
A First-Principle Study of Two-Dimensional Boron Nitride Polymorph with Tunable Magnetism


1
Team of Micro & Nano Sensor Technology and Application in High-Altitude Regions, School of Information Engineering, Xizang Minzu University, Xianyang 712082, China
2
College of Science, Jinling Institute of Technology, Nanjing 211169, China
3
College of Information and Control Engineering, Xi’an University of Architecture and Technology, Xi’an 710055, China
*
Author to whom correspondence should be addressed.
Inorganics 2024, 12(2), 59; https://doi.org/10.3390/inorganics12020059
Submission received: 25 December 2023
/
Revised: 30 January 2024
/
Accepted: 30 January 2024
/
Published: 15 February 2024
(This article belongs to the Special Issue Advanced Inorganic Semiconductor Materials)
Abstract
:Using the first-principles calculation, two doping two-dimensional (2D) BN (boron nitride) polymorphs are constructed in this work. The two doping 2D BN polymorphs B5N6Al and B5N6C sheets are thermally stable under 500 K. All the B6N6, B5N6Al, and B5N6C sheets are semiconductor materials with indirect band gaps on the basis of a hybrid functional. The anisotropic calculation results indicate that Young’s modulus (E) and Poisson’s ratio (v) of the B6N6, B5N6Al, and B5N6C sheets are anisotropic in the xy plane. In addition, the magnetic properties of the B6N6, B5N6Al, and B5N6C sheets have also been investigated. According to the calculation of the magnetic properties, B6N6 sheet does not exhibit magnetism, while it shows weak magnetism after doping carbon atom to the BN sheet. This paper explores the influence mechanism of doping different atoms on the basic physical properties of two-dimensional BN sheets. It not only constructs a relationship between structure and performance but also provides theoretical support for the performance regulation of BN materials.
1. Introduction
Graphene is a planar 2D carbon allotrope with single atomic thickness. Due to its special electronic and magnetic characteristics, graphene is considered to be a revolutionary material for multiple facilities, such as high-speed electronic devices, thermal and conductivity-enhancing composite materials, sensors, RF logic devices, transparent electrodes, etc. [1,2,3,4,5,6]. In recent years, novel theoretical two-dimensional materials based on first-principles calculations have shown many interesting physical and chemical properties [7,8,9,10,11,12]. As isoelectronic bodies, BN and carbon also have rich and colorful physical properties and polymorphs [13,14,15,16,17,18].
Research on low-dimensional boron nitride nanomaterials is important in the field of materials science [19,20,21,22,23,24], especially because their excellent chemical properties and thermal stability have been widely studied [25]. They can enhance the mechanical enhancement and thermal conductivity of various crystal structures, for instance, polymers, ceramics, and metals [26,27,28]. Research has shown that graphene-like h-BN sheets have remarkable electronic and optical characteristics [29] and show a wide band gap and intense absorption capacity in the ultraviolet region. In other studies in the literature, it was shown that at low doping rates, B-N prefers to replace sp hybrid carbon on the chain than hexagonal. At high doping rates, it first attacks the hexagonal structure and then the chain.
Qi et al. [30] predicted 2D BxNy (1 < x/y ≤ 2) sheets using the density functional theory (DFT) [31,32]. B5N3 and B7N5 sheets possess enough low enthalpy of formation and outstanding dynamic stability, which makes them possible to be found in experiments. Unlike previous BN, both B5N3 and B7N5 sheets exhibit narrow band gaps of 1.99 eV and 2.40 eV, respectively. Two-dimensional orthorhombic boron nitride crystal, named o-B2N2, was designed by Demirci et al. [23]. The stability of o-B2N2 at room temperature and ambient pressure has been confirmed. o-B2N2 is a semiconductor with a direct band gap of 1.70 eV. An appropriate band gap makes this structure exhibit higher light absorption in the visible light range along the armchair direction. The in-plane stiffness of o-B2N2 is also very close to that of hexagonal BN. Based on the DFT, Fan et al. [33] first proposed and studied in detail a new 2D Pmma-BN sheet. The stability of Pmma-BN sheet was demonstrated by phonon spectroscopy and ab initio molecular dynamics simulations at 300 and 500 K. Uniaxial strain has weakened the ZT of the Pmma-BN sheet and led to a decrease in thermoelectric conversion efficiency. Anota et al. [34] reported boron nitride nanosheets containing homonuclear boron bonds and found that the proportion of boron atoms in nanosheets is related to conductivity.
By inserting sp-hybridized BN bonds into a monolayer h-BN structure, Li et al. [24] designed BNyne, Bndiyne, and BNtriyne. To explore the influence mechanism on the physical properties of doped 2D boron nitride, B6N6 sheets (called BNyne in ref. [24]) with doping Al or C atoms are proposed in this work, named B5N6Al sheet and B5N6C sheet, respectively. Both carbon and aluminum have the advantages of being cheap and easy to obtain. The doping of C element is selected to improve the mechanical properties of B6N6, and the selection of Al element is to observe the influence of metal elements on the electrical properties of semiconductor materials. We verify the structural stability of B5N6Al sheet and B5N6C sheet from mechanical and thermal perspectives and study their elementary physical characteristics based on first-principles calculations. B5N6Al and B5N6C are both indirect band gap semiconductor materials, which are the same as B6N6. The band gap width of B5N6C is significantly reduced. More importantly, the spin-up and spin-down electronic band structures indicate that B6N6 sheet does not exhibit magnetism. But after adding carbon to the BN sheet, it changes from a nonmagnetic material to a magnetic material.
2. Results and Discussion
Two-dimensional boron nitride structures and the crystal structure model doped with aluminum and carbon atoms are shown in Figure 1, together with the configurations of h-BN, o-B2N2 [23], B5N3 [30], and B7N5 [30]. Here, light blue, light pink, green, and light red represent boron atoms, nitrogen atoms, aluminum atoms, and carbon atoms, respectively. h-BN is a hexagonal network-layered crystal composed of nitrogen atoms and boron atoms; its layered structure is similar to graphite. oB2N2 is a two-dimensional monolayer of boron nitride in an orthorhombic structure with a B-B and N-N double-atom structure. h-BN and oB2N2 are only composed of six-membered rings, while the crystal structure of B5N3 is composed of seven-membered rings and five-membered rings. B7N5 contains seven-membered rings, six-membered rings, and five-membered rings. By inserting B-N bonds into h-BN sheet, Li et al. [24] proposed two-dimensional B6N6, as shown in Figure 1. After being fully optimized, all of them—B6N6 sheet, B5N6Al sheet, and B5N6C sheet—retain their planar structure. All the unit cells of three B6N6, B5N6Al, and B5N6C sheets contain twelve atoms. The boron and nitrogen atoms in B6N6 sheet are half the same, and the other two doping models replace one B atom.
The lattice parameters of the three B6N6, B5N6Al, and B5N6C sheets are listed in Table 1. The optimized lattice parameter of B6N6 is a = 6.20 Å. After doping with Al and C atoms, the symmetry of the crystal structure changes, and the lattice parameters a and b of B5N6Al and B5N6C are no longer equal. In B6N6 sheet structure, due to the larger radius of aluminum atoms, the lattice parameters of B5N6Al increase, while the lattice parameters of B5N6C do not change much. However, the radius difference between carbon atoms, boron atoms, and nitrogen atoms is not significant.
The decrease in lattice parameters is caused by structural distortion in the six-membered ring in the structure, and the details of this are shown in Figure 2. The bond lengths of B6N6, B5N6Al, and B5N6C sheets are also listed in Table 1. The doping of atoms makes the bond lengths in both B5N6Al and B5N6C sheets increase to six different bond lengths, which is three more than in B6N6 sheet, and the angles of the six-membered ring have also become irregular. In B6N6 sheet, the bond length of the six-membered ring is uniform, the bond angle is 120°, the maximum bond length is 1.278 Å, and the shortest bond length is 1.094 Å. For B5N6Al sheet, the six-membered ring has undergone serious distortion, and the bond angle and bond length have changed. The maximum bond length of B5N6Al is 1.796 Å, which is approximately 41% higher than that of B6N6. The shortest bond length is 1.264 Å. Regarding B5N6C sheet, the six-membered ring has a small degree of distortion, and the maximum bond length is 1.451 Å, which is approximately 14% longer than that of B6N6, and the shortest bond length is 1.260 Å.
Ab initio molecular dynamic (AIMD) simulations were performed to evaluate the thermal stabilities of B5N6Al and B5N6C sheets under 500 K. The supercell of B5N6Al and B5N6C sheets before and after 5 ps simulation at a temperature of 500 K is also shown in Figure 3. The supercells of B5N6Al and B5N6C did not change significantly, and the B-N, N-Al and B-C bonds were not broken. The total energy fluctuation was also maintained at a stable level, indicating that B5N6Al and B5N6C have thermal stability at 500 K.
