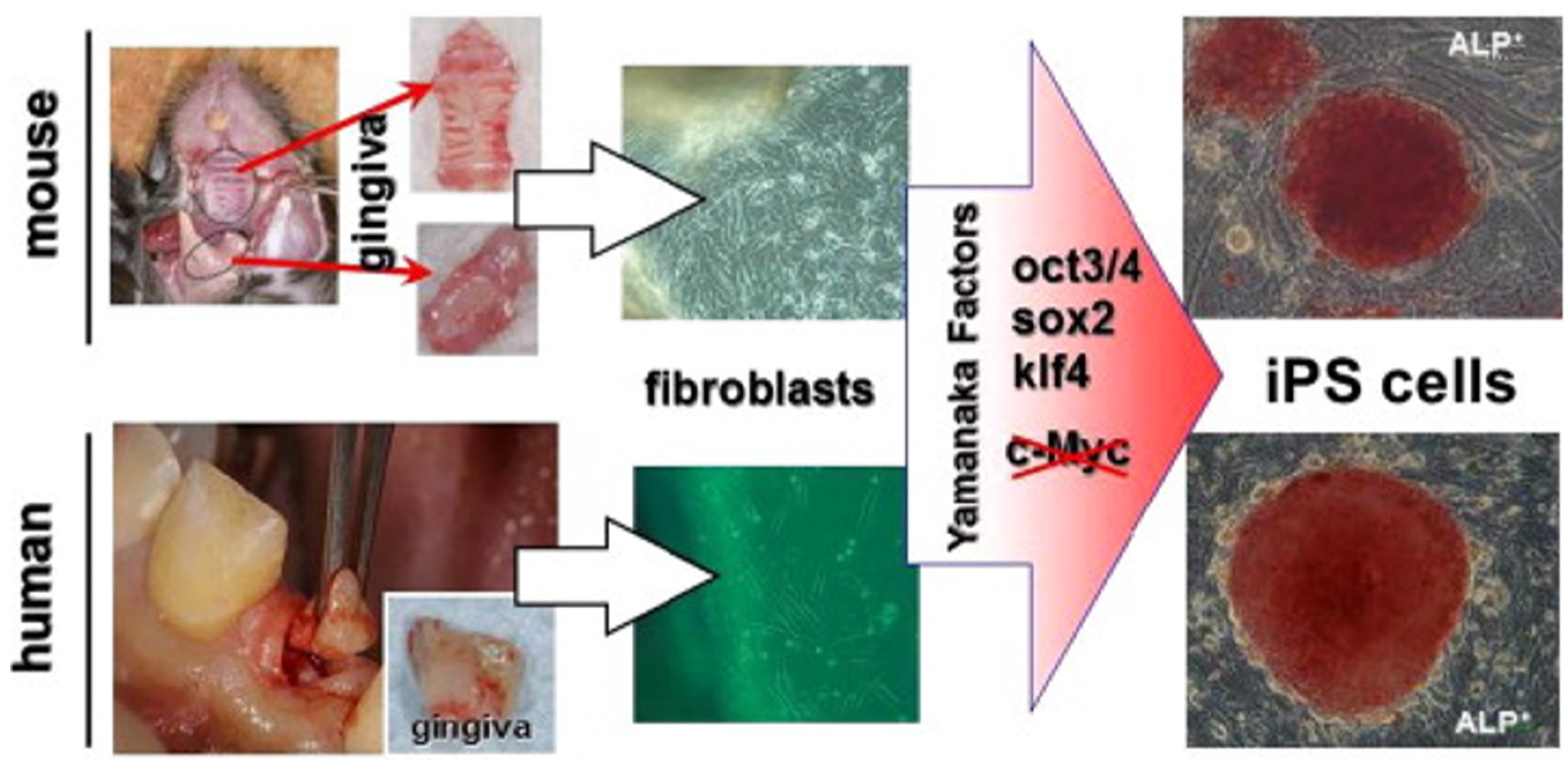
Figure 4.
