
Liquid Chromatography Tandem Mass Spectrometry Analysis of Synthetic Coccidiostats in Eggs

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals, Reagents and Solutions
2.2. HPLC-MS/MS Apparatus and Conditions
2.3. Sample Preparation
3. Result and Discussion
3.1. Optimization of the Chromatographic and Mass Spectrometric Conditions
3.2. Optimization of the Clean-Up Procedure
- optimization of the liquid–liquid extraction (nature and volume of solvent, time and pH of extraction)
- optimization of the purification of the extracts (choice of the SPE cartridge, washing and elution conditions)
4. Validation Study
4.1. Linearity and Matrix Effect
4.2. Specificity
4.3.Recovery
4.4. Precision: Repeatability and Reproducibility Intra-Laboratory
4.5. CCα and CCβ and Robustness (Minor Changes)
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Booth, N.H.; Mc Donald, L.E. Farmacologia e Terapeutica Veterinaria, 1st ed.; EMSI: Roma, Italy, 1991. [Google Scholar]
- Gerhold, R.W., Jr. Overview of coccidiosis in poultry. In Merck Manual—Veterinary Manual, 10th ed.; Merck & Co., Inc.: Kenilworth, NJ, USA, 2015. [Google Scholar]
- Regulation (EC) No. 1831/2003. European Union Register of Feed Additives. Edition 254. Appendixes 3e, 4 23.03.2017 Annex 1 List of Additives. Available online: http://ec.europa.eu/food/safety/animal-feed/feed-additives/index_en.htm (accessed on 28 March 2017).
- The European Agency for the evaluation of Medicinal Products; Veteriny Medicine Evaluation Unit. Committee for Medicinal Products for Veterinary Use Toltrazuril; Summary Report (1). Available online: http://www.eudra.org/emea.html (accessed on 1 February 2017).
- European Medicine Agency, Veterinary Medicines and Inspections; Committee for Medicinal Products for Veterinary Use Toltrazuril. Extension to Cattle and Extrapolation to All Mammalian Food-Producing Species and Poultry; Summary Report (5). Available online: http://www.emea.eu.int (accessed on 30 December 2016).
- Regulation, C. (EU) No. 37/2010 of 22 December 2009 on Pharmacologically Active Substances and Their Classification Regarding Maximum Residue Limits in Foodstuffs of Animal Origin. Available online: http://ec.europa.eu/health//sites/health/files/files/eudralex/vol-5/reg_2010_37/reg_2010_37_en.pdf (accessed on 30 December 2016).
- European medicines agency, Veterinary Medicines Evaluation Unit Committee for Medicinal Products for Veterinary Use Diclazuril Summary Report. Available online: http://www.emea.eu.int (accessed on 30 December 2016).
- Committee for Medicinal Products for Veterinary Use Clazuril Summary Report. Available online: http://www.eudra.org/emea.html (accessed on 30 December 2016).
- European Food safety Authority Question No. EFSA-Q-2005-220K Adopted on 9 April 2008. Available online: http://www.efsa.europa.eu/sites/default/files/scientific_output/files/main_documents/690.pdf (accessed on 30 December 2016).
- European Food safety Authority Question No. EFSA-Q-2005-220g Adopted 19 February 2008. Available online: http://onlinelibrary.wiley.com/doi/10.2903/j.efsa.2008.655/pdf (accessed on 28 December 2016).
- Commission Regulation (EU) No. 610/2012 amending Regulation (EC) No. 124/2009 of 10 February 2009 Setting Maximum Levels for the Presence of Coccidiostats or Histomonostats in Food Resulting from the Unavoidable Carry-over of These Substances in Non-Target Feed. Available online: https://www.fsai.ie/uploadedFiles/Reg610_2012.pdf (accessed on 28 December 2016).
- Commission Regulation (EC) No. 124/2009 Setting Maximum Levels for the Presence of Coccidiostats or Histomonostats in Food Resulting from the Unavoidable Carry-over of These Substances in Non-Target Feed. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2009:040:0007:0011:en:PDF (accessed on 27 December 2016).