The mechanical stability of B6N6, B5N6Al, and B5N6C sheets was also estimated, and the mechanical stability was evaluated by estimating the elastic constant. For 2D materials, the elastic constants can be obtained by the energy–strain method. That is, by applying different strains to the structure, the total energy of the system relative to the ground state energy change can be calculated. The relationship between the strain and the resulting energy change is expressed as [35]:
where E0 and E(ε) represent the ground state configuration and total energy after strain application, respectively. ε denotes the strain; xx and yy denote the direction. By fitting the energy curve corresponding to the strain, C11, C22, C12, and C44 as elastic constants can be obtained. The Born–Huang criteria [36] are a necessary condition for the elastic constants of 2D materials with mechanical stability, that is, C11C22 − > 0 and C44 > 0. The elastic constants of B6N6, B5N6Al, and B5N6C sheets are shown in Table 2, which obviously meet the mechanical stability conditions. The C11 = C22 = 180.98 N/m of B6N6, which is lower than that of h-BN (C11 = C22 = 290 N/m [37]) and pmma BN (C11 = 195 N/m, C22 = 256 N/m).
Compared with B6N6, the C11 of B5N6Al is reduced by approximately 16%, and the C22 is reduced by approximately 30%, which indicates that the ability to resist deformation along the x and y directions is weakened. That is, the Al atom plays a weakening role in the mechanical properties of B6N6. However, the addition of carbon atoms leads to an improvement in mechanical properties. The elastic constants C11 and C22 of B5N6C are increased by approximately 12~13%. Young’s modulus along the x and y directions Ex and Ey of B5N6C is also greater than that of B6N6, which may be due to the fact that carbon atoms are more likely to form stable covalent bonds. With the addition of aluminum and carbon atoms, B5N6Al and B5N6C sheets have lower and higher elastic constants than B6N6 sheets, respectively. Thus, the doping of carbon and aluminum atoms has different influence mechanisms on the mechanical properties of B6N6 sheets. The former significantly improves the elastic constants and elastic modulus, while the latter weakens the ability of B6N6 sheets to resist deformation and makes them easier to compress. Poisson’s ratios of B5N6Al and B5N6C sheets are 0.69 and 0.50 along the x-axis and 0.57 and 0.51 along the y-axis, respectively, which are higher than those of B6N6 sheets.
On the basis of the elastic constants, the in-plane E and v along any direction θ can be taken as [35]:
where α = sinθ; β = cosθ. In order to further explore the effect of carbon and aluminum atoms doping on the elastic anisotropy of B6N6 sheets, the angle-dependent in-plane E and v of B6N6, B5N6Al, and B5N6C sheets are illustrated in Figure 4. Further, 0° and 90° represent the directions of orthorhombic unit cells along the x- and y-axes, respectively. With the addition of aluminum, the maximum value of the E of B5N6Al sheet is lower than that of B6N6 sheet, but the minimum value is greater than that of B6N6 sheet, while the E of B5N6C sheet is larger than that of B6N6 sheet, and it shows an in-plane stiffness superior to B6N6 sheet. For Poisson’s ratio, B6N6 sheet has a higher Poisson ratio than that of B5N6Al and B5N6C sheet, indicating that B6N6 sheet is more likely to expand laterally than B5N6Al and B5N6C sheet when tension is applied. The smallest in-plane Poisson’s ratio of B6N6, B5N6Al, and B5N6C sheets occurs along the x-axis. When they are stretched diagonally, a maximum value occurs.
The orientation dependence of 2D Young’s modulus is closer to the sphere, indicating that the weaker the anisotropy, the smaller the difference between the maximum and minimum values. On the contrary, the more it deviates from the spherical shape, the stronger the anisotropy. The minimum value of Young’s modulus of B6N6 is 2.36 N/m, and the maximum value is 144 N/m, showing strong anisotropy. The minimum value of Young’s modulus of B5N6Al is 4.63 N/m, and the maximum value is 92 N/m; anisotropy is weakened compared with B6N6. The minimum Young’s modulus of B5N6C is 59 N/m, and the maximum is 152 N/m, showing the weakest Young’s modulus anisotropy. The ratio of the maximum to minimum Young’s modulus of B6N6, B5N6Al, and B5N6C sheets is 60.91, 19.94, and 2.57, respectively. These values clearly and intuitively show the influence of atomic doping on the anisotropy of Young’s modulus. Both carbon atoms and aluminum atoms can weaken anisotropy, especially when the effect of carbon atoms is more significant. The ratio of the 2D extreme values of Poisson’s ratio of B6N6, B5N6Al, and B5N6C sheets is 2.2, 1.72, and 1.60, respectively, indicating that the order of Poisson’s ratio anisotropy is B6N6 > B5N6Al > B5N6C, which is consistent with the order of anisotropy of Young’s modulus.
In addition to in-plane stiffness, the stress neutralization of B6N6, B5N6Al, and B5N6C sheets under uniaxial strain is further analyzed, and the detailed results are presented in Figure 5. The ultimate strength of B6N6 sheet is 18.49 N/m loaded along the x (y)-axis with 19.5% uniaxial strain, while with the addition of aluminum atoms, although the ultimate strength has decreased slightly, the strain along the x-axis is not significantly different (18.5%). The x- and y-axis of B5N6C sheet have similar and good uniaxial strain limitations. The ultimate strength along the x- and y-axis of B5N6C sheet under the calculated maximum strength is 17.0%, respectively. For B5N6C sheet, the ultimate strength is 18.14 N/m and 18.50 N/m along the x or y direction, respectively. Therefore, B6N6 sheet and B6N6 sheet doped with carbon atom have good mechanical properties compared to B5N6Al sheet, which may be suitable for nanomechanical applications.
The band structures of the B6N6, B5N6Al, and B5N6C sheets are shown in Figure 6. The related results show that all the B6N6, B5N6Al, and B5N6C sheets exhibit semiconductor characters, and the band gaps of B6N6 and B5N6Al sheets are 5.684 and 5.418 eV, respectively. After doping with Al atoms, the variation in the band gap of B5N6Al sheet is relatively small, while after doping the C atoms, the change in the band gap of B5N6C sheet is significant. To study the magnetism of these materials, we also calculated spin-up and spin-down band structures of B6N6, B5N6Al, and B5N6C sheets. From the band structures analysis, there is no difference between spin-up and spin-down for B6N6 and B5N6Al sheets, which indicates that they do not conform to the characteristics of magnetic materials. Nevertheless, after doping with carbon atoms, there is a significant difference in the band structure of spin-up and spin-down. The conduction band minimum (CBM) and valence band maximum (VBM) of the spin-up band structure are located at the X and Γ points, respectively, with a wide indirect band gap of 5.181 eV, while the CBM and VBM of the -up appear at the Y and X points, with the narrow band gap being only 1.062 eV.
We then explored the electronic properties of B6N6, B5N6Al, and B5N6C sheets by using the density of states (DOSs) and band decomposition charge density (BDCD) at the CBM and VBM. To verify the magnetism of these materials, their spin-up and spin-down DOSs were simulated and are shown in Figure 7. For B6N6 sheet, the spin-up and spin-down density of states of N and B atoms are the same. In the energy range of 0~−20 eV, the contribution of N atoms is more than that of B atoms. In 5~10 eV, the contribution of the energy band is mainly from B atoms, that is, the conduction band is mainly contributed by N atoms, while the valence band is mainly contributed by B atoms. After doping with Al atoms, the spin-up and spin-down DOSs of nitrogen, boron, and aluminum atoms are also the same. It can be seen that the electrons contributed by Al atoms have always been the least, which may be due to the smallest proportion of Al atoms. At 0~−20 eV, N atoms contribute the most, which is similar to B6N6. At 5~8 eV, electrons mainly come from B atoms, while after doping with carbon atoms, both boron, nitrogen, and carbon atoms in B5N6C sheet exhibit differences, which indicates that B5N6C has a modicum of magnetism. For B5N6C sheet, the spin-up electrons come from B and C atoms in the energy window of −2~0 eV, which indicates that the narrowing of the spin-up band gap is mainly due to B and C atoms, independent of N atoms. In the energy ranges of −3~−27 eV and 0~5 eV, the N atom contributes the most to the electronic band, while at 5~10 eV, and the electrons are mainly derived from the B atom.
