
Method for the Determination of Benzene Metabolite t,t-Muconic Acid in Urine by HPLC-UV with an Ion Exclusion Column

Abstract
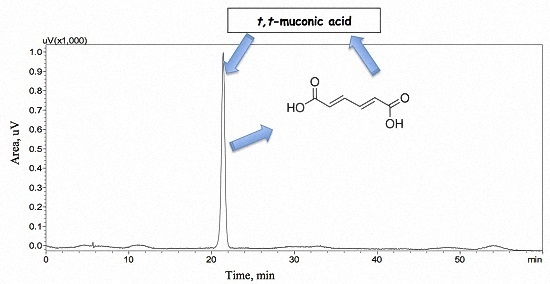
:
1. Introduction
2. Experimental Section
2.1. Instruments and Accessories
2.2. Reagents, Solutions and Samples
2.3. Optimization of the Procedure
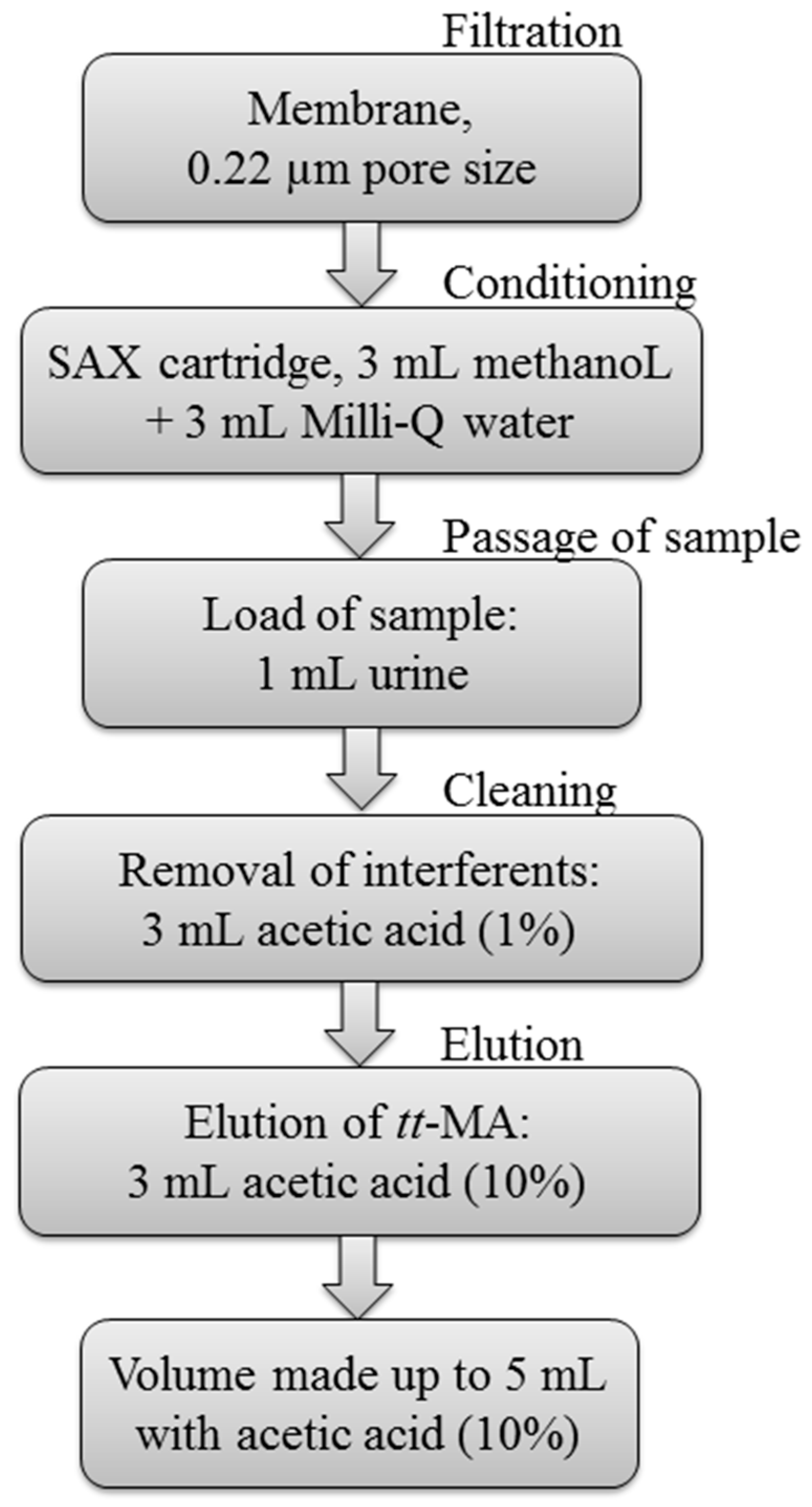
2.3.1. Preparation of Samples
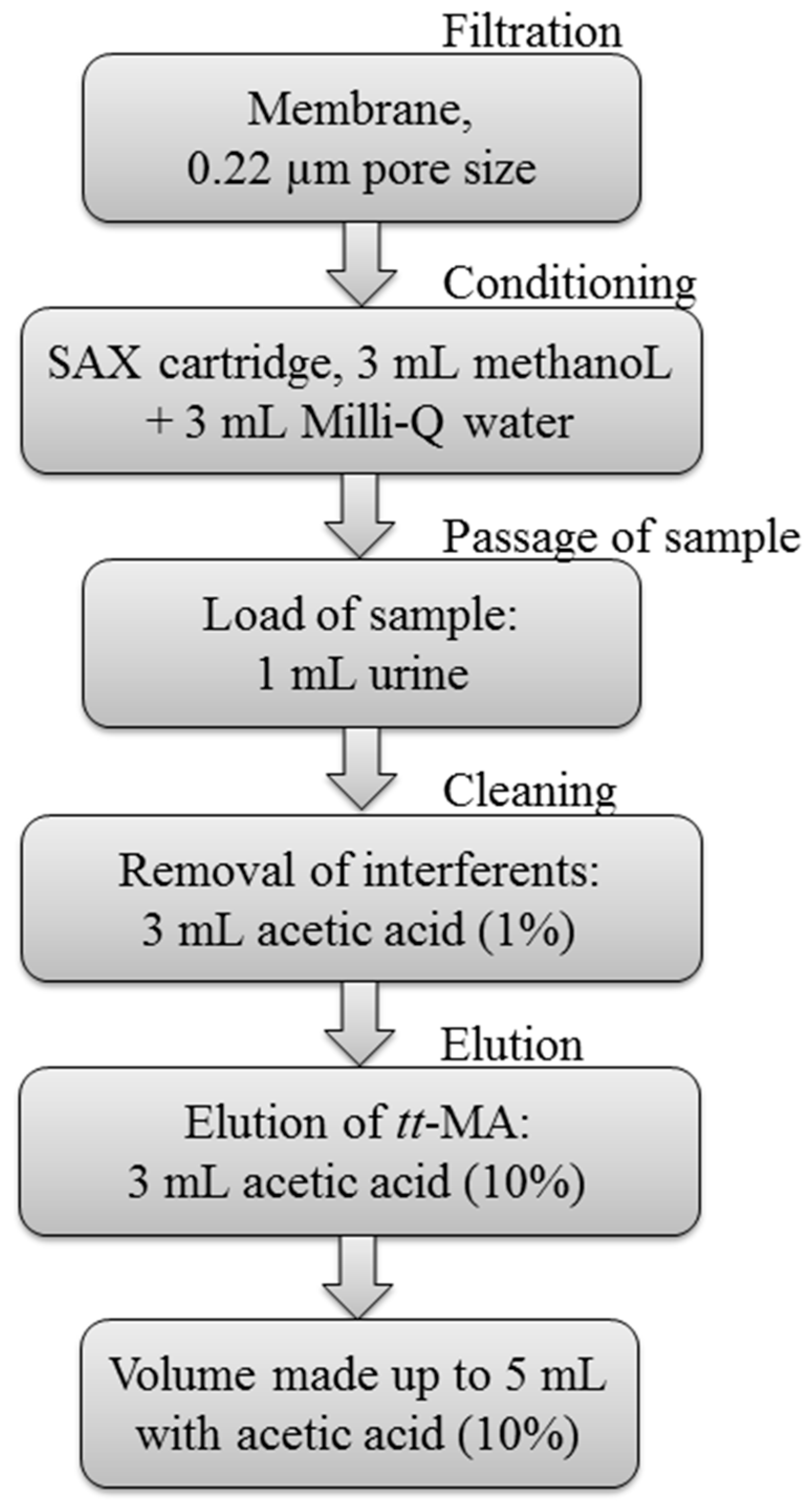
2.3.2. Optimization of the Sample Preparation Procedure
2.3.3. Chromatographic Conditions
2.4. Validation of the Method
2.4.1. Selectivity
2.4.2. Matrix Interferences