- Mortier, L.; Daeseleire, E.; Delahaut, P. Simultaneous detection of five coccidiostats in eggs by liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2003, 483, 27–37. [Google Scholar] [CrossRef]
- Stahl, R.S.; Ver Cauteren, K.; Buettgenbach, T.L.; Johnston, J.J. Determination of 4,4′-Dintrocarbanilide (DNC), a Component of Nicarbazin, in Canada Goose (Brantacanadensis) Eggs hells Using High Performance Liquid Chromatography. J. Agric. Food Chem. 2003, 51, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Wilga, J.; Wasik, A.K.; Namiesnik, J. Comparison of extraction techniques of robenidine from poultry feed Samples. Talanta 2007, 73, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Dowling, G.; Keeffe, M.O.; Smyth, M.R. Determination of robenidine in eggs by liquid chromatography with UV spectrophotometric detection. Anal. Chim. Acta 2005, 539, 31–34. [Google Scholar] [CrossRef]
- Draisci, R.; Lucentini, L.; Boria, P.; Lucarelli, C. Micro high-performance liquid chromatography for the determination of nicarbazin in chicken tissues, eggs, poultry feed and litter. J. Chromatogr. A 1995, 697, 407–414. [Google Scholar] [CrossRef]
- Dubois, M.; Pierret, G.; Delhaut, P. Efficient and sensitive detection of nine coccidiostats in egg by liquid chromatography—Electrospray tandem mass spectrometry. J. Chromatogr. B 2004, 813, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, M.; Szprengier-Juszkiewicz, T.; Jedziniak, P.; Sledzińska, E.; Szymanek-Bany, I.; Korycińska, B.; Pietruk, K.; Zmudzki, J. Residue control of coccidiostats in food of animal origin in Poland during 2007–2010. Food Addit. Contam. Part B 2011, 4, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Mortier, L.; Daeseleire, E.; Van Peteghem, C. Liquid chromatographic tandem mass spectrometry determination of five coccidiostats in poultry eggs and feed. J. Chromatogr. B 2005, 820, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.; Moloney, M.; O’Mahony, I.; O’Kennedy, R.; Danaher, M. Determination of 20 coccidiostats in milk, duck muscle, and non avian muscle tissue. Food Addit. Contam. Part A 2013, 30, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Galarini, R.; Fioroni, L.; Moretti, S.; Pettinacci, L.; Dusi, G. Development and validation of a multi-residue liquid chromatography–tandem mass spectrometry confirmatory method for eleven coccidiostats in eggs. Anal. Chim. Acta 2010, 700, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Broekaert, N.; Van Peteghem, C.; Daeseleire, E.; Sticker, D.C.; Van Poucke, C. Development and validation of an UPLC-MS/MS method for the determination of ionophoric and synthetic coccidiostats in vegetables. Anal. Bioanal. Chem. 2011, 401, 3335–3344. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, M.; Szprengier-juszkiewicz, T.; Jedziniak, P. Confirmatory method for determination of coccidiostats in eggs. Bull. Vet. Inst. Pulawy 2010, 54, 327–333. [Google Scholar]
- Chico, J.; Rúbies, A.; Centrich, F.; Companyó, R.; Prat, M.D.; Granados, M. Use of gel permeation chromatography for clean-up in the analysis of coccidiostats in eggs by liquid chromatography–tandem mass Spectrometry. Anal. Bioanal. Chem. 2013, 405, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Song, G.; Ai, L.F.; Li, J.C. Determination of six polyether antibiotics residues in foods of animal origin by solid phase extraction combined with liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2016, 1017, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Connolly, L.; Fodey, T.L.; Crooks, S.R.; Delahaut, P.; Elliott, C.T.J. The production and characterisation of dinitrocarbanilide antibodies raised using antigen mimics. Immunol. Methods 2002, 264, 45–51. [Google Scholar] [CrossRef]
- Moloney, M.; Clarke, L.; O’Mahony, J.; Gadaj, A.; O’Kennedy, R.; Danaher, M. Determination of 20 coccidiostats in egg and avian muscle tissue using ultra high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2012, 1253, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.; Fodey, T.L.; Crooks, S.R.H.; Moloney, M.; O’Mahony, J.; Delahaut, P.; Gadaj, A.; O’Kennedy, R.; Danaher, M. A review of coccidiostats and the analysis of their residues in meat and other food. Meat Sci. 2014, 97, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Villalba, A.; Moyano, E.; Martins, C.P.; Galceran, M.T. Fast liquid chromatography/tandem mass spectrometry (highly selective selected reaction monitoring) for the determination of toltrazuril and its metabolites in food. Anal. Bioanal. Chem. 2010, 397, 2893–2901. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Sun, H.; Wang, F.; Chen, R.; Guo, C. Determination of diclazuril, toltrazuril and its two metabolites in poultry tissues and eggsby gel permeation chromatography–liquid chromatography-tandem masss pectrometry. J. Chromatogr. B 2011, 879, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.P.J.; Balzer-Rutgers, P.; Brinke, E.M.; Bolck, Y.J.C.; Berendsen, B.J.A.; Gerçek, H.; Schat, B.; van Rhijn, J.A. Deposition and depletion of the coccidiostats toltrazuril and halofuginone in eggs. Anal. Chim. Acta 2005, 529, 331–337. [Google Scholar] [CrossRef]
- Dubreil-Chéneau, E.; Bessiral, M.; Roudaut, B.; Verdon, E.; Sanders, P. Validation of multi-residue liquid chromatography-tandem mass spectrometry confirmatory method for 10 anticoccidials in eggs according to Commission Decision 2002/657/EC. J. Chromatogr. A 2009, 1216, 8149–8157. [Google Scholar] [CrossRef] [PubMed]
- Commission Decision 2002/657/EC of 12 August 2002. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32002D0657&from=it (accessed on 20 December 2016).