The band decomposed charge densities (BDCDs) at the CBM and VBM of B6N6, B5N6Al, and B5N6C sheets are shown in Figure 8. As shown in the BDCDs, the electrons at the VBM of B6N6 sheet are mainly from the B atom, while the electrons at the CBM are mainly from the N atom. With the doping of Al atoms, the main contribution atoms of the CBM and VBM have reversed from before doping, while the Al atoms have no contribution to the CBM and VBM. For B5N6Al sheet, the VBM of B5N6Al sheet mainly comes from N atoms, and the CBM of B6N6 sheet mainly comes from B atoms. However, with the addition of the C atom, the distribution of electrons near the B5N6C sheet’s CBM and VBM is irregular compared to that of the previous B6N6 sheet and B5N6Al sheet. After doping the C atom, the main contribution of the CBM and VBM is participated by doped atoms, but this also occurs mainly by nitrogen and boron atoms. In addition, the charge density at the spin-up VBM and CBM is also significantly different from that at the spin-down VBM and CBM. The charge at the spin-up VBM is mainly contributed by B and C atoms, while the charge at the spin-down VBM is mainly contributed by N atoms. More rarely, negative charge appears at the spin-down CBM, as shown in the green part of Figure 8c.
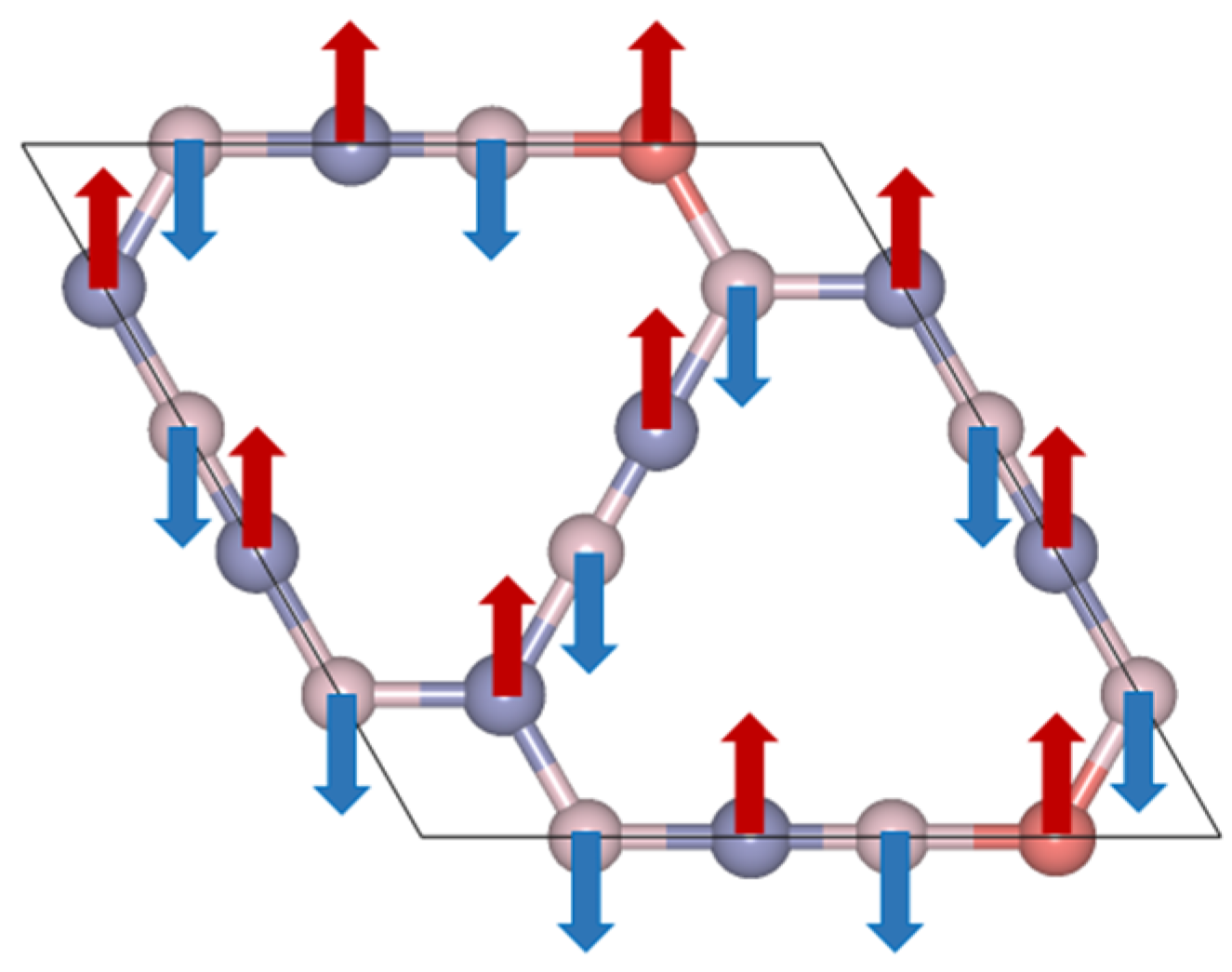
Figure 9 illustrates the electron localization function (ELF) of B6N6, B5N6Al, and B5N6C. The electrons are well localized around the B-N bond, Al-N bond, and C-N bond. The strength of the covalent bond in B6N6 and B5N6Al sheets is similar, indicating that the doping of Al does not have a significant effect on the ELF. Compared with B6N6, the electron localization function of B5N6C is weaker. For B5N6C, the difference between the spin-up and spin-down ELF is less evident, but they are not exactly the same. Careful observation shows that the spin-down charge is slightly more than the spin-up charge. The magnetization direction for each atom of B5N6C is shown in Figure 10, where the blue arrow represents spin-up, and the red arrow represents spin-down. Both B and C atoms are spin-up, while N atoms are spin-down.
3. Materials and Methods
The prediction of all the geometric optimization and properties of B6N6, B5N6Al, and B5N6C in this article was made on the basis of the first-principles calculation of the density functional theory (DFT) implemented in Mede A and VASP (3.3) [38,39]. The cutoff energy was selected to be 500 eV for plane waves. Electron–ion interactions were represented with the projector augmented wave (PAW) [40] pseudopotentials. The generalized gradients approximation proposed by the Perdew–Burke–Ernzerhof (GGA-PBE) functional is employed for the exchange correlation potential [41]. In order to fully optimize the geometry of the unit cell, the total energy convergence was set to 1 × 10−8 eV, and the atomic convergence force was 0.001 eV/Å. The Brillouin zone was sampled with an 8 × 8 × 1 Monkhorst–Pack (MP) [42] special k-point grid for geometric optimization and properties prediction. The hybrid Heyd–Scuseria–Ernzerhof functional (HSE06) [43] was used to simulate the band structures. In order to verify the mechanical stability, the elastic constants of B6N6, B5N6Al, and B5N6C sheets were estimated in vaspkit (1.4.0) [44] using the energy–strain method.
4. Conclusions
Based on density functional theory, we studied the electronic properties, mechanical properties, anisotropy properties, and magnetic properties of B6N6, B5N6Al, and B5N6C sheets. B6N6, B5N6Al, and B5N6C sheets are all composed of six-membered rings and twelve-membered rings. All the B6N6, B5N6Al, and B5N6C sheets are anisotropic materials, and B6N6 sheet has the largest anisotropy in terms of Young’s modulus and Poisson’s ratio. B6N6 and B5N6C sheets have good mechanical properties compared to B5N6Al sheet, making them suitable for nanomechanical applications. B6N6 and B5N6Al sheets have indirect band gaps of 5.684 and 5.418 eV, respectively. Therefore, by calculating the spin-up and spin-down band structures, it was found that B6N6 sheet did not exhibit magnetic properties. With the insertion of aluminum atoms, B5N6Al sheet also did not show magnetism. However, with the doping of carbon atoms, B5N6C sheet shows a modicum of magnetism, and all three atoms (B atom, N atom, and C atom) that were not magnetic atoms themselves showed magnetism.
