
Synthesis, Crystal Structure, Polymorphism, and Magnetism of Eu(CN3H4)2 and First Evidence of EuC(NH)3



Abstract
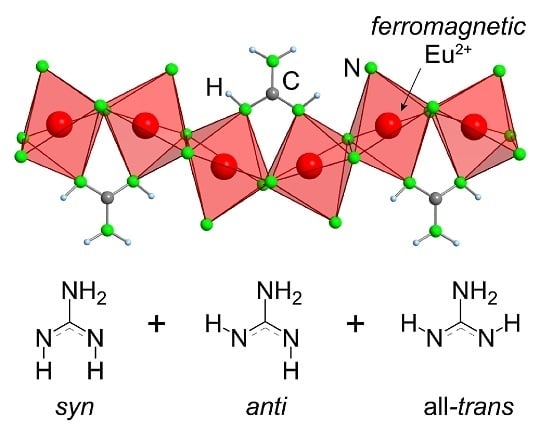
:
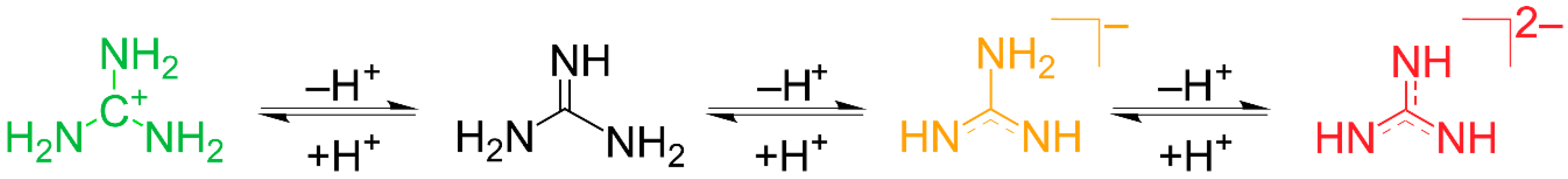
1. Introduction
2. Results and Discussion