- Buiarelli, F.; Di Filippo, P.; Riccardi, C.; Pomata, D.; Giannetti, L.; Neri, B. Analytical method for the determination of mycotoxins in indoor/outdoor airborne particulate matter by HPLC-MS-MS. Int. J. Environ. Anal. Chem. 2015, 95, 713–729. [Google Scholar] [CrossRef]
- Vanhaecke, L.; Gowik, P.; Le Bizec, B.; Van Ginkel, L.; Bichon, E.; Blokland, M.; De Brabander, H.F. European Analytical Criteria: Past, Present, and Future. J. AOAC Int. 2011, 94, 360–372. [Google Scholar]
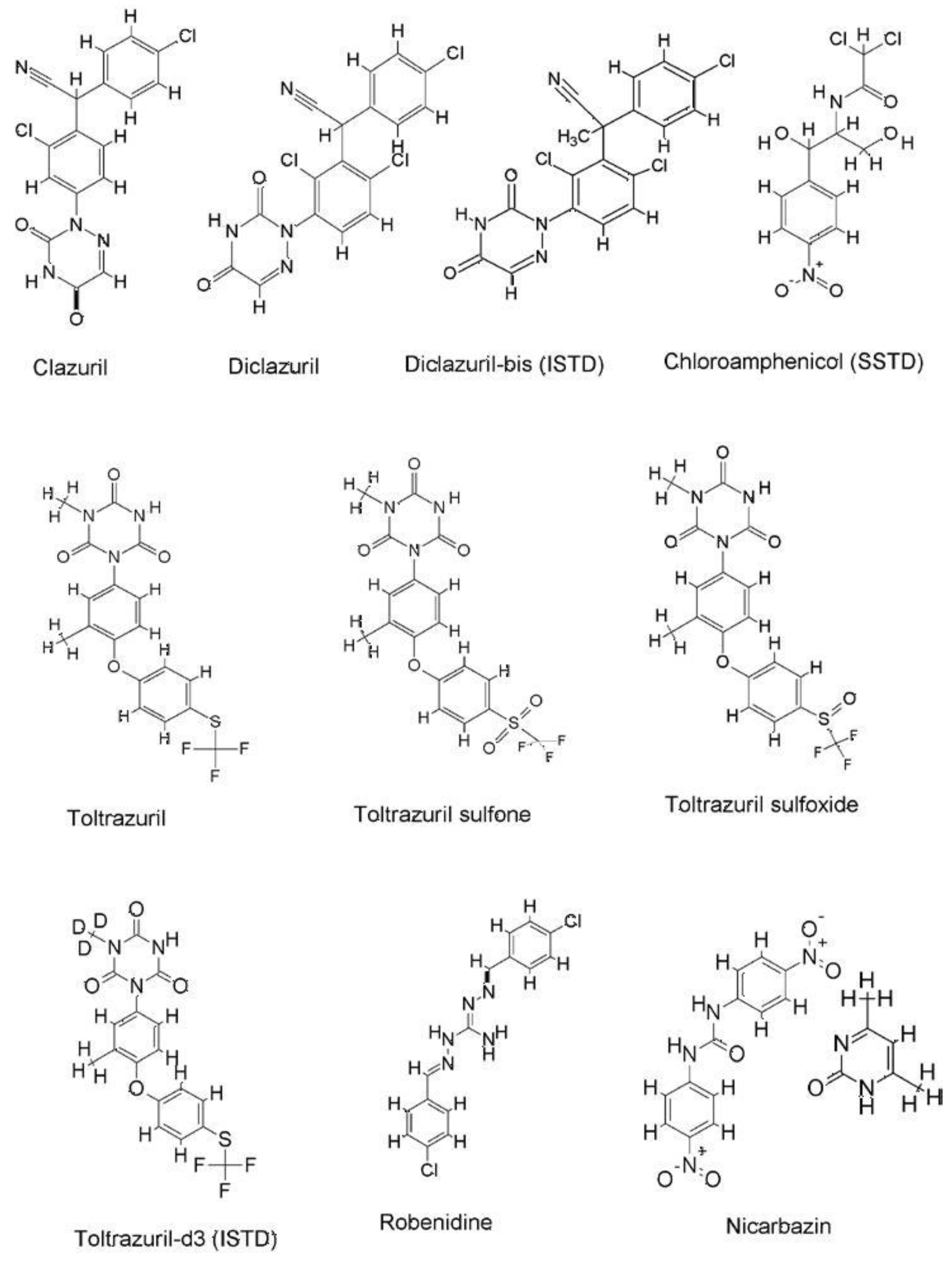

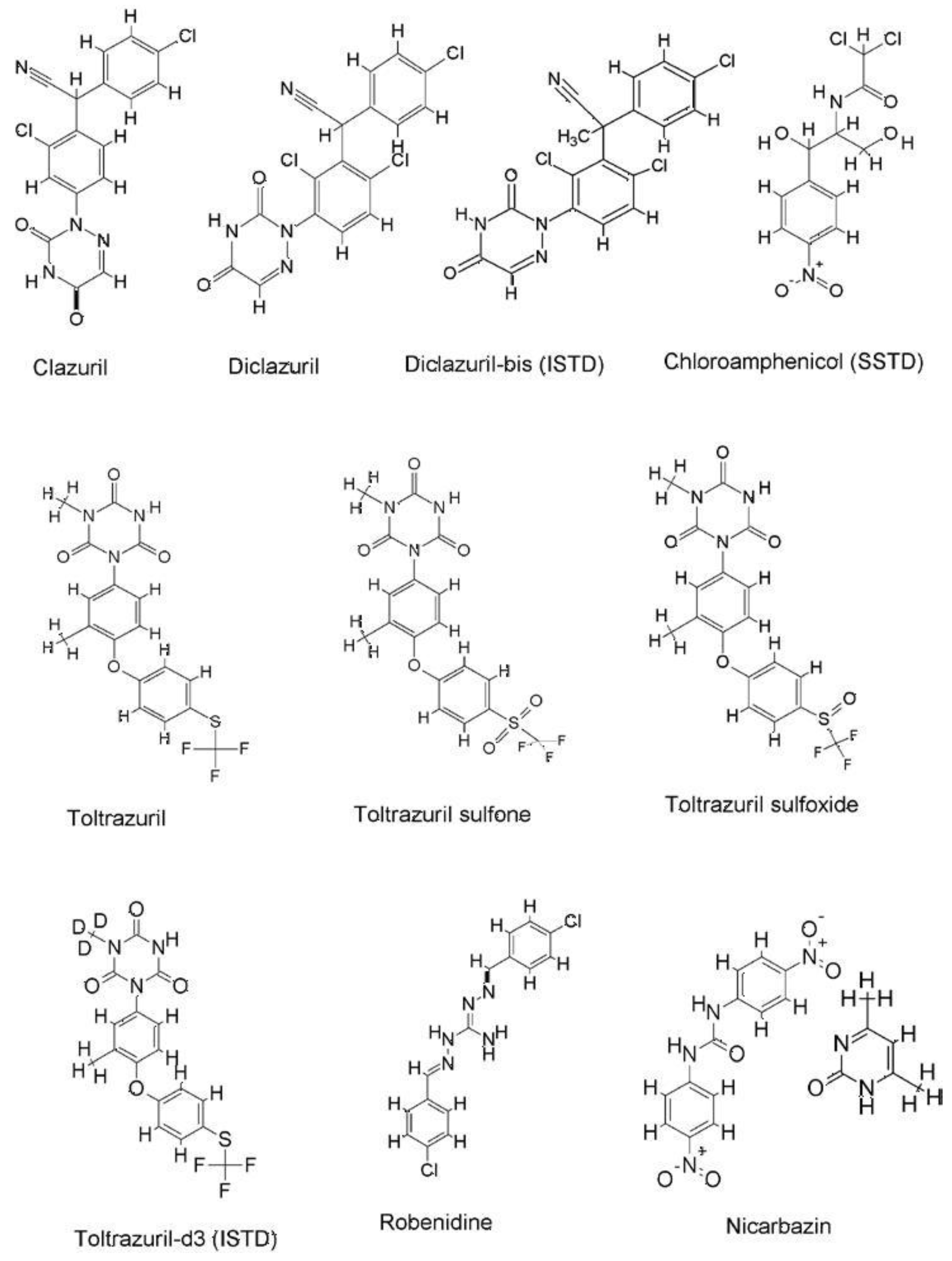{kind=link}
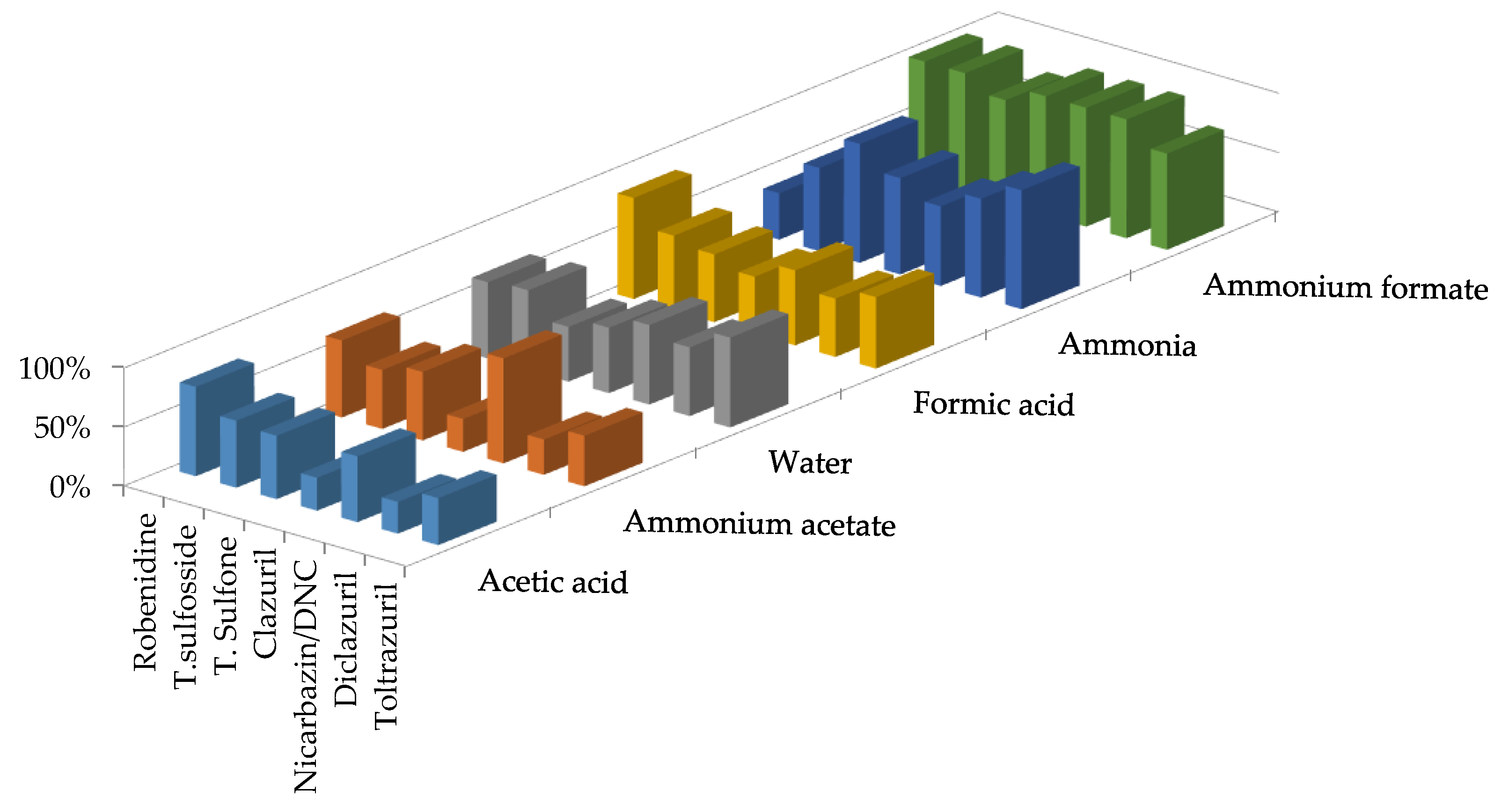
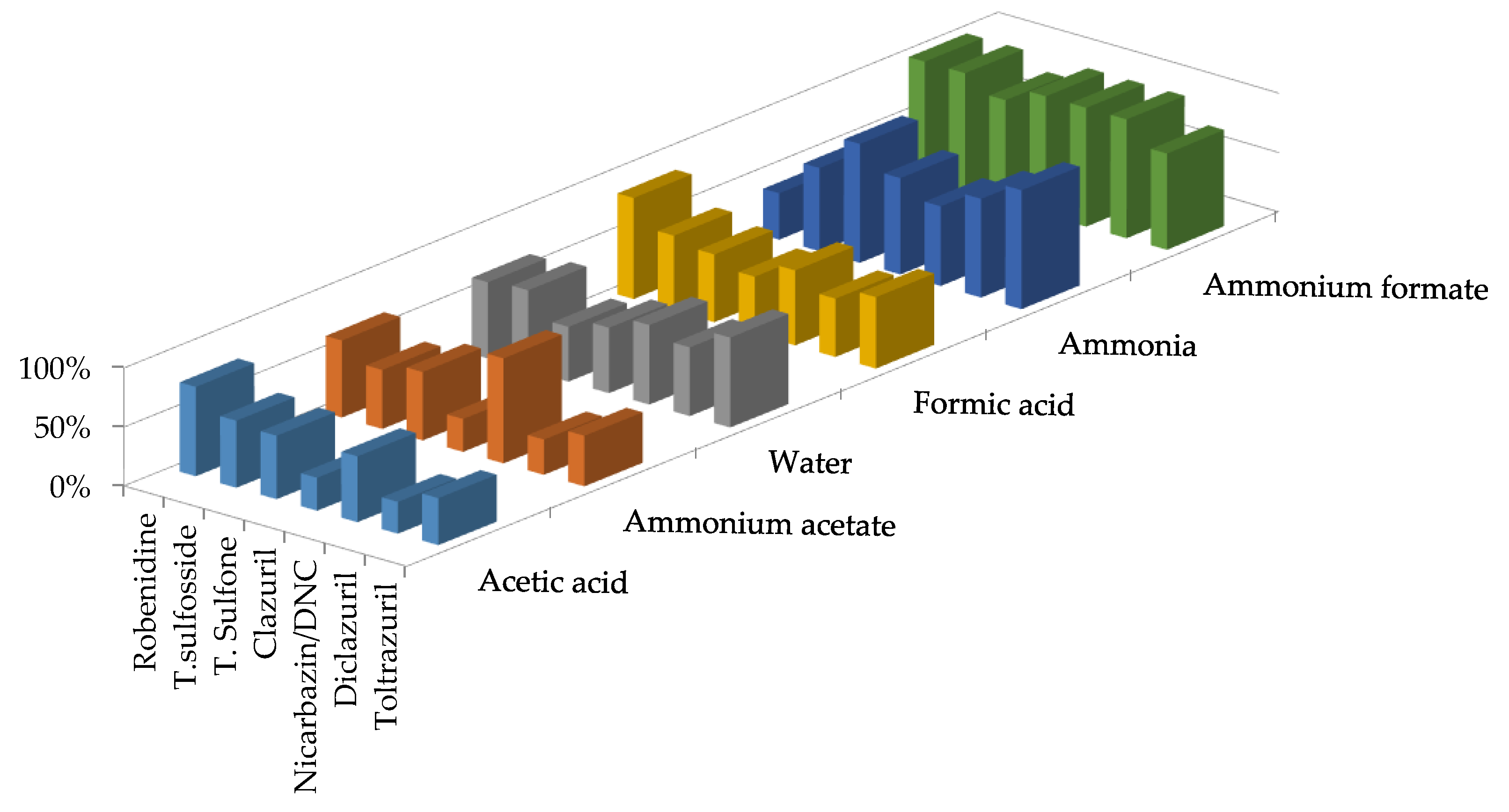{kind=link}
| Compound | Ionization | m/z Precursor Ion (Intensity %) | Capillary (kV) | Cone Voltage (V) | m/z Product Ion (Intensity %) | Collision Energy (eV) | Retention Time Rt (min) |
|---|---|---|---|---|---|---|---|
| Chloroamphenicol CAP (SSTD) | ES− | 325.0 (10) | −4200 | −35 | 194.0 (100) 152.0 (45) | −18 −20 | 5.7 |