Author Contributions
Conceptualization, L.Q.; methodology, L.Q.; software, L.Q.; validation, Z.M.; formal analysis, Z.M.; investigation, F.Y.; resources, Q.F.; data curation, F.Y.; writing—original draft preparation, Q.F.; writing—review and editing, S.W.; visualization, S.W.; supervision, L.Q.; project administration, Q.F.; funding acquisition, L.Q. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 62164011 and 61804120), China Postdoctoral Science Foundation (Nos. 2019TQ0243, 2019M663646), the Natural Science Basic Research Program of Shaanxi Province (No. 2023-JC-YB-567), Key Science and Technology Innovation Team of Shaanxi Province (2022TD-34), the Natural Science Foundation of Jiangsu Province (No. BK20211002), as well as Qinglan Project of Jiangsu Province of China.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bi, J.X.; Du, Z.Z.; Sun, J.M.; Liu, Y.H.; Wang, K.; Du, H.F.; Ai, W.; Huang, W. On the Road to the Frontiers of Lithium-Ion Batteries: A Review and Outlook of Graphene Anodes. Adv. Mater. 2023, 35, e2210734. [Google Scholar] [CrossRef]
- Olabi, A.G.; Abdelkareem, M.A.; Wilberforce, T.; Sayed, E.T. Application of graphene in energy storage device—A review. Renew. Sust. Energ. Rev. 2021, 135, 110026. [Google Scholar] [CrossRef]
- Xiao, Y.Q.; Pang, Y.X.; Yan, Y.X.; Qian, P.; Zhao, H.T.; Manickam, S.; Wu, T.; Pang, C.H. Synthesis and Functionalization of Graphene Materials for Biomedical Applications: Recent Advances, Challenges, and Perspectives. Adv. Sci. 2023, 10, e2205292. [Google Scholar] [CrossRef]
- Yang, H.B.; Zheng, H.J.; Duan, Y.X.; Xu, T.; Xie, H.X.; Du, H.S.; Si, C.L. Nanocellulose-graphene composites: Preparation and applications in flexible electronics. Int. J. Biol. Macromol. 2023, 253, 126903. [Google Scholar] [CrossRef]
- Ghosa, S.; Mondal, N.S.; Chowdhury, S.; Jana, D. Two novel phases of germa-graphene: Prediction, electronic and transport applications. Appl. Surf. Sci. 2023, 614, 156107. [Google Scholar] [CrossRef]
- Asim, N.; Badiei, M.; Samsudin, N.A.; Mohammad, M.; Razali, H.; Soltani, S.; Amin, N. Application of graphene-based materials in developing sustainable infrastructure: An overview. Compos. B. Eng. 2022, 245, 110188. [Google Scholar] [CrossRef]
- Sun, M.; Chou, J.P.; Hu, A.; Schwingenschlögl, U. Point Defects in Blue Phosphorene. Chem. Mater. 2019, 31, 8129–8135. [Google Scholar] [CrossRef]
- Ma, Y.; Yan, Y.; Luo, L.; Pazos, S.; Zhang, C.; Lv, X.; Chen, M.; Liu, C.; Wang, Y.; Chen, A.; et al. High-performance van der Waals antiferroelectric CuCrP2S6-based memristors. Nat. Commun. 2023, 14, 7891. [Google Scholar] [CrossRef]
- Zhang, C.; Ren, K.; Wang, S.; Luo, Y.; Tang, W.; Sun, M. Recent progress on two-dimensional van der Waals heterostructures for photocatalytic water splitting: A selective review. J. Phys. D Appl. Phys. 2023, 56, 483001. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Fan, Q.; Yang, Y.; Sun, M.; Palummo, M.; Schwingenschlögl, U. Two-dimensional borocarbonitrides for photocatalysis and photovoltaics. J. Mater. Chem. C 2023, 11, 3875–3884. [Google Scholar] [CrossRef]
- Ren, K.; Yan, Y.; Zhang, Z.; Sun, M.; Schwingenschlogl, U. A family of LixBy monolayers with a wide spectrum of potential applications. Appl. Surf. Sci. 2022, 604, 154317. [Google Scholar] [CrossRef]
- Sun, M.; Re Fiorentin, M.; Schwingenschlögl, U.; Palummo, M. Excitons and light-emission in semiconducting MoSi2X4 two-dimensional materials. NPJ 2D Mater. Appl. 2022, 6, 81. [Google Scholar] [CrossRef]
- Wang, Y.; Miao, M.; Lv, J.; Zhu, L.; Yin, K.; Liu, H.; Ma, Y. An Effective Structure Prediction Method for Layered Materials Based on 2D Particle Swarm Optimization Algorithm. J. Chem. Phys. 2012, 137, 224108–224114. [Google Scholar] [CrossRef]
- Pakdel, A.; Bando, Y.; Golberg, D. Nano boron nitride flatland. Chem. Soc. Rev. 2014, 43, 934–959. [Google Scholar] [CrossRef]
- Entani, S.; Larionov, K.V.; Popov, Z.I.; Takizawa, M.; Mizuguchi, M.; Watanabe, H.; Li, S.T.; Naramoto, H.; Sorokin, P.B.; Sakai, S. Non-chemical fluorination of hexagonal boron nitride by high-energy ion irradiation. Nanotechnology 2020, 31, 125705. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.G.; Mu, X.J.; Wang, X.X.; Wang, N.; Ma, F.C.; Liang, W.J.; Sun, M.T. The thermal and thermoelectric properties of in-plane C-BN hybrid structures and graphene/h-BN van der Waals heterostructures. Mater. Today Phys. 2018, 5, 29–57. [Google Scholar] [CrossRef]
- Sun, M.; Tang, W.; Ren, Q.; Wang, S.; Yu, J.; Du, Y. A first-principles study of light non-metallic atom substituted blue phosphorene. Appl. Surf. Sci. 2015, 356, 110–114. [Google Scholar] [CrossRef]
- Wang, Z.H.; Zhou, X.F.; Zhang, X.M.; Zhu, Q.; Dong, H.F.; Zhao, M.W.; Oganov, A.R. Phagraphene: A Low-Energy Graphene Allotrope Composed of 5–6-7 Carbon Rings with Distorted Dirac Cones. Nano Lett. 2015, 15, 6182–6186. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.B.; Bhattacharya, B.; Sarkar, U. A first principle study of pristine and BN-doped graphyne family. Struct. Chem. 2014, 25, 1695–1710. [Google Scholar] [CrossRef]
- Enyashin, A.N.; Ivanovskii, A.L. Graphene-like BN allotropes: Structural and electronic properties from DFTB calculations. Chem. Phys. Lett. 2011, 509, 143–147. [Google Scholar] [CrossRef]
- Bu, H.; Zhao, M.; Zhang, H.; Wang, X.; Xi, Y.; Wang, Z. Isoelectronic Doping of Graphdiyne with Boron and Nitrogen: Stable Configurations and Band Gap Modification. J. Phys. Chem. A 2012, 116, 3934–3939. [Google Scholar] [CrossRef]
- Shahrokhi, M.; Mortazavi, B.; Berdiyorov, R.G. New two-dimensional Boron Nitride allotropes with attractive electronic and optical properties. Solid State Commun. 2017, 253, 51–56. [Google Scholar] [CrossRef]
- Demirci, S.; Rad, S.E.; Kazak, S.; Nezir, S.; Jahangirov, S. Monolayer diboron dinitride: Direct band-gap semiconductor with high absorption in the visible range. Phys. Rev. B 2020, 101, 125408. [Google Scholar] [CrossRef]
- Li, X.D.; Cheng, X.L. Predicting the structural and electronic properties of two-dimensional single layer boron nitride sheets. Chem. Phys. Lett. 2018, 694, 102–106. [Google Scholar] [CrossRef]
- Chen, Y.; Zou, J.; Campbell, S.J.; Le Caer, G. Boron nitride nanotubes: Pronounced resistance to oxidation. Appl. Phys. Lett. 2004, 84, 2430–2432. [Google Scholar] [CrossRef]
- Wang, X.; Zhi, C.; Li, L.; Zeng, H.; Li, C.; Mitome, M.; Golberg, D.; Bando, Y. “Chemical Blowing” of Thin-Walled Bubbles: High-Throughput Fabrication of Large-Area, Few-Layered BN and Cx-BN Nanosheets. Adv. Mater. 2011, 23, 4072–4076. [Google Scholar] [CrossRef] [PubMed]
- Zhi, C.; Bando, Y.; Tang, C.; Kuwahara, H.; Golberg, D. Large-Scale Fabrication of Boron Nitride Nanosheets and Their Utilization in Polymeric Composites with Improved Thermal and Mechanical Properties. Adv. Mater. 2009, 21, 2889–2893. [Google Scholar] [CrossRef]
- Anota, E.C.; Hernández, A.B.; Morales, A.E.; Castro, M. Design of the magnetic homonuclear bonds boron nitride nanosheets using DFT methods. J. Mol. Graph. Model. 2017, 74, 135–142. [Google Scholar] [CrossRef]
- Cao, X.; Li, Y.; Cheng, X.; Zhang, Y. Structural analogues of graphyne family: New types of boron nitride sheets with wide band gap and strong UV absorption. Chem. Phys. Lett. 2011, 502, 217–221. [Google Scholar] [CrossRef]
- Qi, J.; Wang, S.; Wang, J.; Umezawa, N.; Blatov, V.A.; Hosono, H. B5N3 and B7N5 Monolayers with High Carrier Mobility and Excellent Optical Performance. J. Phys. Chem. Lett. 2021, 12, 4823–4832. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1956, 140, A1133. [Google Scholar] [CrossRef]
- Fan, Q.; Zhou, H.; Zhao, Y.; Yun, S. Predicting a novel two-dimensional BN material with a wide band gap. Energy Mater. 2022, 2, 200022. [Google Scholar] [CrossRef]
- Anota, E.C. 2D boron nitride incorporating homonuclear boron bonds: Stabilized in neutral, anionic and cationic charge. SN Appl. Sci. 2022, 4, 295. [Google Scholar] [CrossRef]
- Cadelano, E.; Palla, P.L.; Giordano, S.; Colombo, L. Elastic properties of hydrogenated graphene. Phys. Rev. B 2010, 82, 235414. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, Y. Density functional theory study of the silicene-like SiX and XSi3 (X = B, C, N, Al, P) honeycomb lattices: The various buckled structures and versatile electronic properties. J. Phys. Chem. C 2013, 117, 18266–18278. [Google Scholar] [CrossRef]
- Andrew, R.C.; Mapasha, R.E.; Ukpong, A.M.; Chetty, N. Mechanical properties of graphene and boronitrene. Phys. Rev. B 2012, 85, 125428. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouinzone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.; Liu, J.C.; Tang, G.; Geng, W.T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
Figure 1.