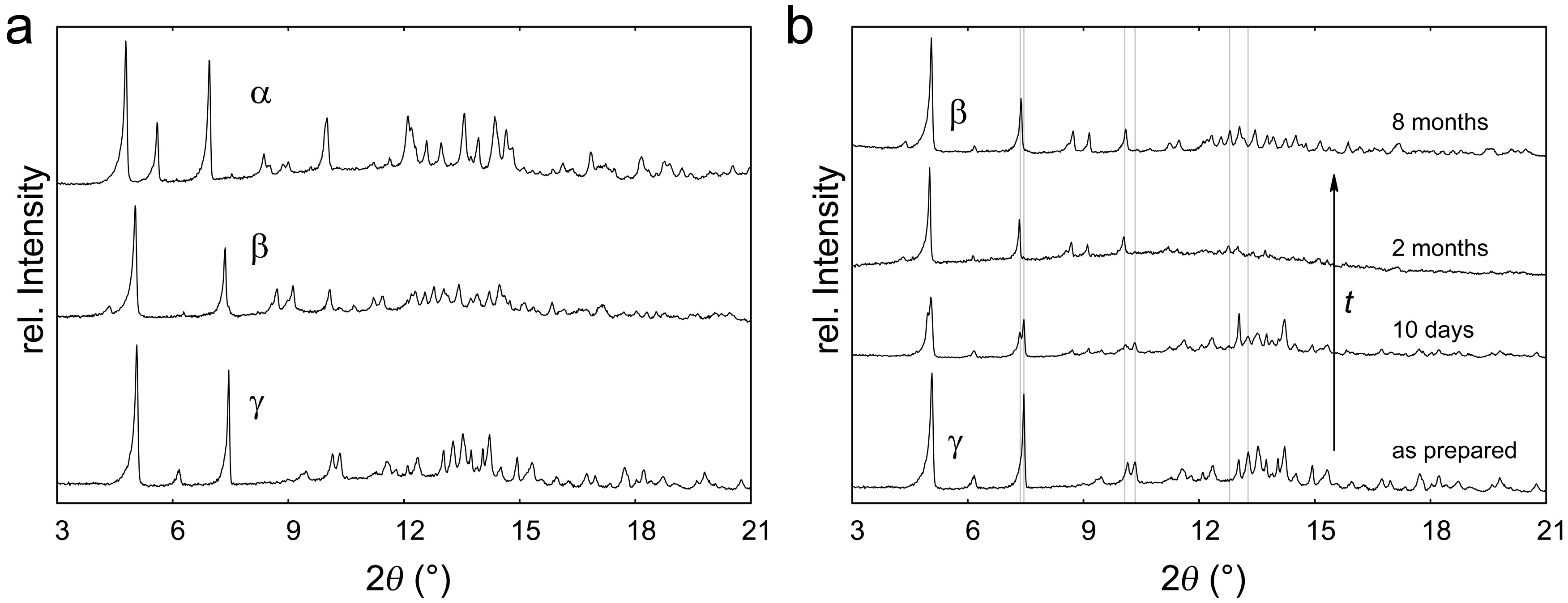
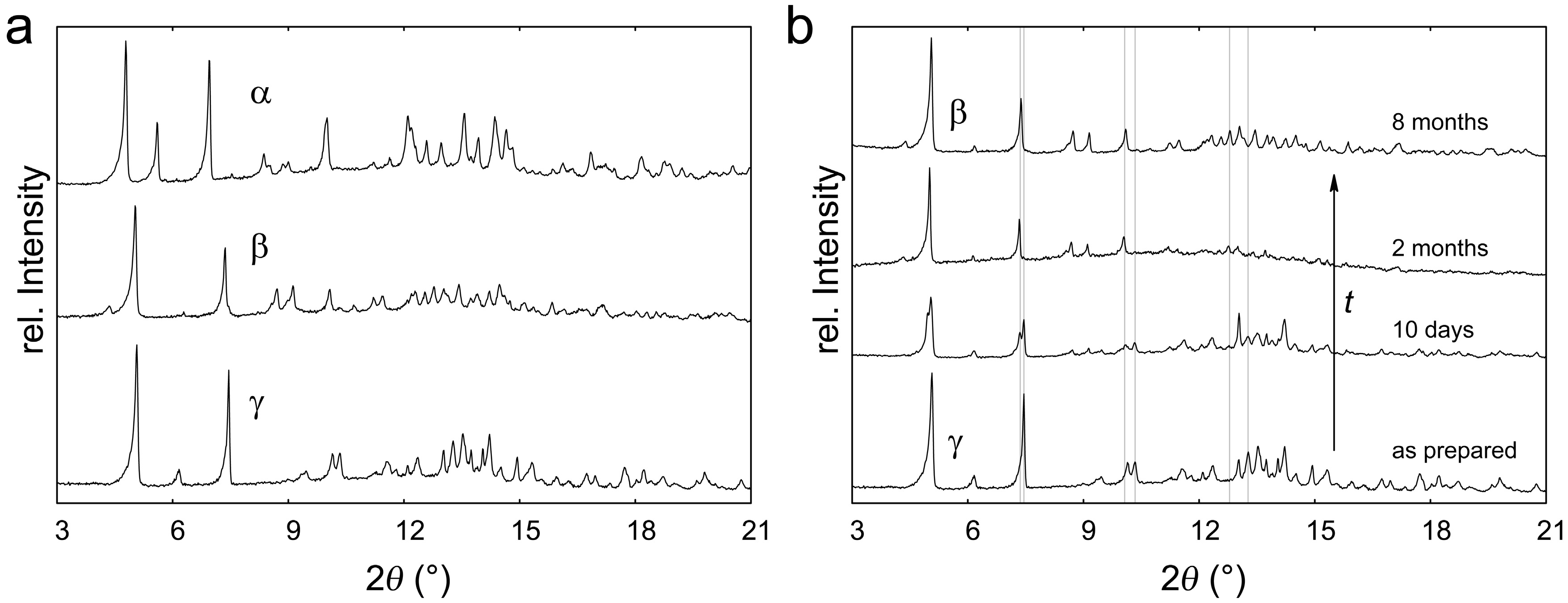
2.1. Polymorphism of Eu(CN3H4)2
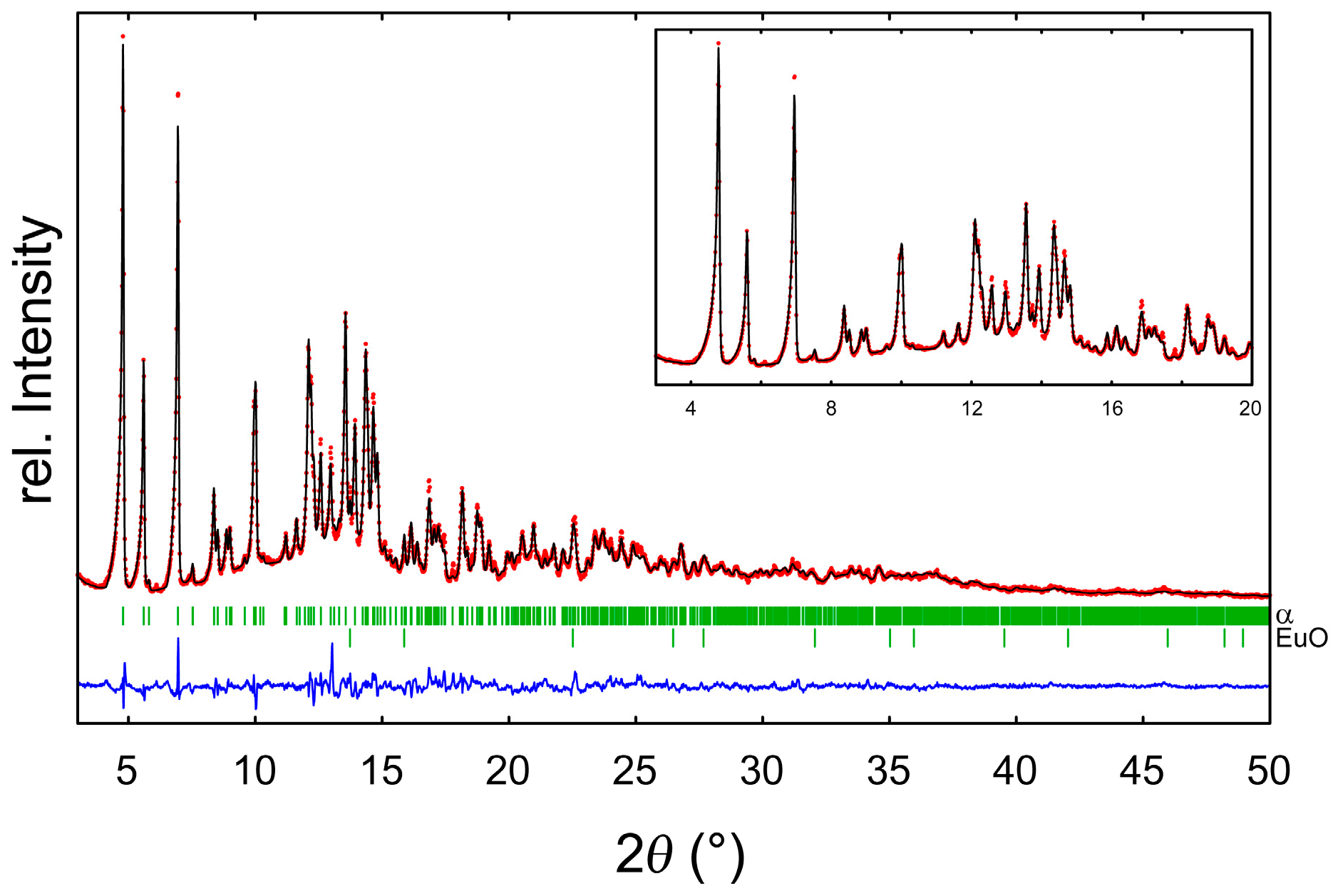
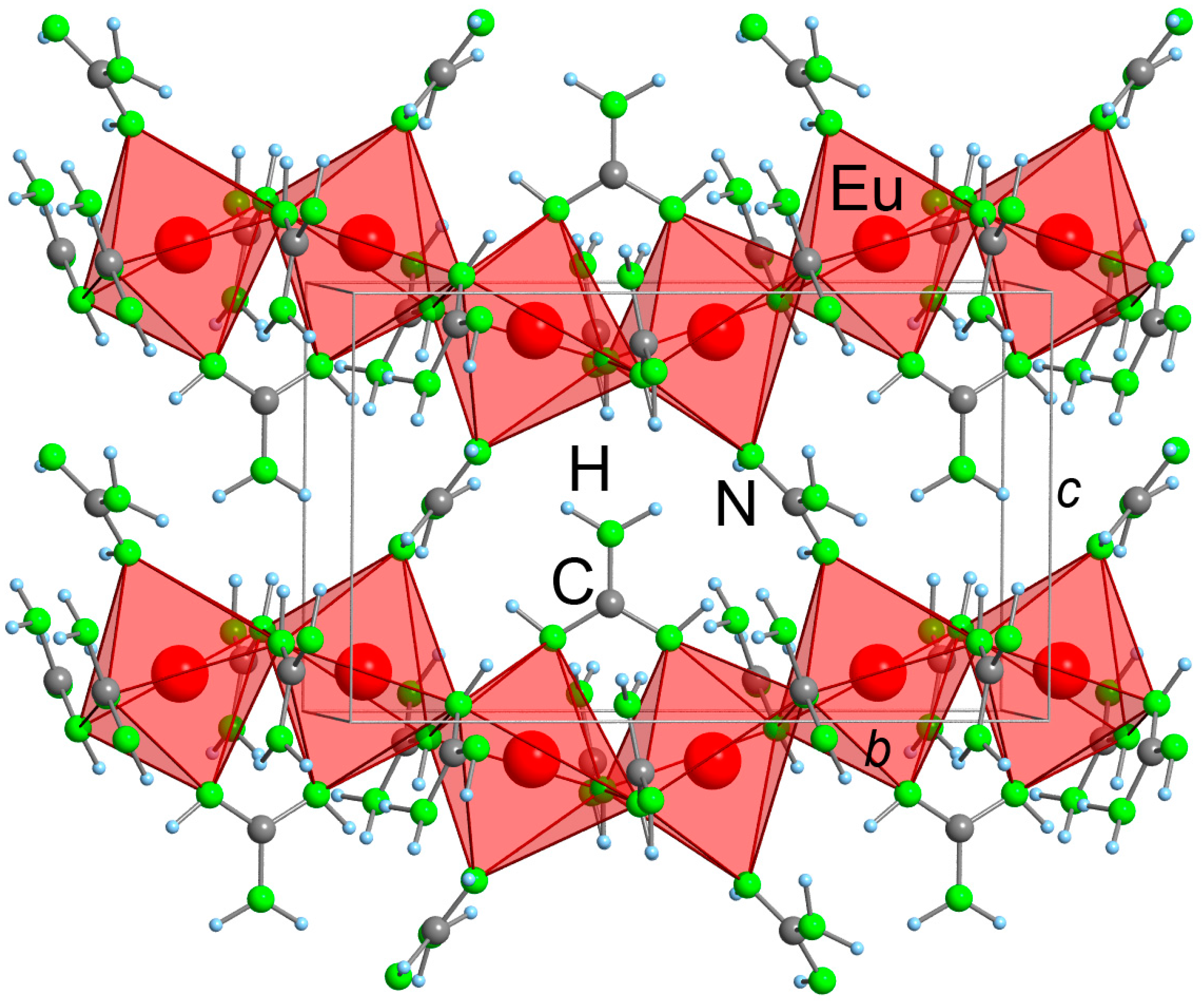
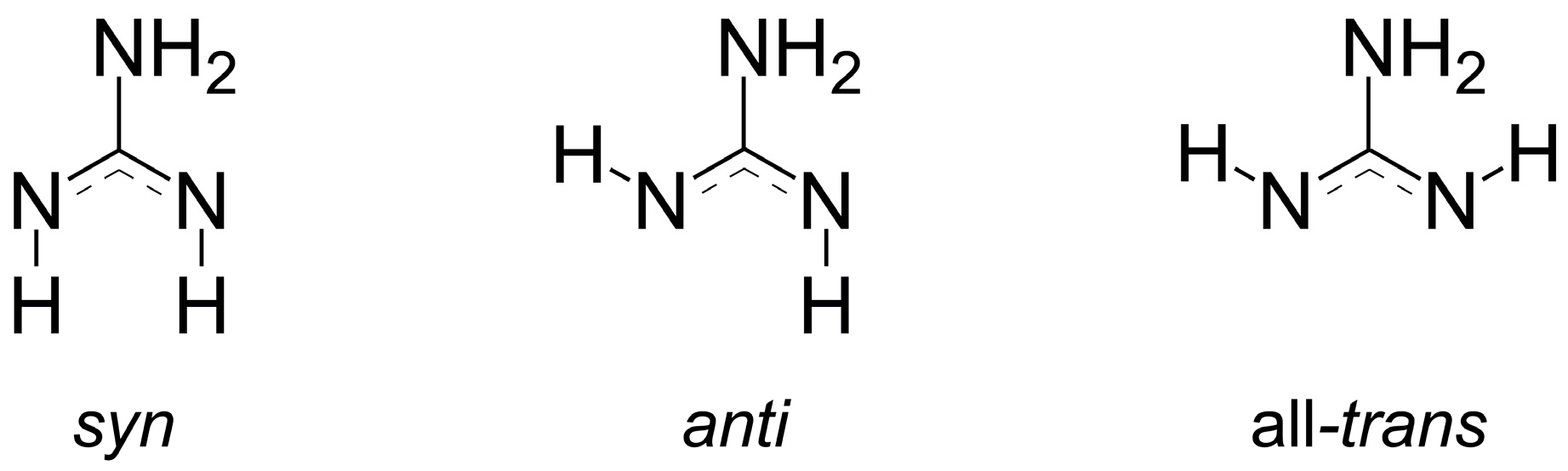