General schematic representation of the current strategy for whole tooth regeneration using iPSs. The patient’s somatic cells are harvested. Reprogramming conditions/factors are introduced to induce self-renewal and pluripotency, and patient-specific iPSCs are established. iPSCs are induced to form ectodermal epithelial cells and neural crest-derived mesenchymal cells, and they are further induced to form odontogenic cells in vitro. The two cell populations are combined by direct contact, mimicking the in vitro arrangement. Interaction of these cells leads to formation of a nearly-stage tooth germ. Once transplanted into the mouth, the recombinants develop and lead to functional recovery from tooth loss. This figure is reproduced from Otsu et al. [148] under open-access license policies.
Figure 4.
General schematic representation of the current strategy for whole tooth regeneration using iPSs. The patient’s somatic cells are harvested. Reprogramming conditions/factors are introduced to induce self-renewal and pluripotency, and patient-specific iPSCs are established. iPSCs are induced to form ectodermal epithelial cells and neural crest-derived mesenchymal cells, and they are further induced to form odontogenic cells in vitro. The two cell populations are combined by direct contact, mimicking the in vitro arrangement. Interaction of these cells leads to formation of a nearly-stage tooth germ. Once transplanted into the mouth, the recombinants develop and lead to functional recovery from tooth loss. This figure is reproduced from Otsu et al. [148] under open-access license policies.
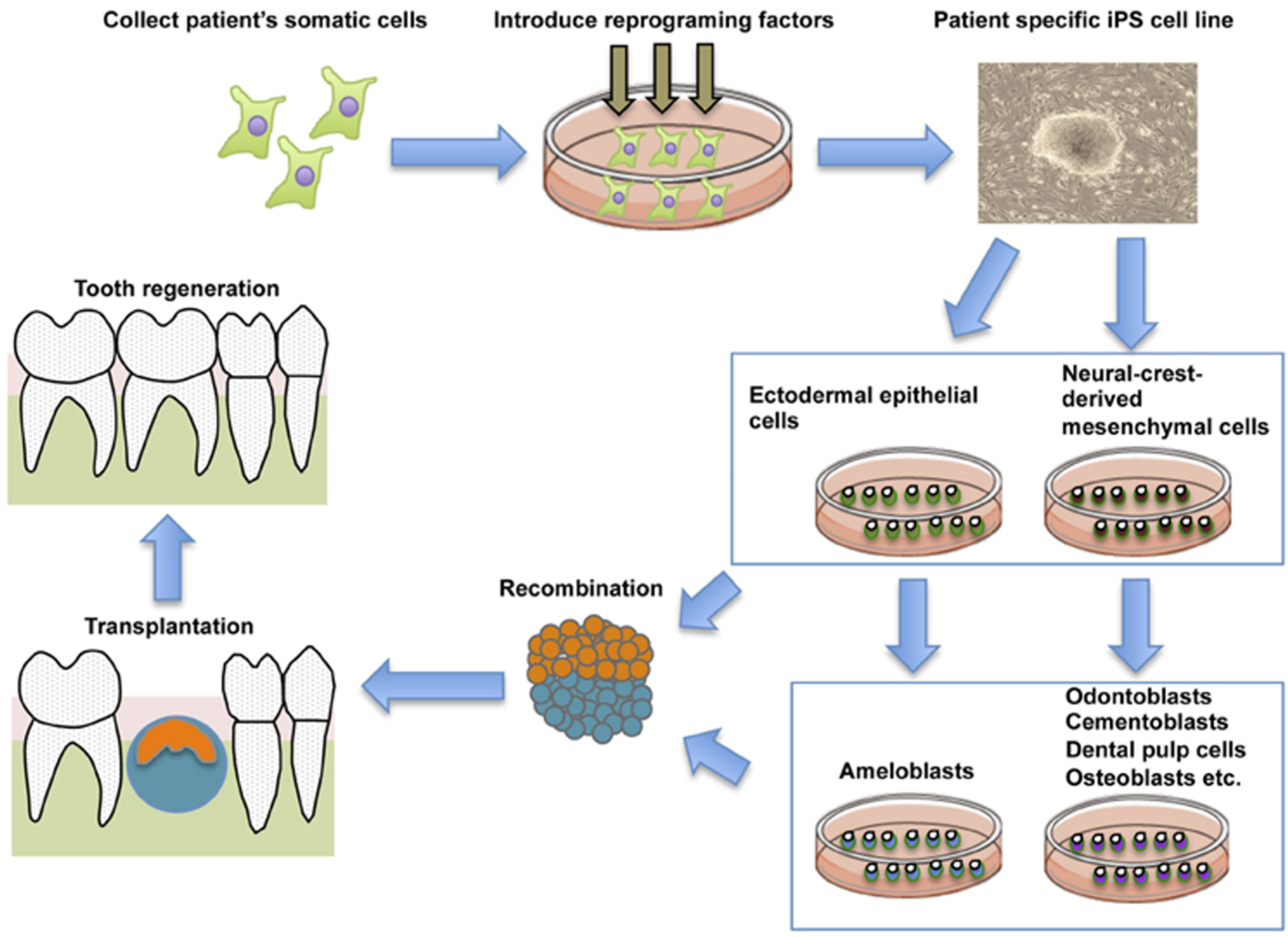

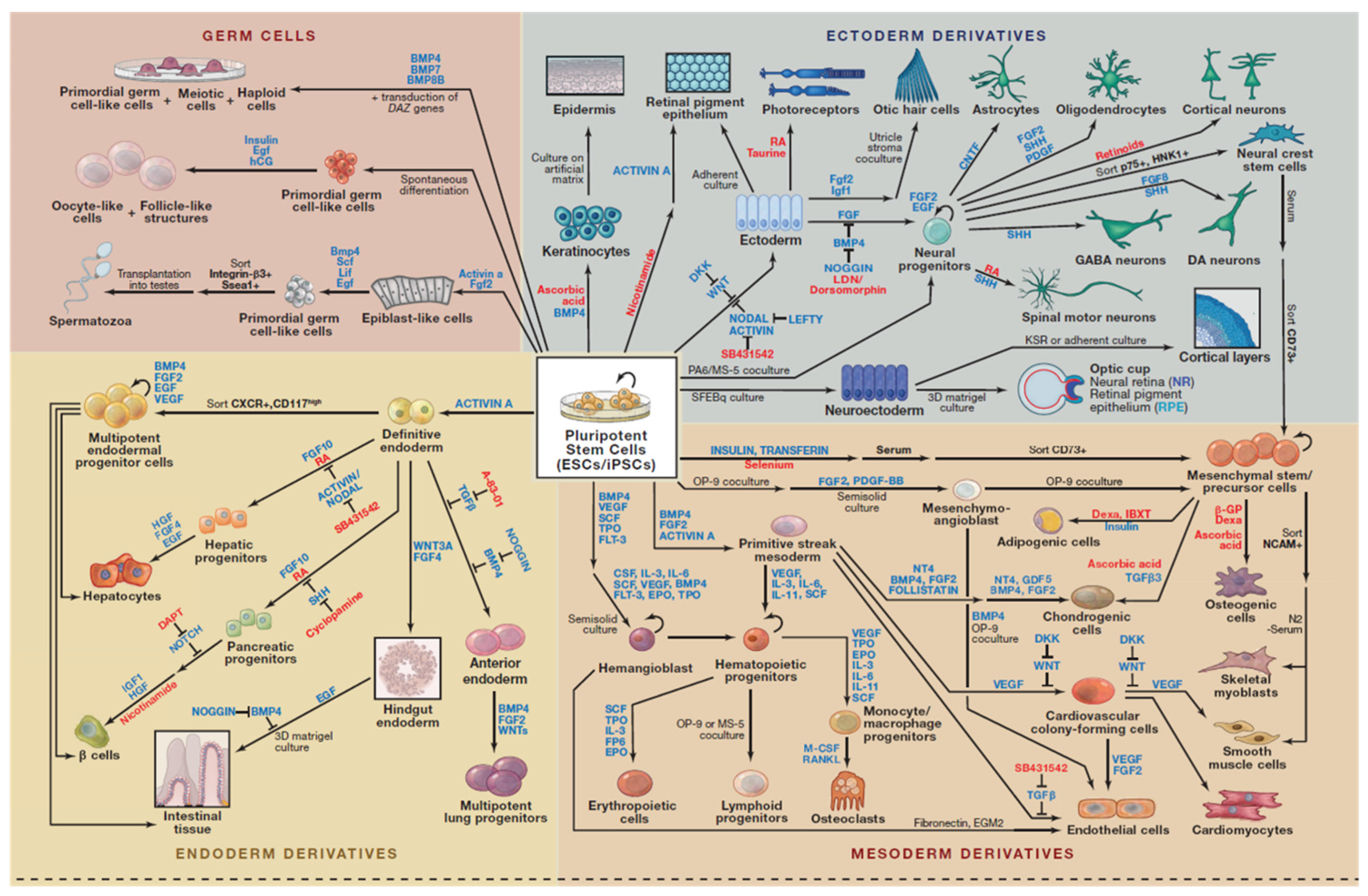{kind=link}
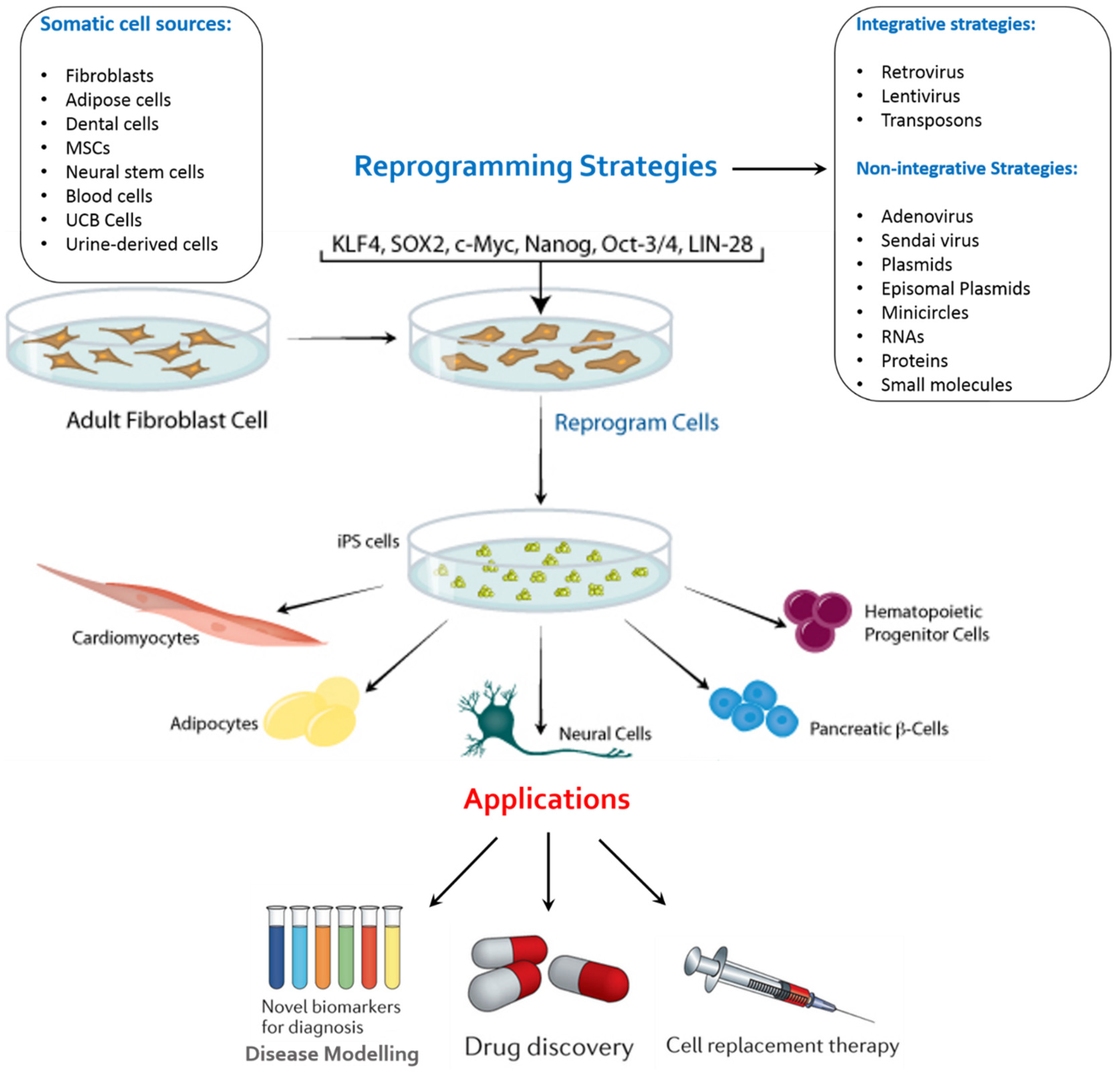{kind=link}
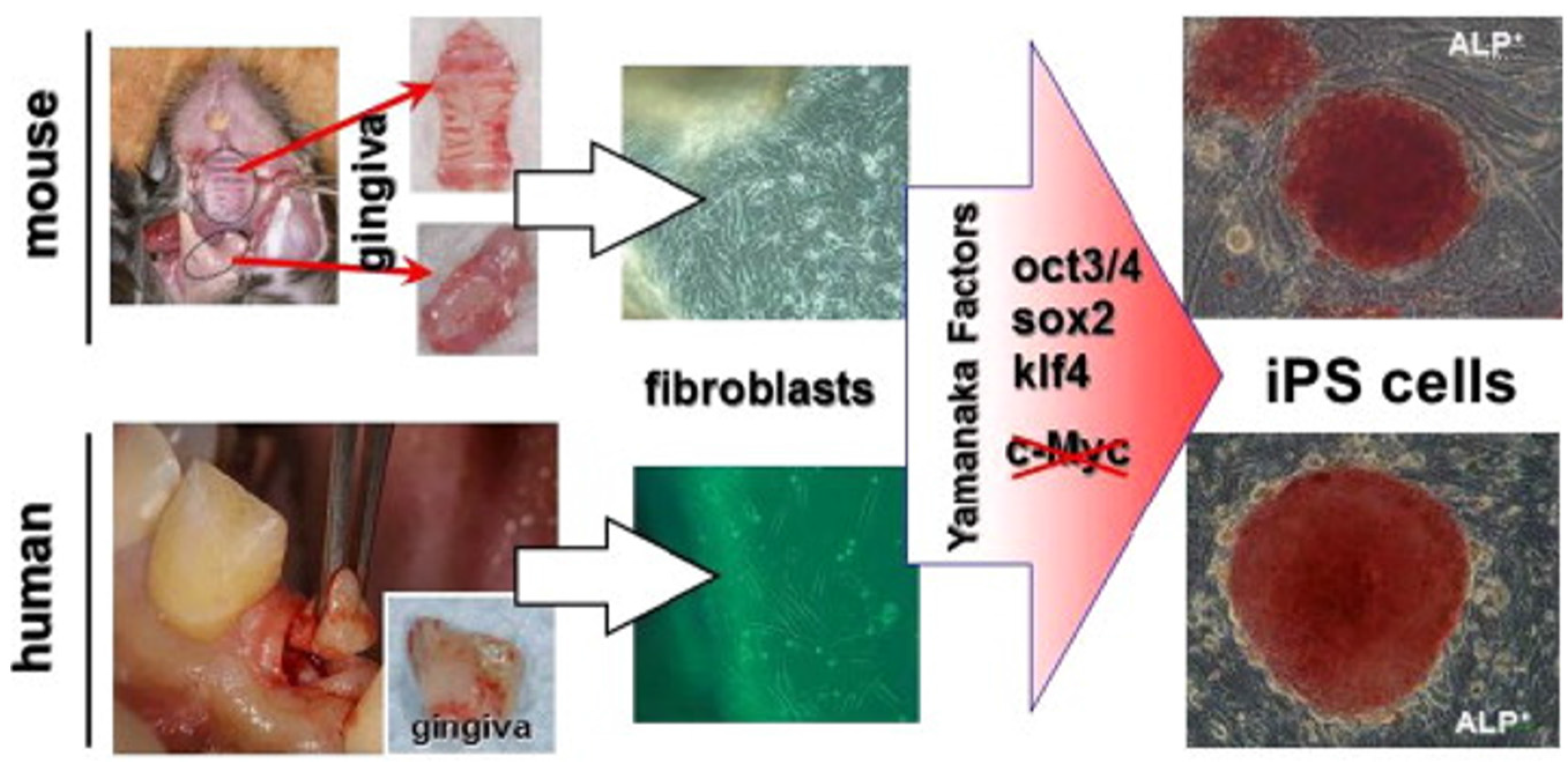{kind=link}
{kind=link}
Table 1.
Comparison of different cell origins used for reprogramming [111].
| Cell Source | Derivation | In vitro Expansion | Reprogramming Efficiency (4 factor) | Reprogramming Speed | Reprogramming Factors | References |