| Robenidine | ES+ | 334.1 (58) | +5300 | 50 | 138.0 (90) 155.0 (100) | 35 28 | 6.7 |
| Toltrazuril Sulfoxide | ES− | 440.3 (60) | −4200 | −40 | 42.1 (80) 371.0(100) | −25 −22 | 8.6 |
| Toltrazuril Sulfone | ES− | 456.2 (10) 456.2 (10) | −4200 | −40 | 42.1 (100) 456.0 (100) | −30 −10 | 9.8 |
| Clazuril | ES− | 371.2 (9) | −4200 | −40 | 299.9 (100) 265.0 (39) | −22 −22 | 9.8 |
| Nicarbazin/DNC | ES− | 301.0 (37) | −4200 | −40 | 107.0(5) 137.0 (100) | −43 −20 | 10.1 |
| Diclazuril | ES− | 405.0 (6) 406.9 (6) | −4200 | −40 | 333.8 (100) 335.8 (100) | −25 −25 | 10.5 |
| Diclazuril-bis (ISTD) | ES− | 419.0 (20) | −4200 | −40 | 321.0 (80) 348.0(100) | −22 −20 | 10.8 |
| Toltrazuril | ES− | 424.0 (37) 424.0 (37) | −4200 | −40 | 42.1 (100) 424.0 (100) | −38 −9 | 10.9 |
| Toltrazuril-d3 (ISTD) | ES− | 427.0 (30) | −4200 | −40 | 42.1 (100) | −38 | 10.9 |
| Compound | Ethyl Acetate (xm ± σ)% | Acetonitrile (xm ± σ)% | Ethyl Acetate–Acetone = 50:50 (xm ± σ)% |
|---|---|---|---|
| Robenidine | 10 ± 3.2 | 82 ± 1.0 | 33 ± 2.0 |