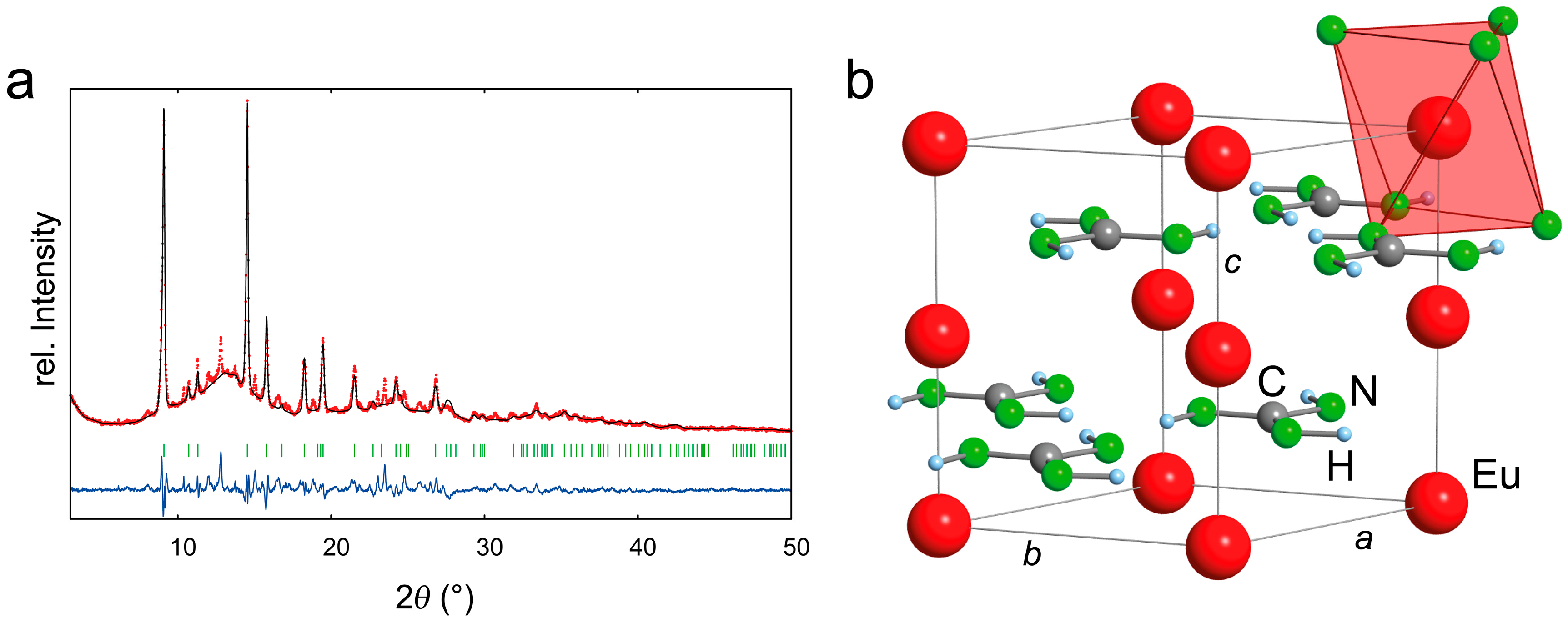
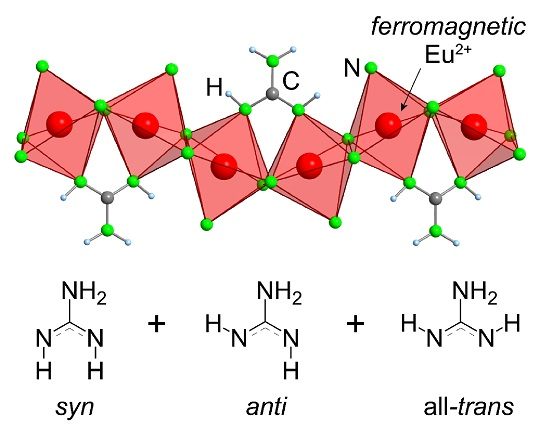
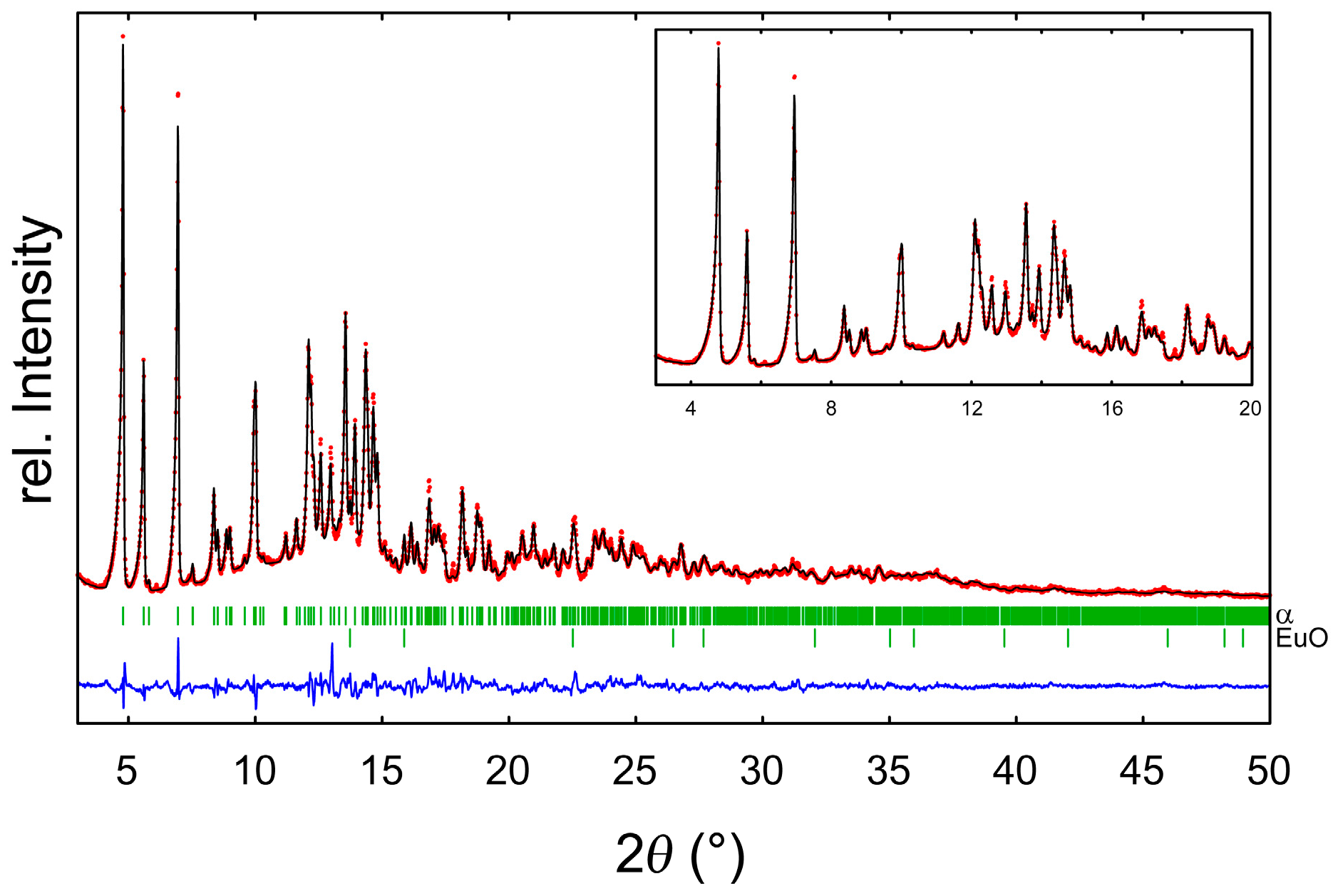
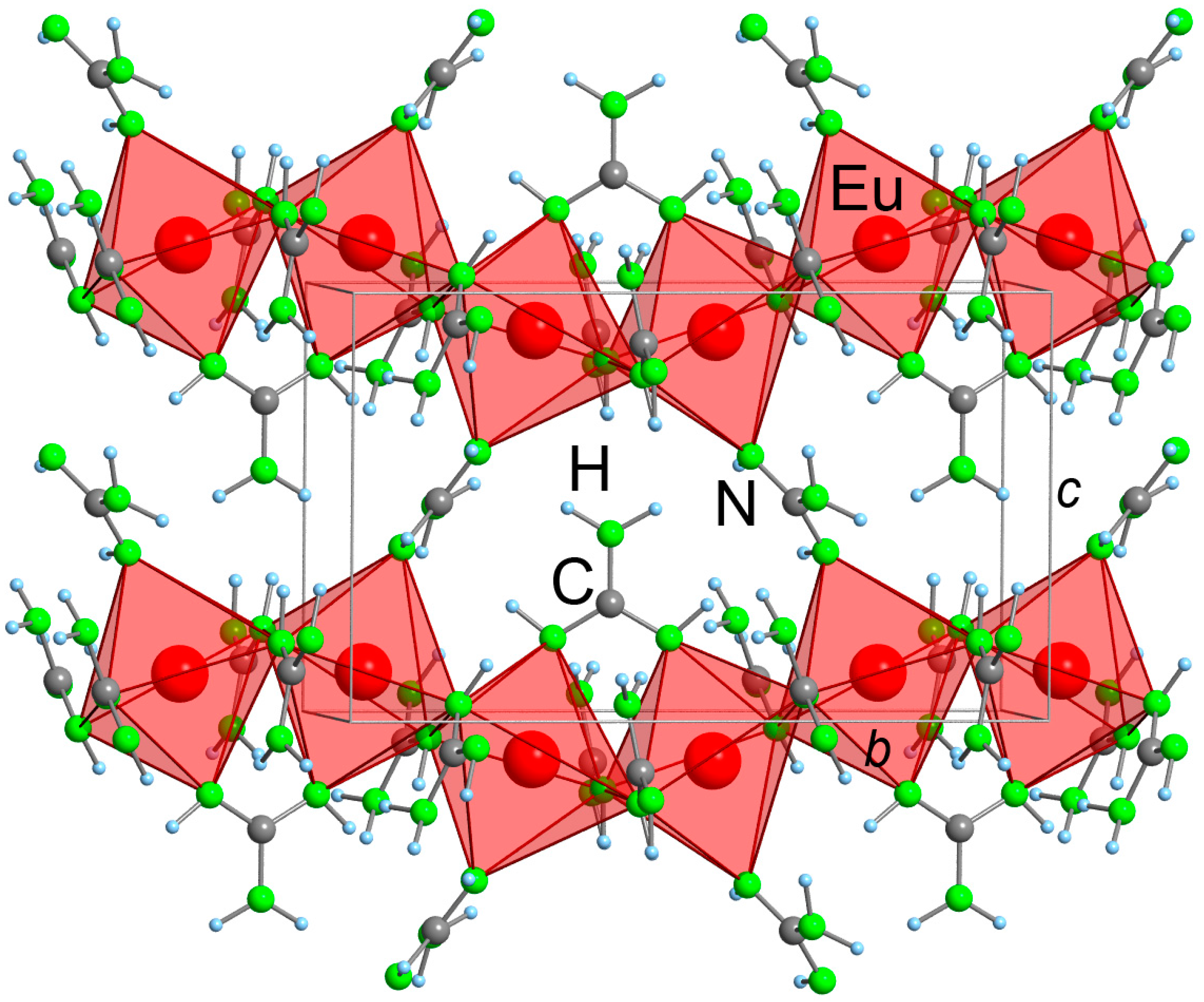
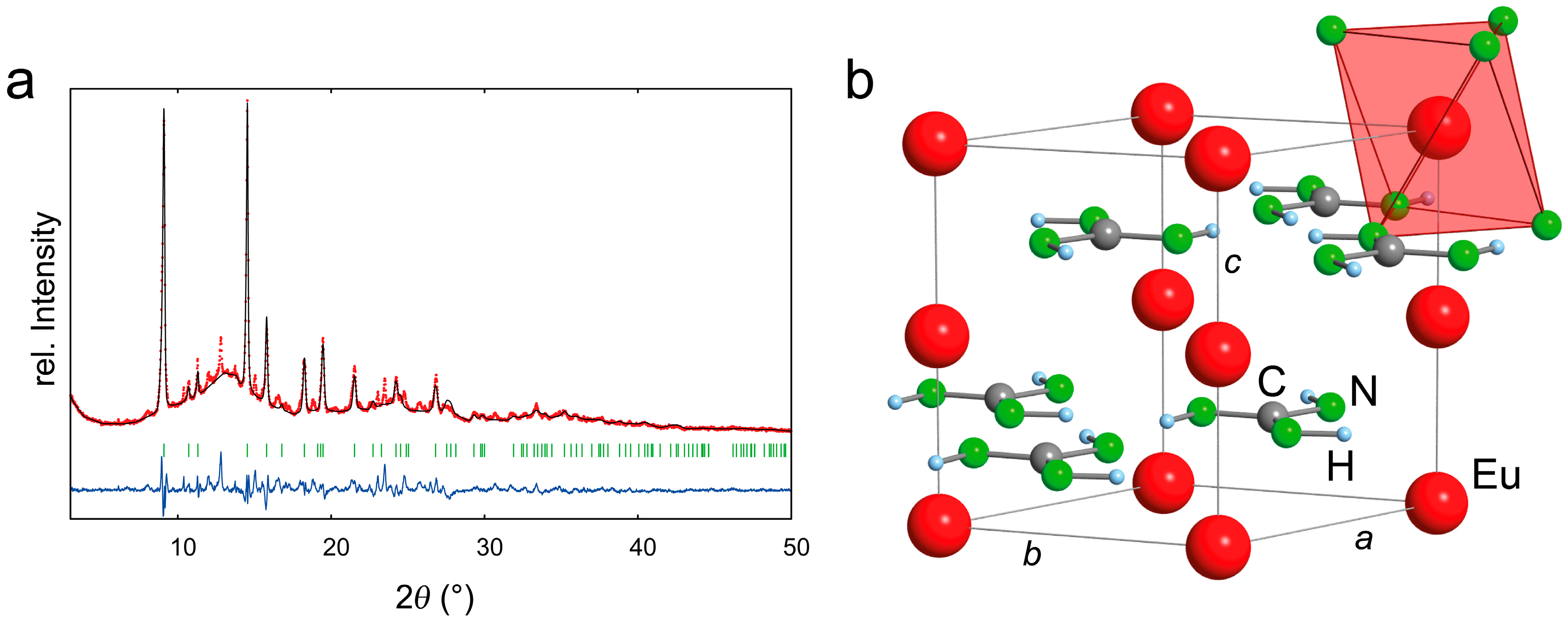
2.2. Refinement and Crystal Structure of α-Eu(CN3H4)2