|---|---|---|---|---|---|---|
| Skin Fibroblasts | Skin Biopsy | Yes | ~0.01% (Adult cells) | >21 days | OSKM, OSK, OSNL | [5] |
| Keratinocytes | Skin Biopsy | Yes | ~1% (neonatal and juvenile cells) | >10days | OSKM, OSK | [112] |
| CD34 Blood Cells | Peripheral Blood undergo G-CSF stimulation | No | ~0.01-0.02% (Adult Cells) | >14days | OSKM | [22] |
| Adipose Stem Cells | Lipoaspiration | No | ~0.2% (Adult Cells) | >13–14 days | OSKM | [113] |
| Melanocytes | Skin Biopsy | Yes | ~0.05% (not known) | >10 days | OSKM, OKM | [114] |
| Cord Blood Cells | Collected at birth from cord cells | No | ~0.01% (neonatal cells) | >12–15 days | OSKM, OSNL, OSK, OS | [115,116] |
| Neural Stem Cells | NA | Yes | 0.004% (1 Factor, Fetal cells) | >7–8 Weeks (1Factor) | OK, O | [117] |
O: Oct4, S:Sox2, K: Klf4, M: c-MYC, N: Nanog, L: Lin-28, NA: Not Applicable.
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sharma, R. iPS Cells—The Triumphs and Tribulations. Dent. J. 2016, 4, 19. https://doi.org/10.3390/dj4020019
AMA Style
Sharma R. iPS Cells—The Triumphs and Tribulations. Dentistry Journal. 2016; 4(2):19. https://doi.org/10.3390/dj4020019
Chicago/Turabian StyleSharma, Riddhi. 2016. "iPS Cells—The Triumphs and Tribulations" Dentistry Journal 4, no. 2: 19. https://doi.org/10.3390/dj4020019
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.