| ToltrazurilSulfoxide | 75 ± 2.2 | 72 ± 0.9 | 81 + 2.1 |
| ToltrazurilSulfone | 73 ± 1.5 | 70 ± 1.3 | 85 ± 1.5 |
| Clazuril | 95 ± 3.1 | 95 ± 0.8 | 45 ± 1.0 |
| Nicarbazin/DNC | 90 ± 2.8 | 98 ± 2.1 | 25 ± 4.2 |
| Diclazuril | 88 ± 1.0 | 95 ± 1.1 | 31 ± 0.5 |
| Toltrazuril | 75 ± 0.5 | 70 ± 1.5 | 82 ± 3.0 |
| Analyte | Validation Level | Experimental Concentration | Recovery | Repeatability | Reproducibility | ||
|---|---|---|---|---|---|---|---|
| (µg·kg−1) | (µg·kg−1) ± σ | (%) | (CV%) (n = 6) | (CV%) (n = 18) | |||
| (n = 18) | 1 | 2 | 3 | ||||
| Robenedine | 5.0 | 3.1 ± 0.4 | 62 | 13.7 | 14.7 | 11.9 | 13.9 |
| 12.5 | 8.5 ± 0.8 | 68 | 11.5 | 10.1 | 9.0 | 11 | |
| 25.0 | 18.5 ± 1.2 | 74 | 7.1 | 11 | 7.0 | 9 | |
| 37.5 | 28.5 ± 2.5 | 76 | 8.0 | 10 | 13 | 12.0 | |
| Toltrazuril Sulfoxide | 5.0 | 4.0 ± 0.5 | 80 | 13.0 | 7.5 | 7.5 | 13.6 |
| 7.5 | 6.4 ± 0.6 | 85 | 5.7 | 4.6 | 10.5 | 10.2 | |
| 10.0 | 7.5 ± 0.6 | 75 | 9.4 | 8.8 | 8.4 | 9.2 | |
| 15.0 | 11.4 ± 1.3 | 76 | 8.2 | 13.2 | 12.5 | 11.5 | |