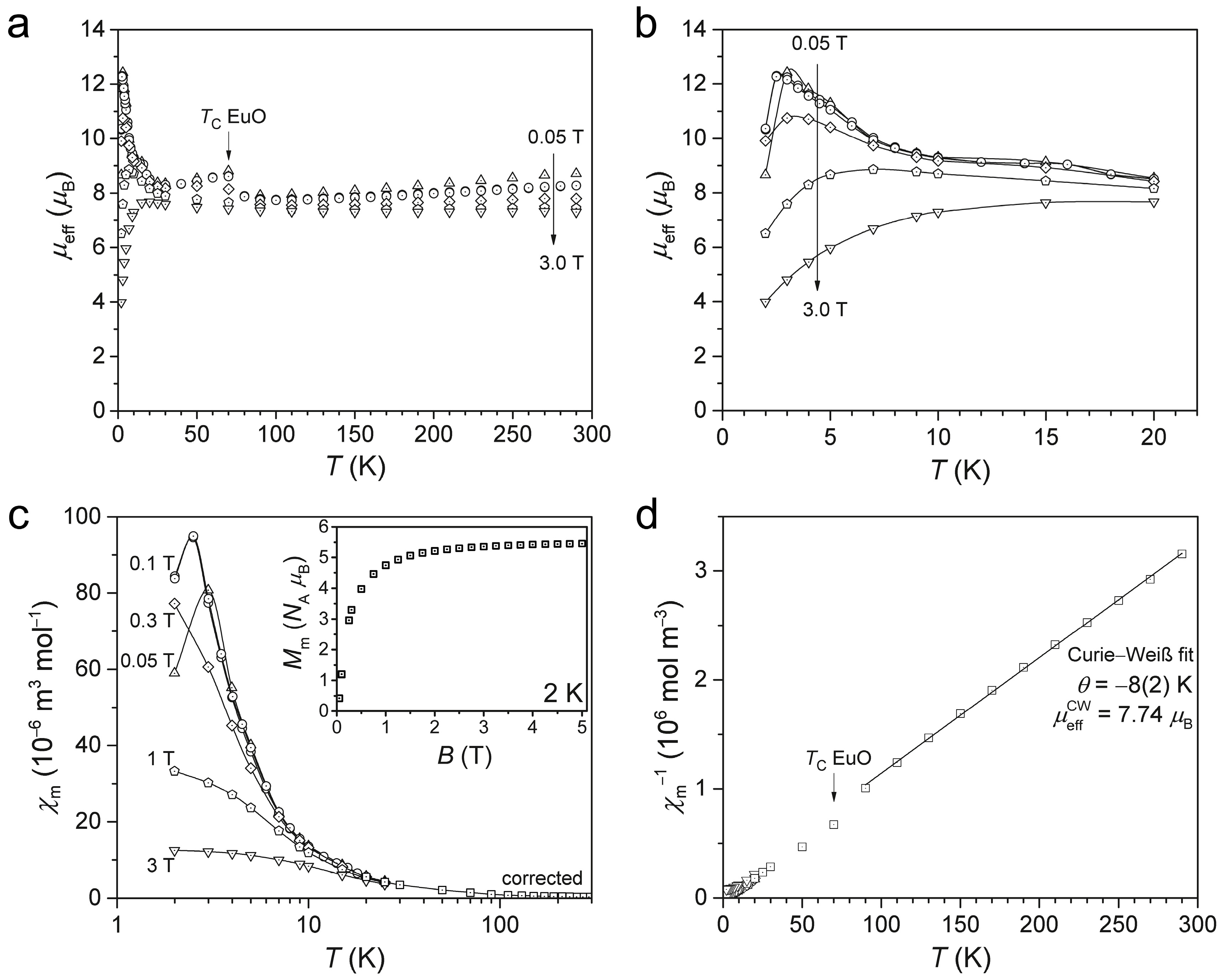
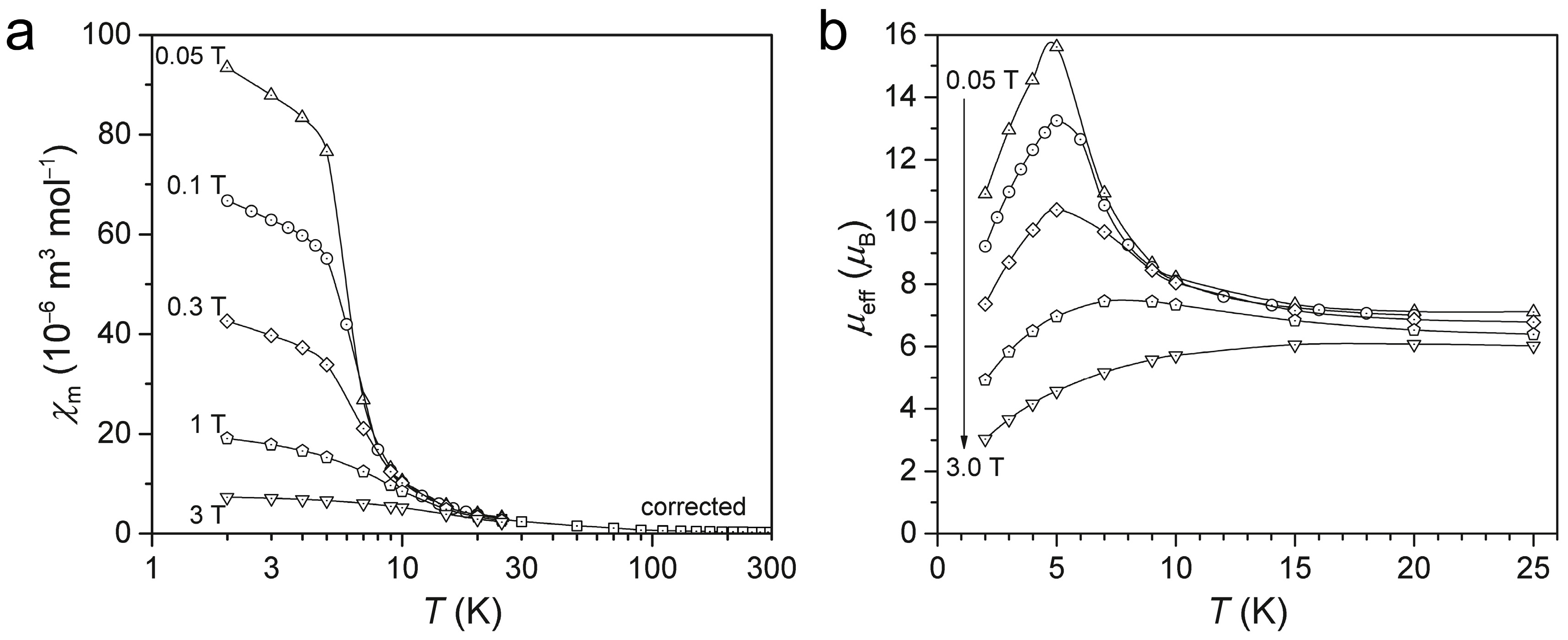
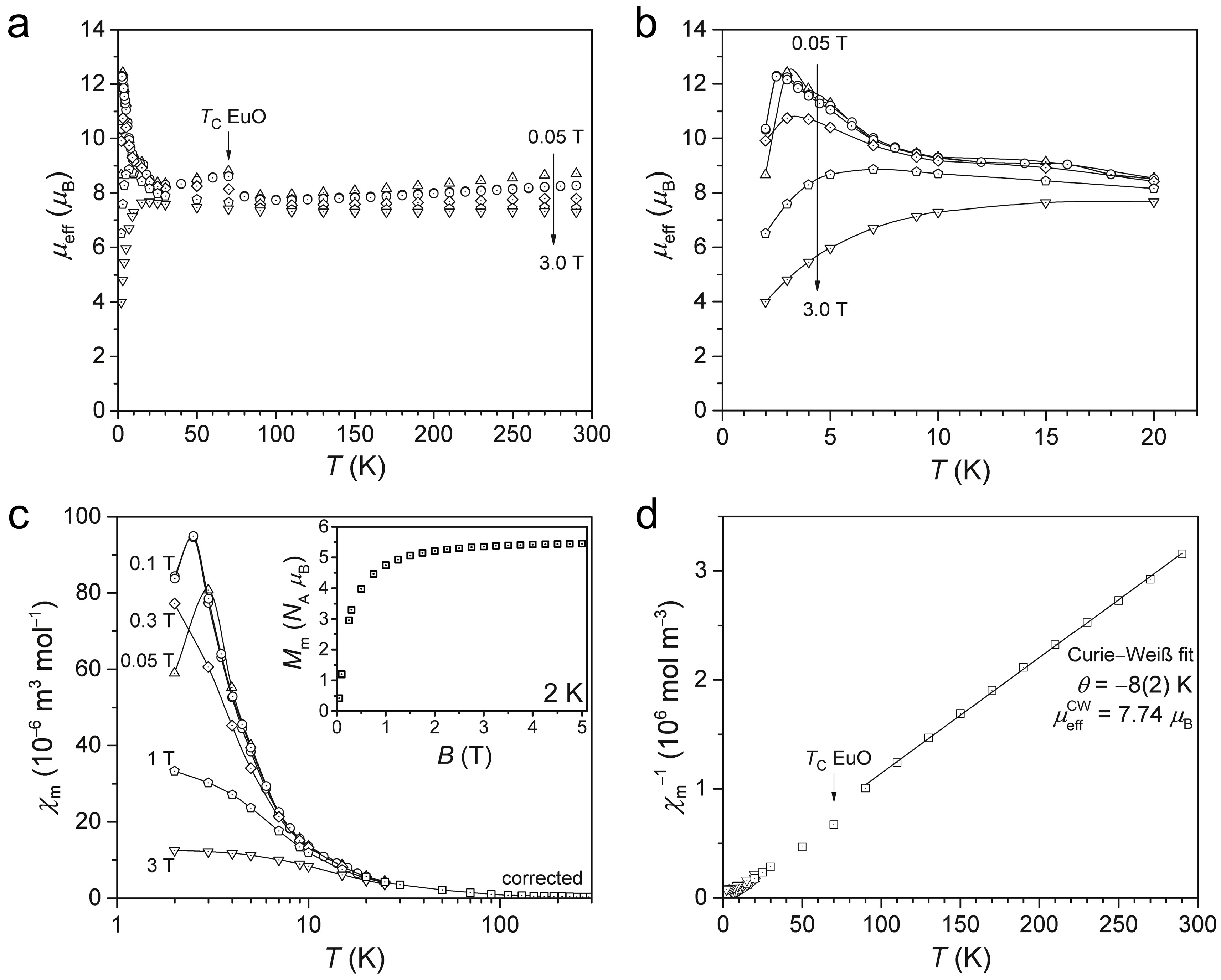
2.3. Magnetism of α-Eu(CN3H4)2
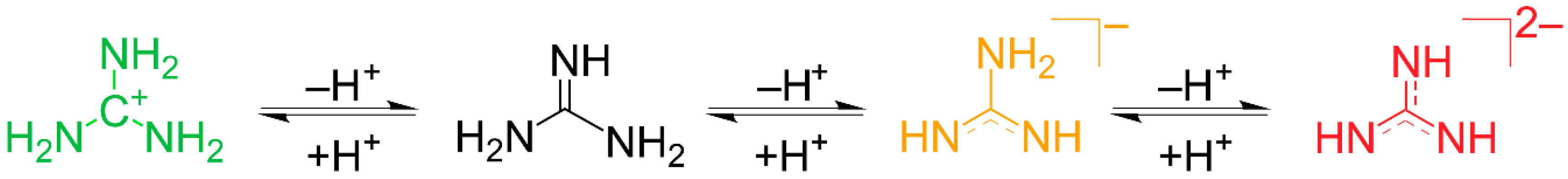
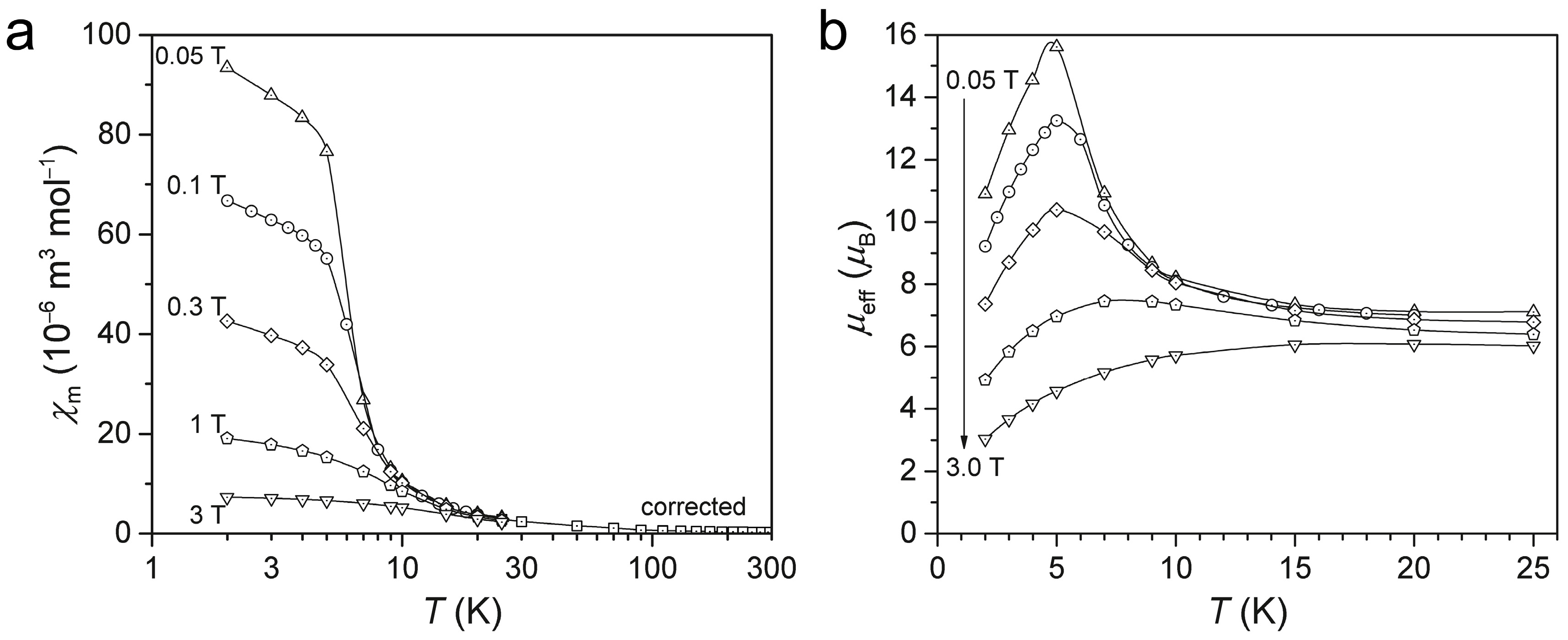