Crystal structures of the h-BN (a), o-B2N2 (b), B5N3 (c), B7N5 (d), B6N6 (e), B5N6Al (f), and B5N6C (g). Light blue, light pink, green, and light red represent boron atoms, nitrogen atoms, aluminum atoms, and carbon atoms, respectively.
Figure 1.
Crystal structures of the h-BN (a), o-B2N2 (b), B5N3 (c), B7N5 (d), B6N6 (e), B5N6Al (f), and B5N6C (g). Light blue, light pink, green, and light red represent boron atoms, nitrogen atoms, aluminum atoms, and carbon atoms, respectively.
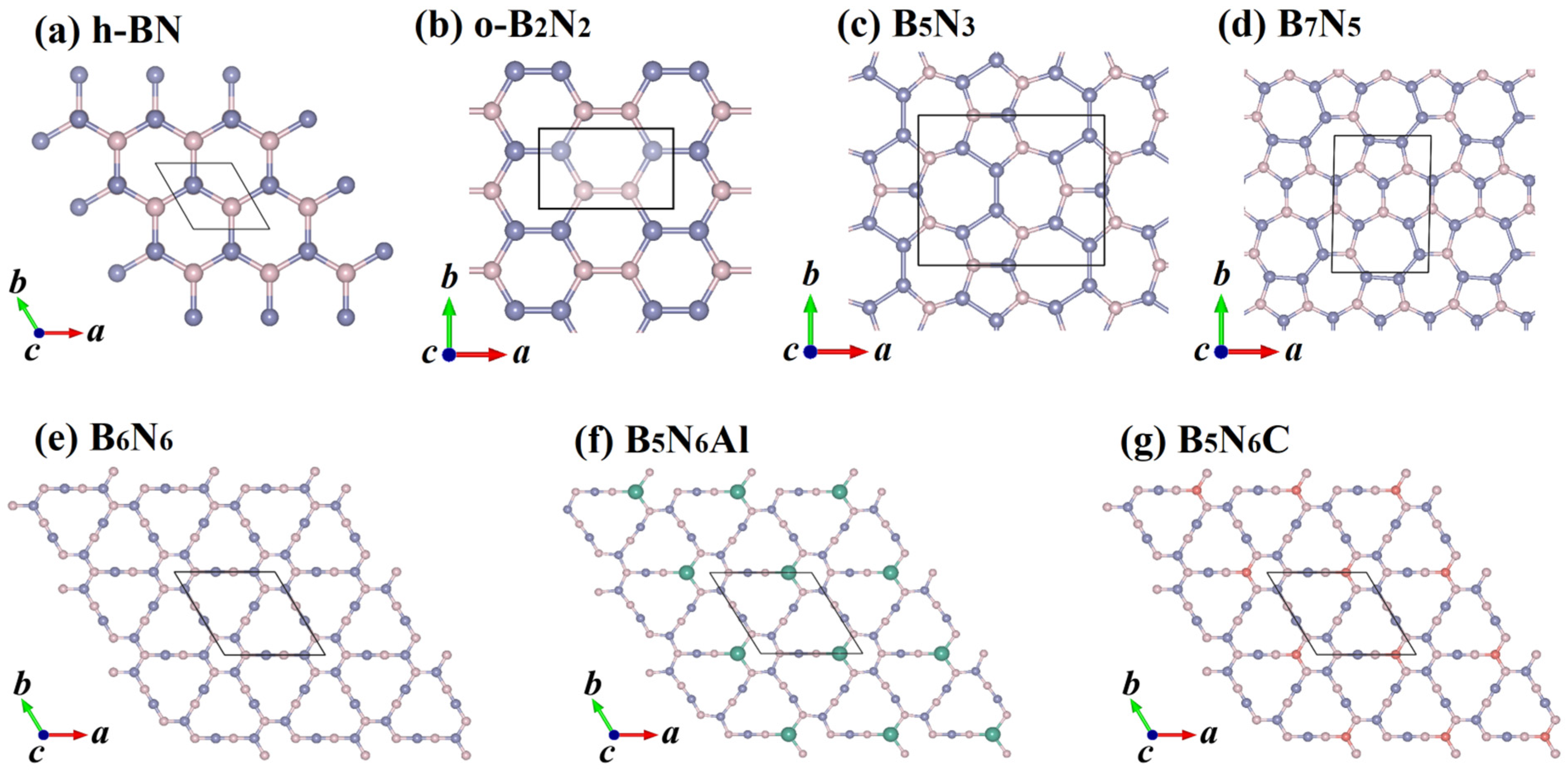
Figure 2.
The bond length and bond angle of B6N6 (a), B5N6Al (b), and B5N6C (c).
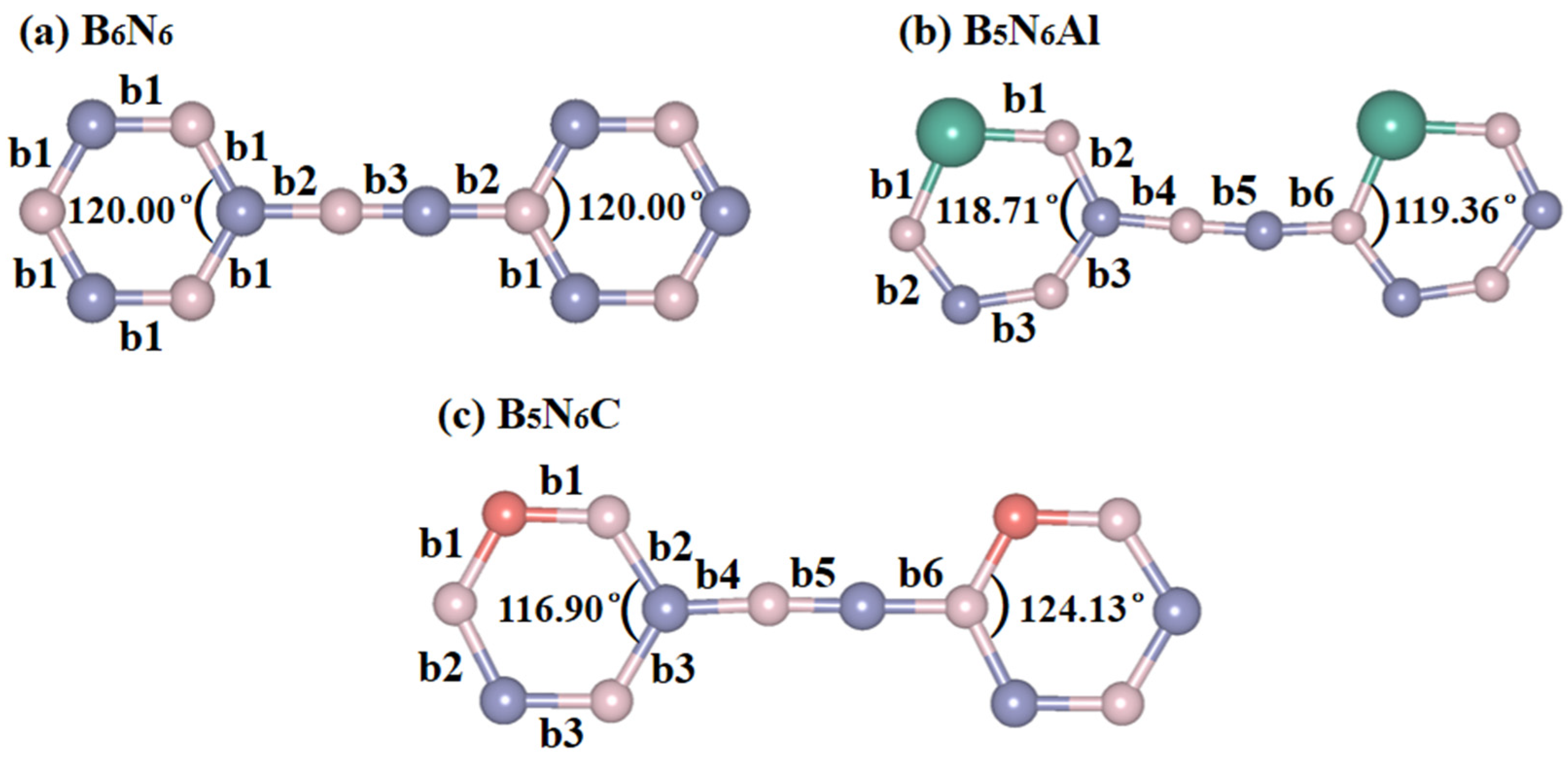
Figure 3.