| Toltrazuril Sulfone | 5.0 | 4.4 ± 0.5 | 88 | 9.6 | 4.2 | 11.5 | 11.6 |
| 7.5 | 6.6 ± 0.6 | 88 | 9.1 | 5.4 | 11.4 | 9.2 | |
| 10.0 | 8.1 ± 0.9 | 81 | 12.2 | 7.8 | 8.4 | 10.5 | |
| 15.0 | 12.7 ± 1.1 | 85 | 5.0 | 10.1 | 11.4 | 8.6 | |
| Clazuril | 1.0 | 0.8 ± 0.1 | 80 | 12.5 | 11.9 | 12.1 | 12.0 |
| 2.0 | 1.7 ± 0.2 | 85 | 12.4 | 7.9 | 11.0 | 11.0 | |
| 3.0 | 2.4 ± 0.2 | 80 | 6.1 | 8.4 | 8.5 | 8.9 | |
| 4.0 | 3.2 ± 0.3 | 80 | 3.2 | 10.3 | 10.3 | 10.2 | |
| 6.0 | 5.4 ± 0.3 | 90 | 2.9 | 4.8 | 8.0 | 8.7 | |
| Nicarbazin | 100.0 | 75.0 + 9.0 | 75 | 11.0 | 9.0 | 13.0 | 12.0 |
| 150.0 | 110.0 + 11.0 | 73 | 9.0 | 10.0 | 12.0 | 11.0 | |
| 300.0 | 234.0 + 10.0 | 78 | 5.0 | 8.0 | 7.0 | 8.0 | |
| 450.0 | 315.0 + 28 | 70 | 9.0 | 8.0 | 12.0 | 9.0 | |
| 100.0 | 75.0 + 9.0 | 75 | 11.0 | 9.0 | 13.0 | 12.0 | |
| Diclazuril | 1.0 | 0.8 ± 0.1 | 80 | 11.0 | 13.0 | 12.1 | 12.1 |
| 2.00 | 1.8 ± 0.3 | 90 | 7.0 | 5.6 | 8.8 | 13.7 | |
| 3.0 | 2.6± 0.2 | 87 | 7.2 | 5.5 | 7.0 | 10.2 | |
| 4.0 | 3.2 ± 0.3 | 80 | 9.2 | 9.1 | 8.9 | 9.5 | |
| 6.0 | 5.1 ± 0.5 | 85 | 9.9 | 12.2 | 8.4 | 11.5 | |