2.4.3. Analytical Curve Adjustment and Working Range
2.4.4. Limits of Detection and Quantification
2.4.5. Precision and Accuracy
3. Results and Discussion
3.1. Optimization of Sample Preparation
3.2. Evaluation of Analytical Conditions
3.3. Validation of the Method
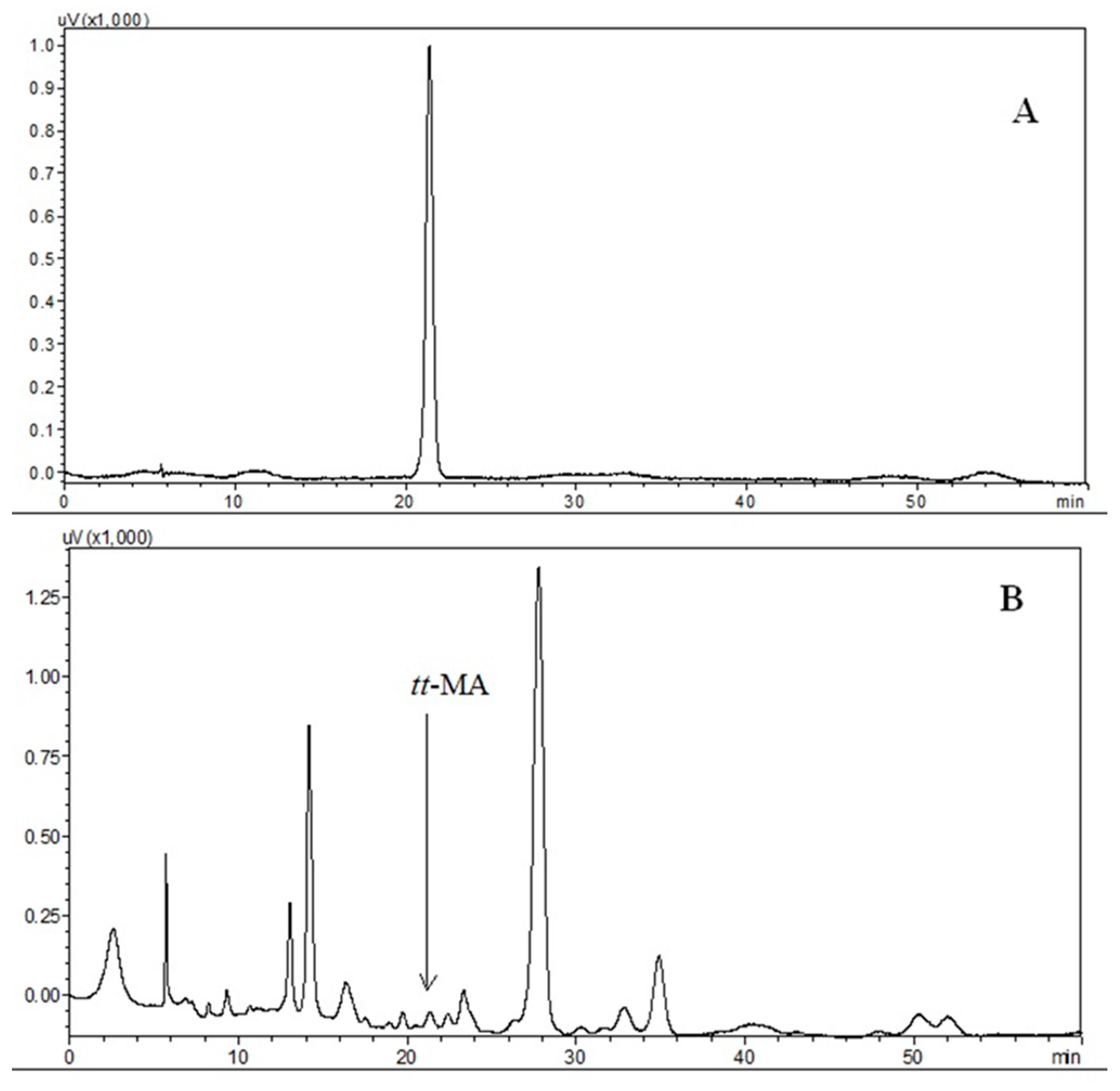