Total energy fluctuations of B5AlN6 and B5N6C as a function of the AIMDs simulation at 500 K.
Figure 3.
Total energy fluctuations of B5AlN6 and B5N6C as a function of the AIMDs simulation at 500 K.
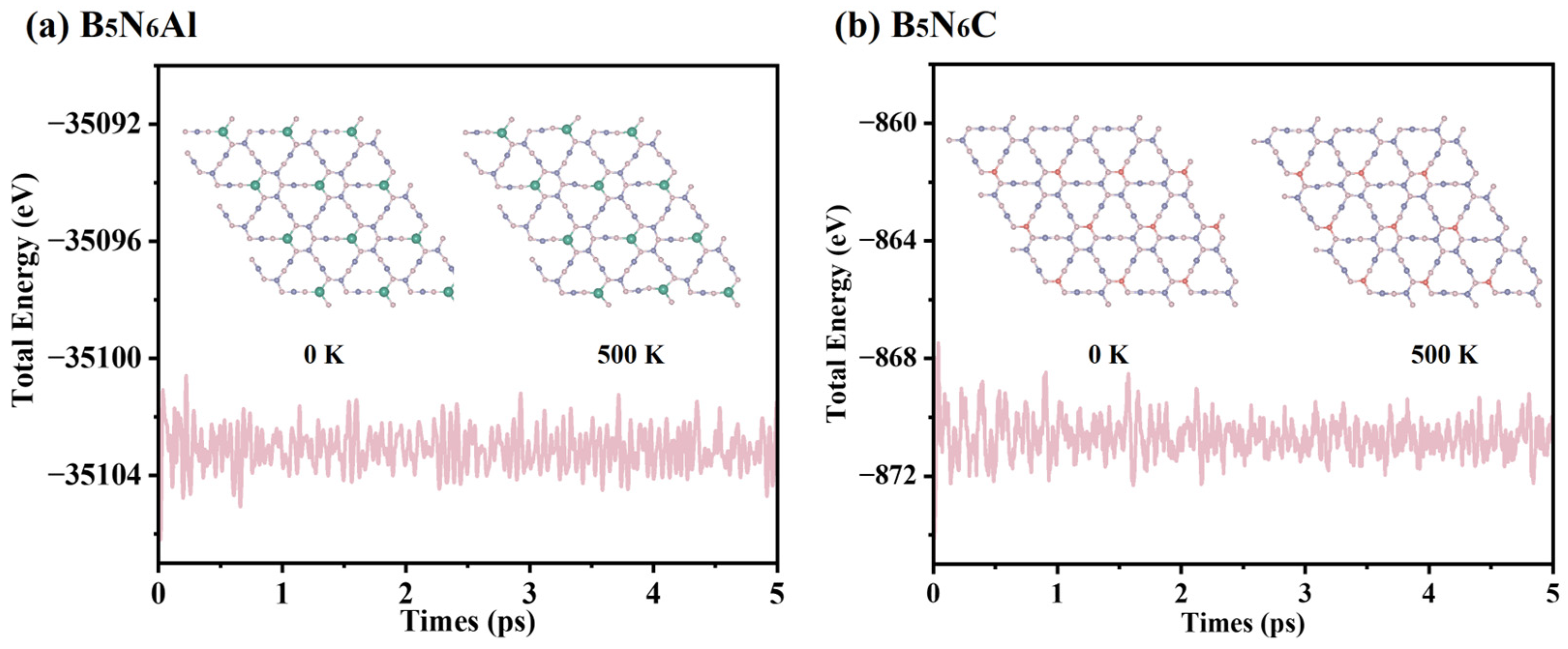
Figure 4.
Orientation dependencies of Young’s modulus (a) and Poisson’s ratio (b) for B6N6, B5N6Al, and B5N6C.
Figure 4.
Orientation dependencies of Young’s modulus (a) and Poisson’s ratio (b) for B6N6, B5N6Al, and B5N6C.
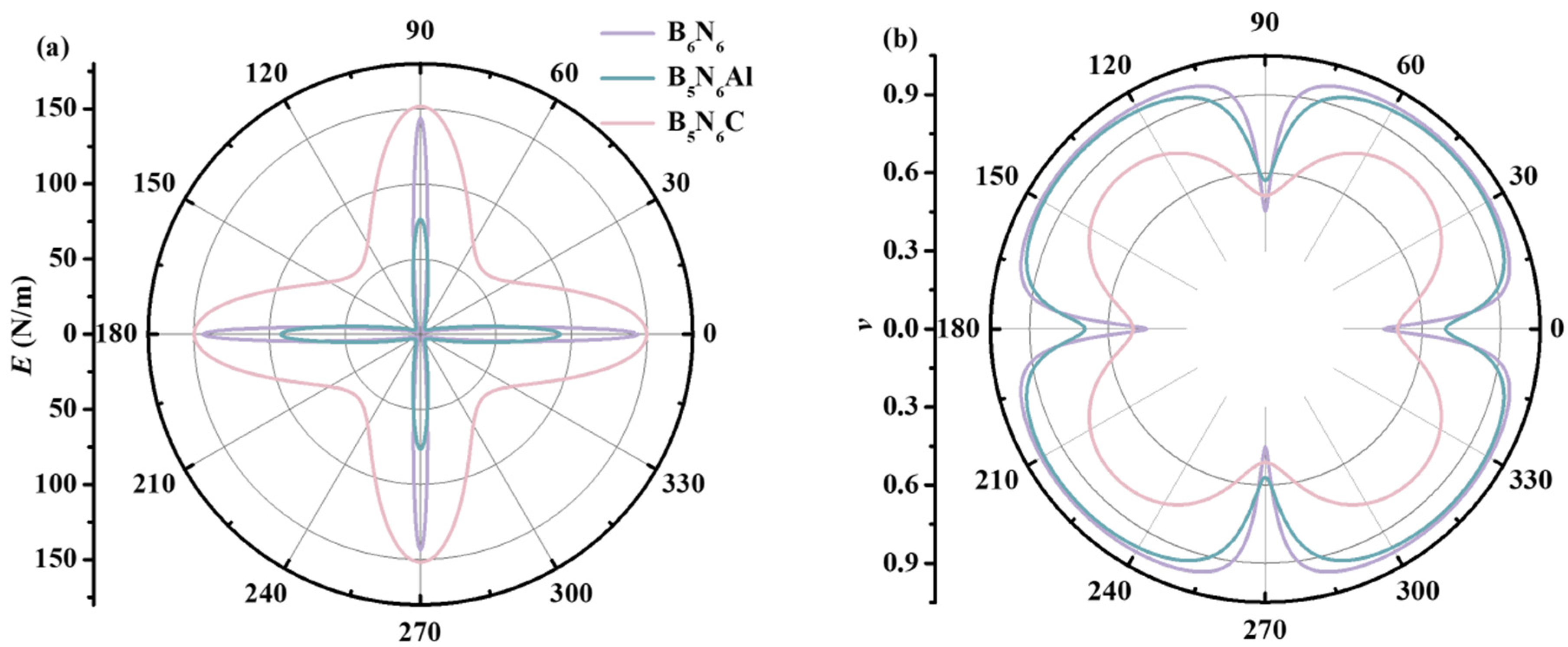
Figure 5.
Strain–stress curves for the uniaxial tensile strains of B6N6 (a), B5N6Al (b), and B5N6C (c).
Figure 5.
Strain–stress curves for the uniaxial tensile strains of B6N6 (a), B5N6Al (b), and B5N6C (c).