| Toltrazuril | 5.0 | 4.4 ± 0.3 | 88 | 9.3 | 7.7 | 8.8 | 14.1 |
| 7.5 | 7.1 ± 0.5 | 95 | 5.5 | 4.1 | 9.5 | 6.4 | |
| 10.0 | 8.7 ± 0.9 | 87 | 11.7 | 12.4 | 10.1 | 10.9 | |
| 15.0 | 12.7 ± 1.2 | 85 | 8.1 | 9.2 | 11.3 | 9.8 | |
| Analyte | CCα (µg·kg−1) | CCβ (µg·kg−1) |
|---|---|---|
| Robenidine | 28.0 | 30.0 |
| Toltrazuril sulfoxide | 5.1 | 6.1 |
| Toltrazuril sulfone | 5.8 | 6.7 |
| Clazuril | 2.2 | 2.6 |
| Nicarbazin/DNC | 320.0 | 350.0 |
| Diclazuril | 2..2 | 2.6 |
| Toltrazuril | 6.0 | 6.9 |
| Variable Number | Experiment Number | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Lot of SPE | lotA | lotA | lotA | lotA | lotA | lotA | lotA | lotA |
| Extraction Volume Acetonitrile (mL) | 5.5 | 5.5 | 4.5 | 4.5 | 5.5 | 5.5 | 4.5 | 4.5 |
| Dilution Volume with Water (mL) | 30 | 20 | 30 | 20 | 30 | 20 | 30 | 20 |
| Washing Volume for SPE with Water (mL) | 5.5 | 5.5 | 4.5 | 4.5 | 4.5 | 4.5 | 5.5 | 5.5 |
| Washing Volume for SPE with MeOH 5% (mL) | 5.5 | 4.5 | 5.5 | 4.5 | 4.5 | 5.5 | 4.5 | 5.5 |