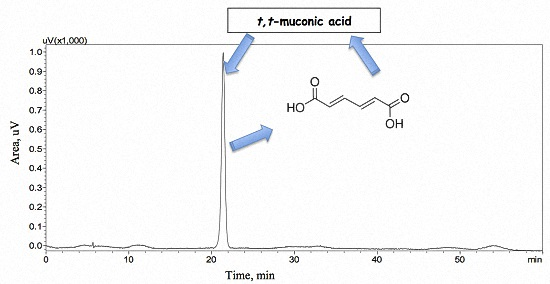
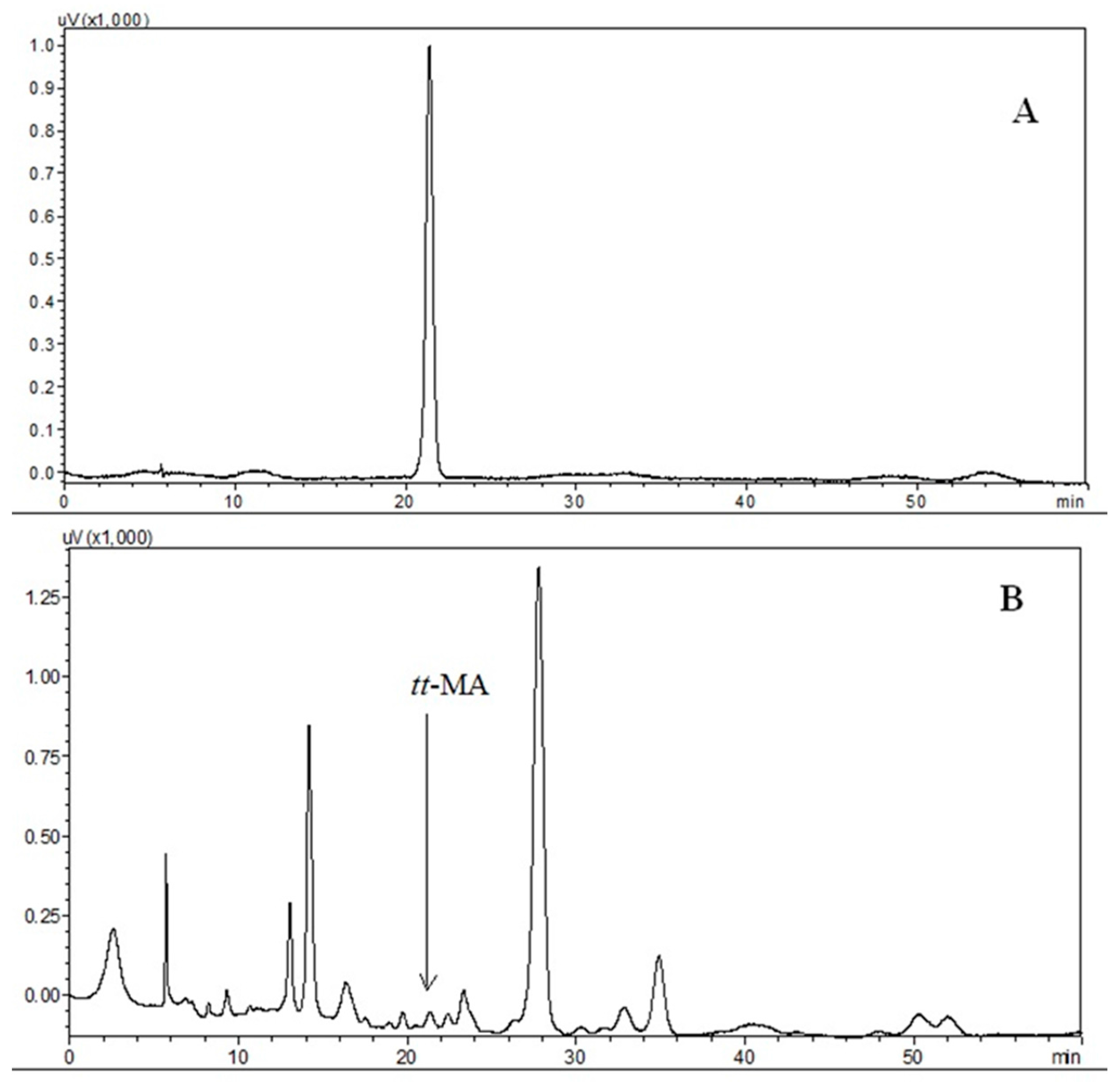
3.3.1. Selectivity
3.3.2. Interferences due to the Biological Matrix
- X1: tt-MA peak area for the analytical curve prepared using aqueous standards;
- X2: tt-MA peak area for the analytical curve prepared using pooled urine.