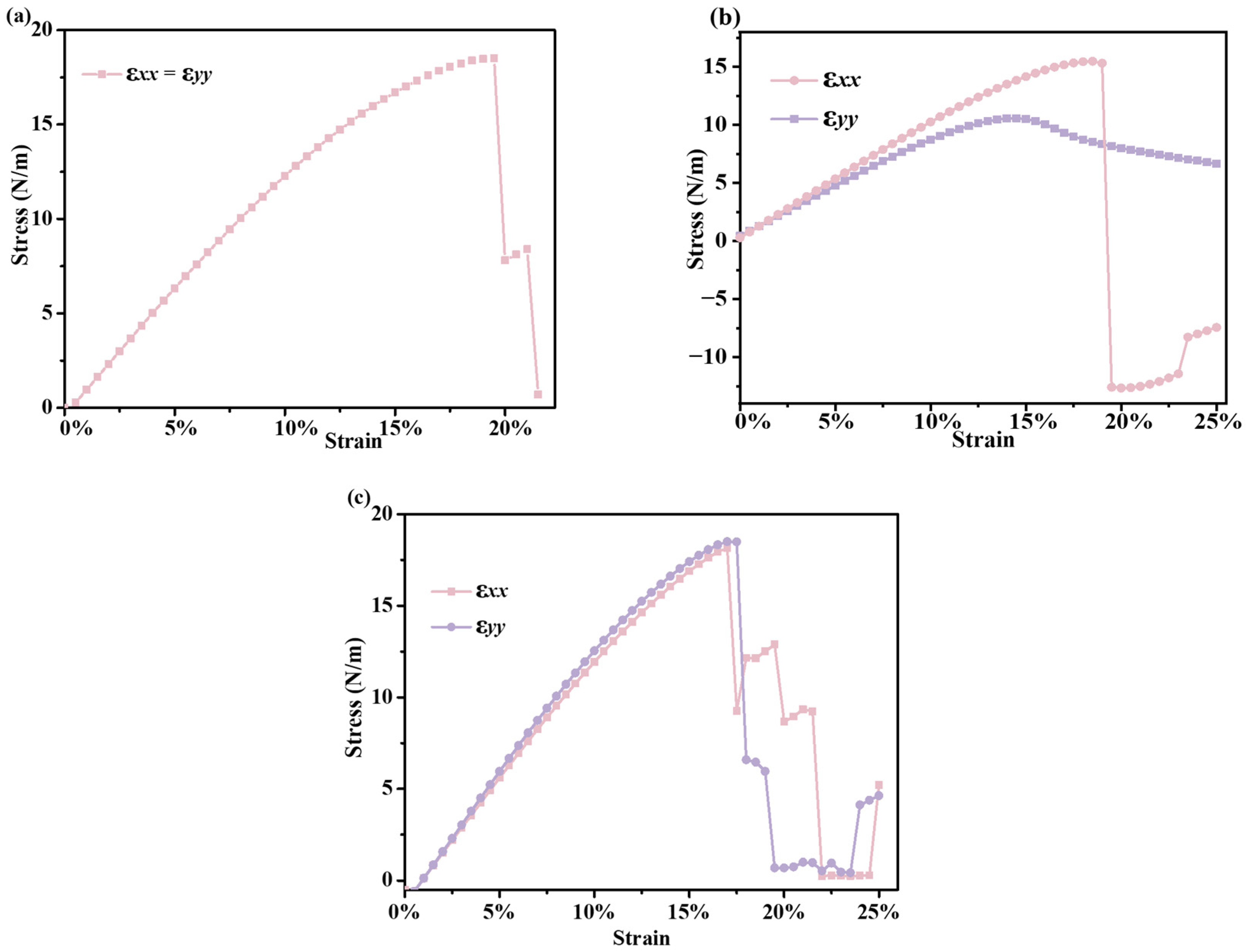
Figure 6.
Electronic band structures of B6N6 (a), B5N6Al (b), and B5N6C (c). Black and red lines in the band structure present spin-up electrons and spin-down electrons, respectively.
Figure 6.
Electronic band structures of B6N6 (a), B5N6Al (b), and B5N6C (c). Black and red lines in the band structure present spin-up electrons and spin-down electrons, respectively.
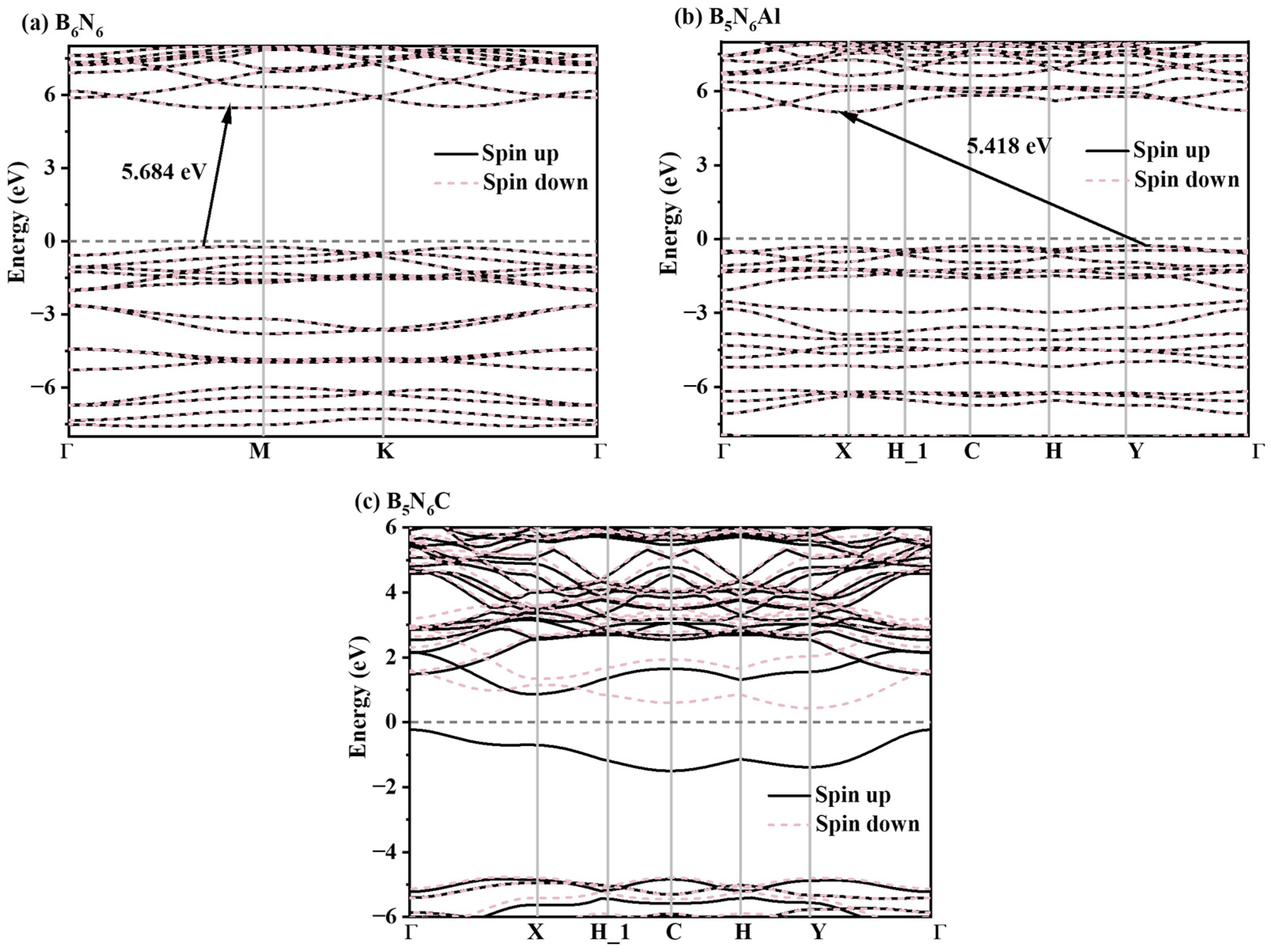
Figure 7.
Spin-up and spin-down density of states for B6N6 (a), B5N6Al (b), and B5N6C (c).

Figure 8.
The band decomposed charge densities at the CBM and VBM of B6N6 (a), B5N6Al (b), and B5N6C (c).
Figure 8.
The band decomposed charge densities at the CBM and VBM of B6N6 (a), B5N6Al (b), and B5N6C (c).