| Elution Volume for SPE (mL) | 5.5 | 4.5 | 4.5 | 5.5 | 5.5 | 4.5 | 4.5 | 5.5 |
| Drying temperature (°C) | 44 | 36 | 36 | 44 | 36 | 44 | 44 | 36 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buiarelli, F.; Di Filippo, P.; Riccardi, C.; Pomata, D.; Giannetti, L.; Neri, B.; Rago, D. Liquid Chromatography Tandem Mass Spectrometry Analysis of Synthetic Coccidiostats in Eggs. Separations 2017, 4, 15. https://doi.org/10.3390/separations4020015
Buiarelli F, Di Filippo P, Riccardi C, Pomata D, Giannetti L, Neri B, Rago D. Liquid Chromatography Tandem Mass Spectrometry Analysis of Synthetic Coccidiostats in Eggs. Separations. 2017; 4(2):15. https://doi.org/10.3390/separations4020015
Chicago/Turabian StyleBuiarelli, Francesca, Patrizia Di Filippo, Carmela Riccardi, Donatella Pomata, Luigi Giannetti, Bruno Neri, and Daniela Rago. 2017. "Liquid Chromatography Tandem Mass Spectrometry Analysis of Synthetic Coccidiostats in Eggs" Separations 4, no. 2: 15. https://doi.org/10.3390/separations4020015