2.4. Introduction of EuC(NH)3
3. Materials and Methods
3.1. Syntheses
3.2. Powder X-Ray Diffraction
3.3. DFT Calculations
3.4. Magnetometry
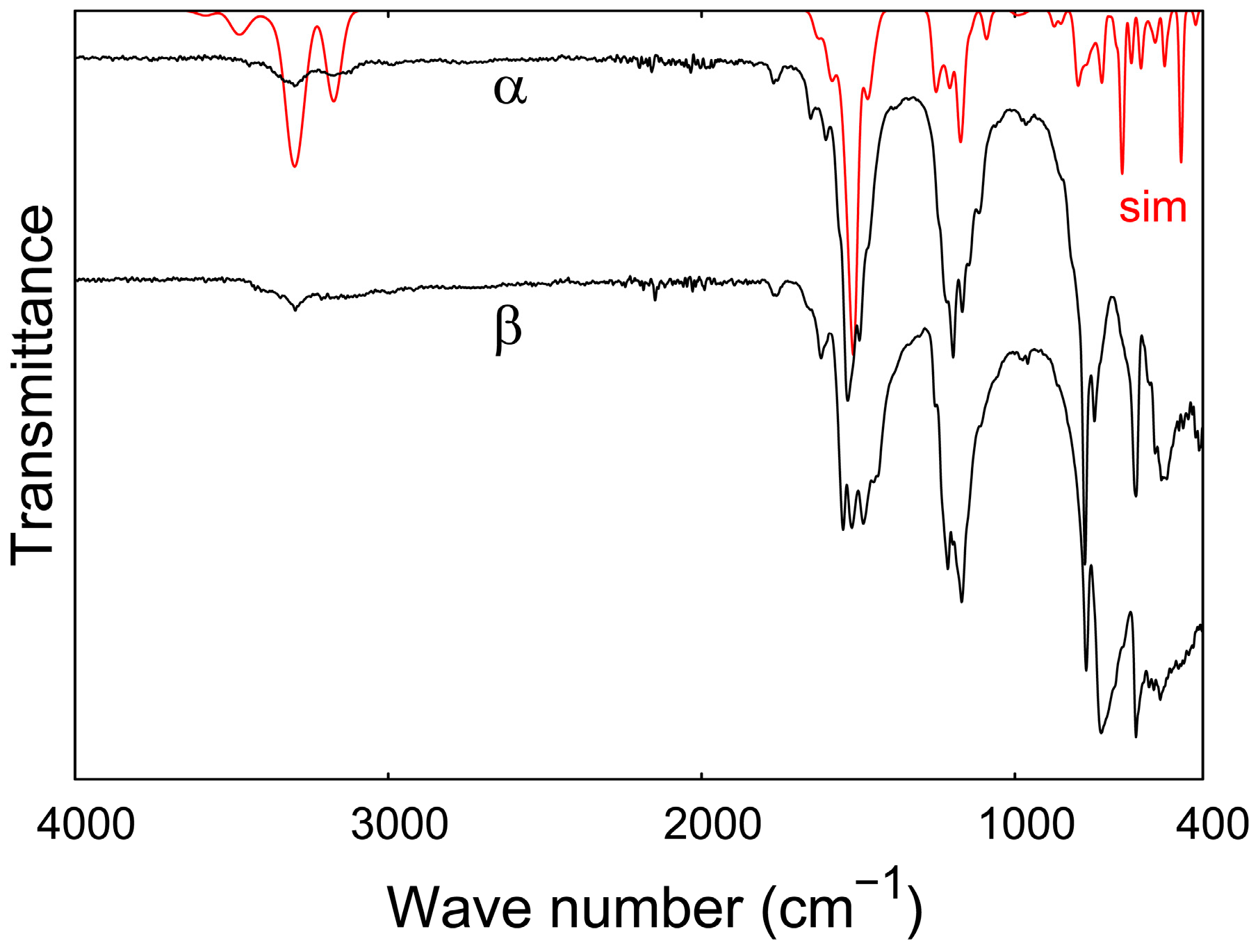
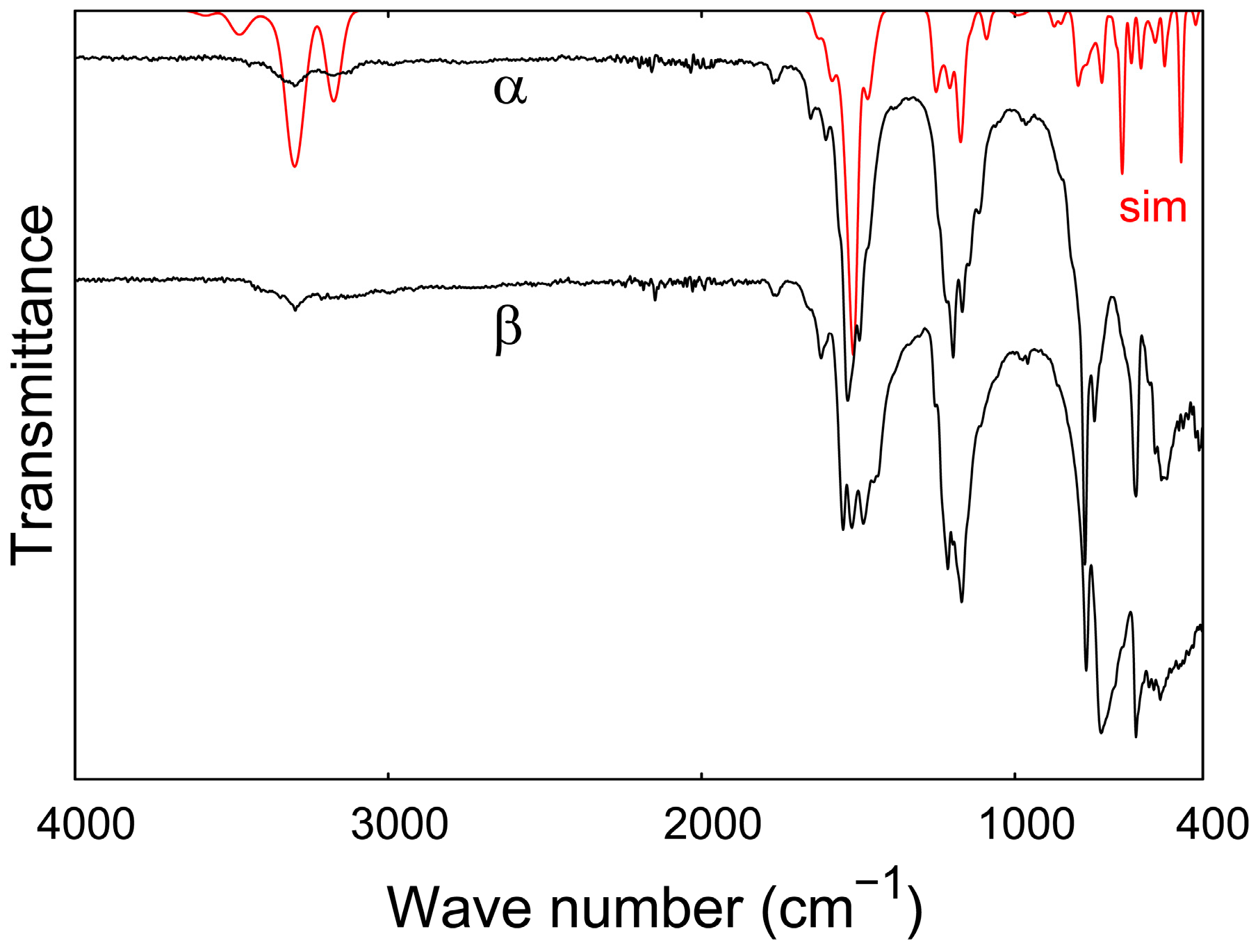
3.5. IR Spectroscopy
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Abraham, D.S. The Next Resource Shortage? The New York Times, 20 November 2015. [Google Scholar]
- Natural Environment Research Council. Rare Earth Elements; British Geological Survey: Nottingham, UK, 2011.