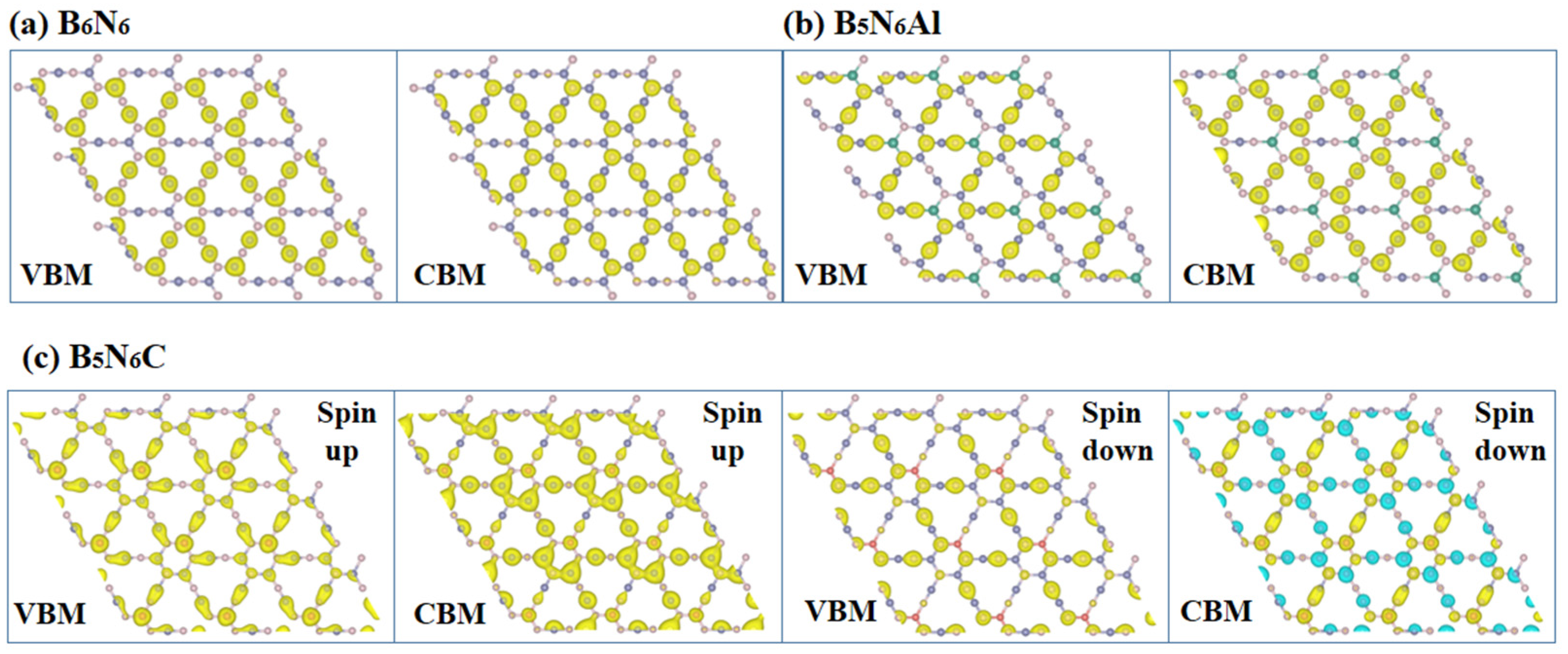
Figure 9.
The electron localization function (ELF) of B6N6 (a), B5N6Al (b), and B5N6C (c,d).
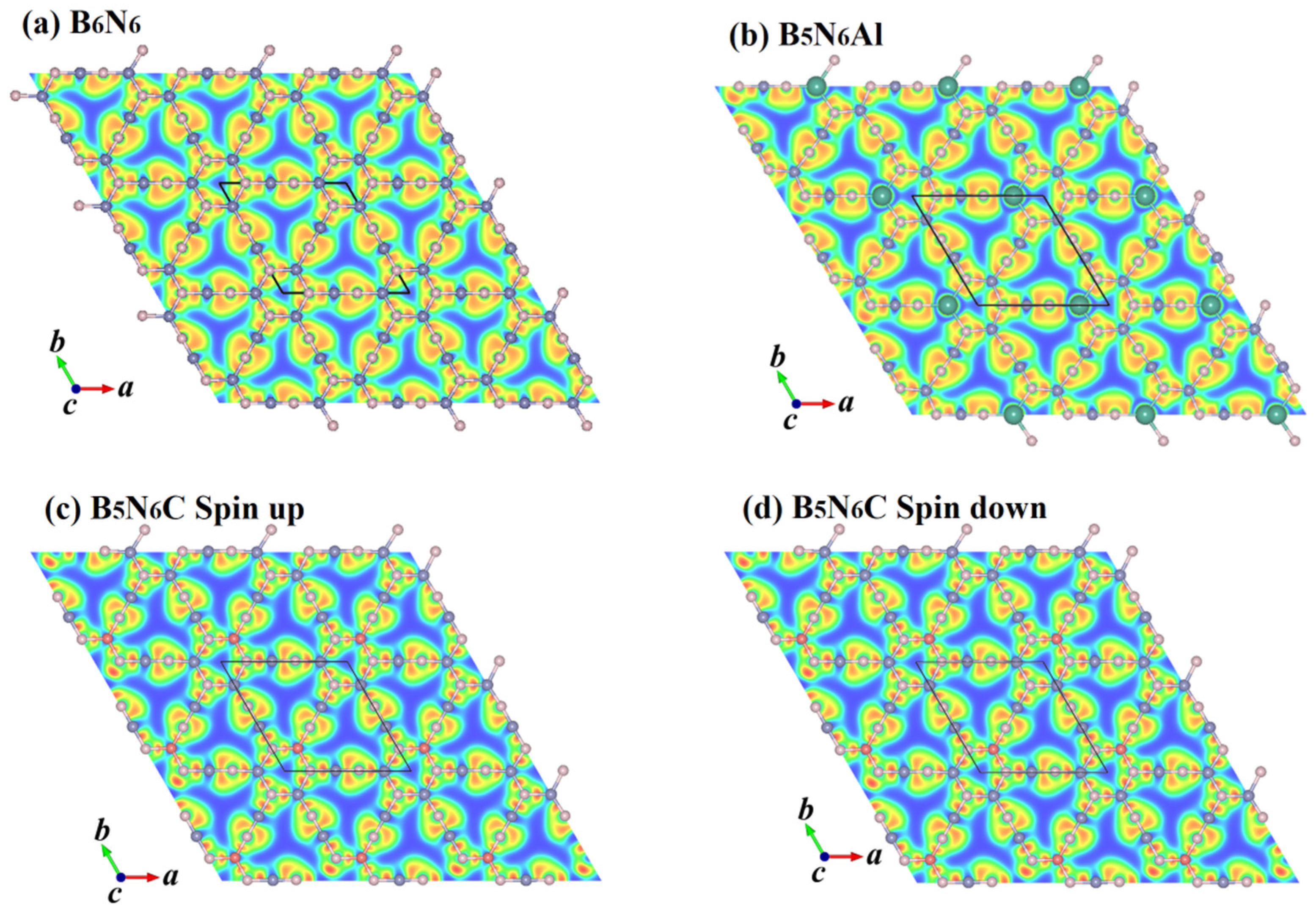
Figure 10.
The magnetization direction for each atom of B5N6C; arrows denote spins.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Lattice constant (Å) and bond length (Å) of B6N6, B5N6Al, and B5N6C.
| a | b | b1 | b2 | b3 | b4 | b5 | b6 | |
|---|---|---|---|---|---|---|---|---|
| B6N6 | 6.20 | 1.275 | 1.278 | 1.094 | ||||
| B5N6Al | 7.40 | 7.18 | 1.796 | 1.451 | 1.465 | 1.390 | 1.264 | 1.374 |
| B5N6C | 6.86 | 5.93 | 1.381 | 1.451 | 1.441 | 1.380 | 1.260 | 1.391 |
Table 2.
Elastic constants Cij (N/m), Young’s modulus E (N/m), and Poisson’s ratio v of B6N6, B5N6Al, and P-62m B5N6C.
Table 2.
Elastic constants Cij (N/m), Young’s modulus E (N/m), and Poisson’s ratio v of B6N6, B5N6Al, and P-62m B5N6C.
| C11 | C12 | C22 | C66 | Ex | Ey | vx | vy | |
|---|---|---|---|---|---|---|---|---|
| B6N6 | 180.98 | 82.09 | 180.98 | 49.45 | 141.18 | 141.18 | 0.46 | 0.46 |
| B5N6Al | 152.14 | 86.84 | 126.11 | 44.78 | 97.83 | 76.19 | 0.69 | 0.57 |
| B5N6C | 201.78 | 103.48 | 204.66 | 53.35 | 149.46 | 151.59 | 0.50 | 0.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Qiao, L.; Ma, Z.; Yan, F.; Wang, S.; Fan, Q. A First-Principle Study of Two-Dimensional Boron Nitride Polymorph with Tunable Magnetism. Inorganics 2024, 12, 59. https://doi.org/10.3390/inorganics12020059
AMA Style
Qiao L, Ma Z, Yan F, Wang S, Fan Q. A First-Principle Study of Two-Dimensional Boron Nitride Polymorph with Tunable Magnetism. Inorganics. 2024; 12(2):59. https://doi.org/10.3390/inorganics12020059
Chicago/Turabian StyleQiao, Liping, Zhongqi Ma, Fulong Yan, Sake Wang, and Qingyang Fan. 2024. "A First-Principle Study of Two-Dimensional Boron Nitride Polymorph with Tunable Magnetism" Inorganics 12, no. 2: 59. https://doi.org/10.3390/inorganics12020059
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.