- Zurawski, A.; Mai, M.; Baumann, D.; Feldmann, C.; Müller-Buschbaum, K. Homoleptic imidazolate frameworks 3∞[Sr1–xEux(Im)2]-hybrid materials with efficient and tuneable luminescence. Chem. Commun. 2011, 47, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Rybak, J.-C.; Hailmann, M.; Matthes, P.R.; Zurawski, A.; Nitsch, J.; Steffen, A.; Heck, J.G.; Feldmann, C.; Götzendörfer, S.; Meinhardt, J.; et al. Metal–Organic Framework Luminescence in the Yellow Gap by Codoping of the Homoleptic Imidazolate 3∞[Ba(Im)2] with Divalent Europium. J. Am. Chem. Soc. 2013, 135, 6896–6902. [Google Scholar] [CrossRef] [PubMed]
- Pust, P.; Weiler, V.; Hecht, C.; Tücks, A.; Wochnik, A.S.; Henß, A.-K.; Wiechert, D.; Scheu, C.; Schmidt, P.J.; Schnick, W. Narrow-band red-emitting Sr[LiAl3N4]:Eu2+ as a next-generation LED-phosphor material. Nat. Mater. 2014, 13, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Kuda-Wedagedara, A.N.W.; Wang, C.; Martin, P.D.; Allen, M.J. Aqueous EuII-Containing Complex with Bright Yellow Luminescence. J. Am. Chem. Soc. 2015, 137, 4960–4963. [Google Scholar] [CrossRef] [PubMed]
- Slabon, A.; Mensing, C.; Kubata, C.; Cuervo-Reyes, E.; Nesper, R. Field-Induced Inversion of the Magnetoresistive Effect in the Zintl Phase Eu5+xMg18−xSi13 (x = 2.2). Angew. Chem. Int. Ed. 2013, 52, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Rushchanskii, K.Z.; Kamba, S.; Goian, V.; Vaněk, P.; Savinov, M.; Prokleška, J.; Nuzhnyy, D.; Knížek, K.; Laufek, F.; Eckel, S.; et al. A multiferroic material to search for the permanent electric dipole moment of the electron. Nat. Mater. 2010, 9, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Niehaus, O.; Ryan, D.H.; Flacau, R.; Lemoine, P.; Chernyshov, D.; Svitlyk, V.; Cuervo-Reyes, E.; Slabon, A.; Nesper, R.; Schellenberg, I.; et al. Complex physical properties of EuMgSi—A complementary study by neutron powder diffraction and 151Eu Mossbauer spectroscopy. J. Mater. Chem. C 2015, 3, 7203–7215. [Google Scholar] [CrossRef]
- Lueken, H. Magnetochemie: Eine Einführung in Theorie und Anwendung; Teubner Verlag: Stuttgart, Leipzig, 1999. [Google Scholar]
- Matthias, B.T.; Bozorth, R.M.; Van Vleck, J.H. Ferromagnetic Interaction in EuO. Phys. Rev. Lett. 1961, 7, 160–161. [Google Scholar] [CrossRef]
- Kornblit, A.; Ahlers, G.; Buehler, E. Heat capacity of RbMnF3 and EuO near the magnetic phase transitions. Phys. Lett. A 1973, 43, 531–532. [Google Scholar] [CrossRef]
- Wachter, P. Europium chalcogenides: EuO, EuS, EuSe and EuTe. In Handbook on the Physics and Chemistry of Rare Earths; Elsevier: Amsterdam, The Netherlands, 1979; Volume 2, pp. 507–574. [Google Scholar]
- Juza, R.; Hadenfeldt, C. Darstellung und Eigenschaften von Europium(II)-amid. Naturwissenschaften 1968, 55, 229. [Google Scholar] [CrossRef]
- Hulliger, F. Ferromagnetism of europium amide Eu(NH2)2. Solid State Commun. 1970, 8, 1477–1478. [Google Scholar] [CrossRef]
- Wickleder, C. M(SCN)2 (M = Eu, Sr, Ba): Kristallstruktur, thermisches Verhalten, Schwingungsspektroskopie. Z. Anorg. Allg. Chem. 2001, 627, 1693–1698. [Google Scholar] [CrossRef]
- Reckeweg, O.; DiSalvo, F.J. EuCN2—The First, but Not Quite Unexpected Ternary Rare Earth Metal Cyanamide. Z. Anorg. Allg. Chem. 2003, 629, 177–179. [Google Scholar] [CrossRef]
- Huppertz, H.; Schnick, W. Eu2Si5N8 and EuYbSi4N7. The First Nitridosilicates with a Divalent Rare Earth Metal. Acta Crystallogr. C 1997, 53, 1751–1753. [Google Scholar] [CrossRef]
- Höppe, H.A.; Trill, H.; Mosel, B.D.; Eckert, H.; Kotzyba, G.; Pöttgen, R.; Schnick, W. Hyperfine interactions in the 13 K ferromagnet Eu2Si5N8. J. Phys. Chem. Solids 2002, 63, 853–859. [Google Scholar] [CrossRef]
- Carrillo-Cabrera, W.; Somer, M.; Peters, K.; Schnering, H.G.V. Crystal structure of trieuropium bis(dinitridoborate), Eu3[BN2]2. Z. Kristallogr. New Cryst. Struct. 2001, 216, 43–44. [Google Scholar] [CrossRef]
- Liao, W.; Dronskowski, R. Carbodiimides with Extended Structures by an Azide-Cyanide Route: Synthesis and Crystal Structure of M2Cl2NCN (M = Eu and Sr). Z. Anorg. Allg. Chem. 2005, 631, 496–498. [Google Scholar] [CrossRef]
- Stadler, F.; Oeckler, O.; Höppe, H.A.; Möller, M.H.; Pöttgen, R.; Mosel, B.D.; Schmidt, P.; Duppel, V.; Simon, A.; Schnick, W. Crystal Structure, Physical Properties and HRTEM Investigation of the New Oxonitridosilicate EuSi2O2N2. Chem. Eur. J. 2006, 12, 6984–6990. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, G.; Thuéry, P.; Rivière, E.; Ephritikhine, M. Europium(II) compounds: Simple synthesis of a molecular complex in water and coordination polymers with 2,2′-bipyrimidine-mediated ferromagnetic interactions. Chem. Commun. 2010, 46, 9143–9145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Hu, C.; Kremer, R.K.; Dronskowski, R. Formation of Complex Three- and One-Dimensional Interpenetrating Networks within Carbodiimide Chemistry: NCN2–-Coordinated Rare-Earth-Metal Tetrahedra and Condensed Alkali-Metal Iodide Octahedra in Two Novel Lithium Europium Carbodiimide Iodides, LiEu2(NCN)I3 and LiEu4(NCN)3I3. Inorg. Chem. 2004, 43, 5884–5890. [Google Scholar] [PubMed]
- Yamada, T.; Liu, X.; Englert, U.; Yamane, H.; Dronskowski, R. Solid-State Structure of Free Base Guanidine Achieved at Last. Chem. Eur. J. 2009, 15, 5651–5655. [Google Scholar] [CrossRef] [PubMed]
- Sawinski, P.K.; Meven, M.; Englert, U.; Dronskowski, R. Single-Crystal Neutron Diffraction Study on Guanidine, CN3H5. Cryst. Growth Des. 2013, 13, 1730–1735. [Google Scholar] [CrossRef]
- Hoepfner, V.; Dronskowski, R. RbCN3H4: The First Structurally Characterized Salt of a New Class of Guanidinate Compounds. Inorg. Chem. 2011, 50, 3799–3803. [Google Scholar] [CrossRef] [PubMed]
- Sawinski, P.K.; Dronskowski, R. Solvothermal Synthesis, Crystal Growth, and Structure Determination of Sodium and Potassium Guanidinate. Inorg. Chem. 2012, 51, 7425–7430. [Google Scholar] [CrossRef] [PubMed]
- Sawinski, P.K.; Deringer, V.L.; Dronskowski, R. Completing a family: LiCN3H4, the lightest alkali metal guanidinate. Dalton Trans. 2013, 42, 15080–15087. [Google Scholar] [CrossRef] [PubMed]
- Missong, R.; George, J.; Houben, A.; Hoelzel, M.; Dronskowski, R. Synthesis, Structure, and Properties of SrC(NH)3, a Nitrogen-Based Carbonate Analogue with the Trinacria Motif. Angew. Chem. Int. Ed. 2015, 54, 12171–12175. [Google Scholar] [CrossRef] [PubMed]
- Görne, A.L.; George, J.; van Leusen, J.; Dück, G.; Jacobs, P.; Chogondahalli Muniraju, N.K.; Dronskowski, R. Ammonothermal Synthesis, Crystal Structure, and Properties of the Ytterbium(II) and Ytterbium(III) Amides and the First Two Rare-Earth-Metal Guanidinates, YbC(NH)3 and Yb(CN3H4)3. Inorg. Chem. 2016, 55, 6161–6168. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, H.; Fink, U. Untersuchung des Systems Kalium/Europium/Ammoniak. Z. Anorg. Allg. Chem. 1978, 438, 151–159. [Google Scholar] [CrossRef]
- Karhánek, D.; Bučko, T.; Hafner, J. A density-functional study of the adsorption of methane-thiol on the (111) surfaces of the Ni-group metals: II. Vibrational spectroscopy. J. Phys. Condens. Matter 2010, 22, 265006. [Google Scholar] [CrossRef] [PubMed]
- Baroni, S.; de Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Hoepfner, V. Synthese und quantenchemische Untersuchung von Alkalimetallguanidinaten. Dissertation, RWTH Aachen University, Aachen, Germany, 2012. [Google Scholar]
- Krott, M.; Liu, X.; Fokwa, B.P.T.; Speldrich, M.; Lueken, H.; Dronskowski, R. Synthesis, Crystal–Structure Determination and Magnetic Properties of Two New Transition-Metal Carbodiimides: CoNCN and NiNCN. Inorg. Chem. 2007, 46, 2204–2207. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Stork, L.; Speldrich, M.; Lueken, H.; Dronskowski, R. FeNCN and Fe(NCNH)2: Synthesis, Structure, and Magnetic Properties of a Nitrogen-Based Pseudo-oxide and -hydroxide of Divalent Iron. Chem. Eur. J. 2009, 15, 1558–1561. [Google Scholar] [CrossRef] [PubMed]
- Pöttgen, R.; Johrendt, D. Equiatomic Intermetallic Europium Compounds: Syntheses, Crystal Chemistry, Chemical Bonding, and Physical Properties. Chem. Mater. 2000, 12, 875–897. [Google Scholar] [CrossRef]
- Van de Streek, J.; Neumann, M.A. Validation of experimental molecular crystal structures with dispersion-corrected density functional theory calculations. Acta Crystallogr. B 2010, 66, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Hoepfner, V.; Deringer, V.L.; Dronskowski, R. Hydrogen-Bonding Networks from First-Principles: Exploring the Guanidine Crystal. J. Phys. Chem. A 2012, 116, 4551–4559. [Google Scholar] [CrossRef] [PubMed]
- Hoepfner, V.; Jacobs, P.; Sawinski, P.K.; Houben, A.; Reim, J.; Dronskowski, R. RbCN3H4 and CsCN3H4: A Neutron Powder and Single-Crystal X-ray Diffraction Study. Z. Anorg. Allg. Chem. 2013, 639, 1232–1236. [Google Scholar] [CrossRef]
- Bailey, P.J.; Blake, A.J.; Kryszczuk, M.; Parsons, S.; Reed, D. The first triazatrimethylenemethane dianion: crystal structure of dilithio-triphenylguanidine Li2[C(NPh)3] as its tetrahydrofuran solvate. J. Chem. Soc. Chem. Commun. 1995, 1647–1648. [Google Scholar] [CrossRef]
- Bailey, P.J.; Mitchell, L.A.; Parsons, S. Guanidine anions as chelating ligands; syntheses and crystal structures of [Rh(η-C5Me5){η2-(NPh)2CNHPh}Cl] and [Ru(η-MeC6H4Pri-p)-{η2-(NPh)2CNHPh}Cl]. J. Chem. Soc. Dalton Trans. 1996, 2839–2841. [Google Scholar] [CrossRef]
- Shuskus, A.J. Electron Spin Resonance of Gd3+ and Eu2+ in Single Crystals of CaO. Phys. Rev. 1962, 127, 2022–2024. [Google Scholar] [CrossRef]
- Baker, J.M.; Williams, F.I.B. Electron Nuclear Double Resonance of the Divalent Europium Ion. Proc. R. Soc. A 1962, 267, 283–294. [Google Scholar] [CrossRef]
- Deringer, V.L.; Hoepfner, V.; Dronskowski, R. Accurate Hydrogen Positions in Organic Crystals: Assessing a Quantum-Chemical Aide. Cryst. Growth Des. 2011, 12, 1014–1021. [Google Scholar] [CrossRef]
- Missong, R. Synthese und Charakterisierung von Strontium- und Bariumguanidinat. Dissertation, RWTH Aachen University, Aachen, Germany, 2016. [Google Scholar]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Kristallogr. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- McWhan, D.B.; Souers, P.C.; Jura, G. Magnetic and Structural Properties of Europium Metal and Europium Monoxide at High Pressure. Phys. Rev. 1966, 143, 385–389. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vibration | α-Eu(CN3H4)2 (cm−1) |
|---|---|
| νs, as(N–H) | 3299, 3172 |
| δsciss(N–H) | 1652, 1602 |
| νs, δsciss(C–N); δsciss(N–H) | 1533, 1495 |
| δrock(N–H) | 1197–1167 |
| δwagg(N–H) | 1147–1114 |
| νbreath(CN3) | 965 |
| δtwist(N–H) | 776 |
| C-inversion by CN3 plane | 746 |
| δrock(C–N), δrock(N–H) | 614 |
| δsciss(C–N), δsciss(N–H) | 532–514 |
| Formula | α-Eu(CN3H4)2 | EuC(NH)3 |
|---|---|---|
| Formula weight (g·mol−1) | 268.09 | 209.02 |
| Crystal system | Monoclinic | Hexagonal |
| Space group | P21 (Nr. 4) | P63/m (Nr. 176) |
| Temperature (K) | 298 | 298 |
| a (Å) | 5.8494(3) | 5.1634(7) |
| b (Å) | 14.0007(8) | = a |
| c (Å) | 8.4887(4) | 7.1993(9) |
| β (°) | 91.075(6) | 90 |
| V (Å3) | 695.07(6) | 166.22(4) |
| Z | 4 | 2 |
| Cryst. density (g·cm−3) | 2.4848(2) | 4.1176(9) |
| Radiation | Mo Kα1 | Mo Kα1 |
| No. reflections | 870 | 108 |
| No. restraints/constraints | 24/4 | 0/1 |
| No. refined parameters | 71 | 14 |
| Rp, wRpa | 3.7/4.9 | 5.1/7.3 |
| RBragg, RFb | 11.3/6.8 | 18.7/11.7 |
| Atom | x | y | z |
|---|---|---|---|
| Eu1 | 0.5358 | 0.7086 | 0.0995 |
| Eu2 | 0.5091 | 0.4477 | 0.1043 |
| C1 | 0.4778 | 0.5939 | −0.2622 |
| C2 | 0.0096 | 0.5851 | 0.1186 |
| C3 | 0.9894 | 0.8521 | 0.0723 |
| C4 | 0.4115 | 0.8357 | 0.5014 |
| N1 | 0.4261 | 0.6722 | −0.1801 |
| N2 | 0.5249 | 0.5123 | −0.1822 |
| N3 | 0.4775 | 0.5942 | −0.4249 |
| N4 | 0.2256 | 0.5851 | 0.1783 |
| N5 | −0.1886 | 0.5721 | 0.1963 |
| N6 | −0.0171 | 0.6030 | −0.0391 |
| N7 | 0.8237 | 0.8338 | −0.0357 |
| N8 | 1.2141 | 0.8350 | 0.0569 |
| N9 | 0.9070 | 0.8881 | 0.2132 |
| N10 | 0.5037 | 0.7872 | 0.3822 |
| N11 | 0.5109 | 0.8953 | 0.6061 |
| N12 | 0.1747 | 0.8252 | 0.5170 |
| H1 | 0.3877 | 0.7278 | −0.2540 |
| H2 | 0.5761 | 0.4617 | −0.2613 |
| H3 | 0.4914 | 0.6572 | −0.4856 |
| H4 | 0.5196 | 0.533 | −0.4836 |
| H5 | 0.2196 | 0.5831 | 0.2991 |
| H6 | −0.1546 | 0.5563 | 0.3121 |
| H7 | 0.1267 | 0.6207 | −0.1006 |
| H8 | −0.1558 | 0.5741 | −0.0972 |
| H9 | 0.8935 | 0.7988 | −0.1291 |
| H10 | 1.2891 | 0.8532 | 0.1636 |
| H11 | 0.7685 | 0.9334 | 0.2022 |
| H12 | 1.0256 | 0.9096 | 0.2948 |
| H13 | 0.6727 | 0.8069 | 0.3758 |
| H14 | 0.6814 | 0.8996 | 0.5808 |
| H15 | 0.1090 | 0.7649 | 0.4674 |
| H16 | 0.1149 | 0.8394 | 0.6260 |
| Atom | Wyckoff position | x | y | z | Uiso or Ueq (Å2) |
|---|---|---|---|---|---|
| Eu | 2b | 0 | 0 | 0 | 0.058(1) |
| C | 2c | ⅓ | ⅔ | ¼ | 0.005(7) |
| N | 6h | 0.065(4) | 0.422(3) | ¼ | 0.005(7) |
| H (DFT) | 6h | −0.092 | 0.486 | ¼ | – |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Görne, A.L.; George, J.; Van Leusen, J.; Dronskowski, R. Synthesis, Crystal Structure, Polymorphism, and Magnetism of Eu(CN3H4)2 and First Evidence of EuC(NH)3. Inorganics 2017, 5, 10. https://doi.org/10.3390/inorganics5010010
Görne AL, George J, Van Leusen J, Dronskowski R. Synthesis, Crystal Structure, Polymorphism, and Magnetism of Eu(CN3H4)2 and First Evidence of EuC(NH)3. Inorganics. 2017; 5(1):10. https://doi.org/10.3390/inorganics5010010
Chicago/Turabian StyleGörne, Arno L., Janine George, Jan Van Leusen, and Richard Dronskowski. 2017. "Synthesis, Crystal Structure, Polymorphism, and Magnetism of Eu(CN3H4)2 and First Evidence of EuC(NH)3" Inorganics 5, no. 1: 10. https://doi.org/10.3390/inorganics5010010
APA StyleGörne, A. L., George, J., Van Leusen, J., & Dronskowski, R. (2017). Synthesis, Crystal Structure, Polymorphism, and Magnetism of Eu(CN3H4)2 and First Evidence of EuC(NH)3. Inorganics, 5(1), 10. https://doi.org/10.3390/inorganics5010010