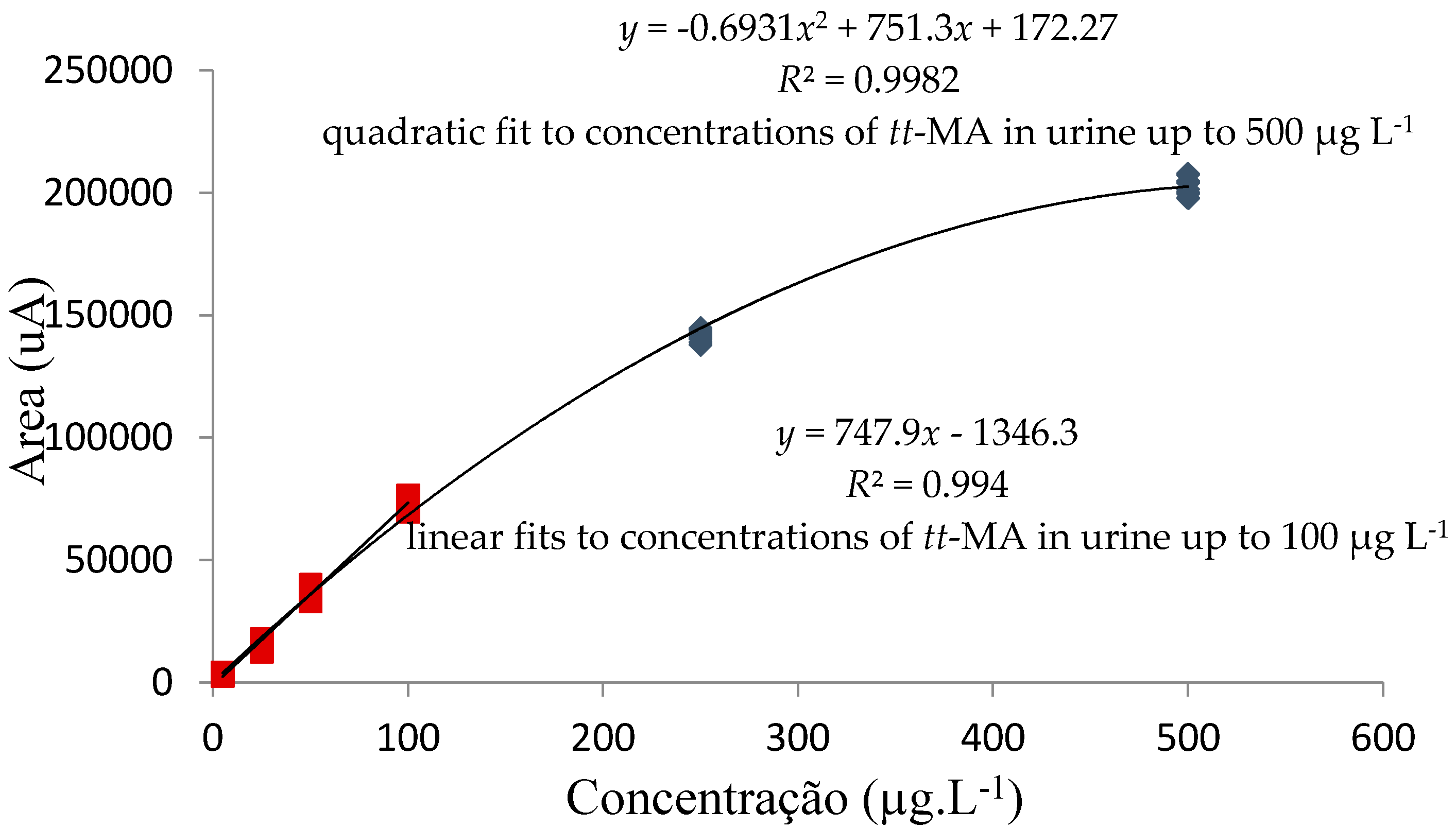
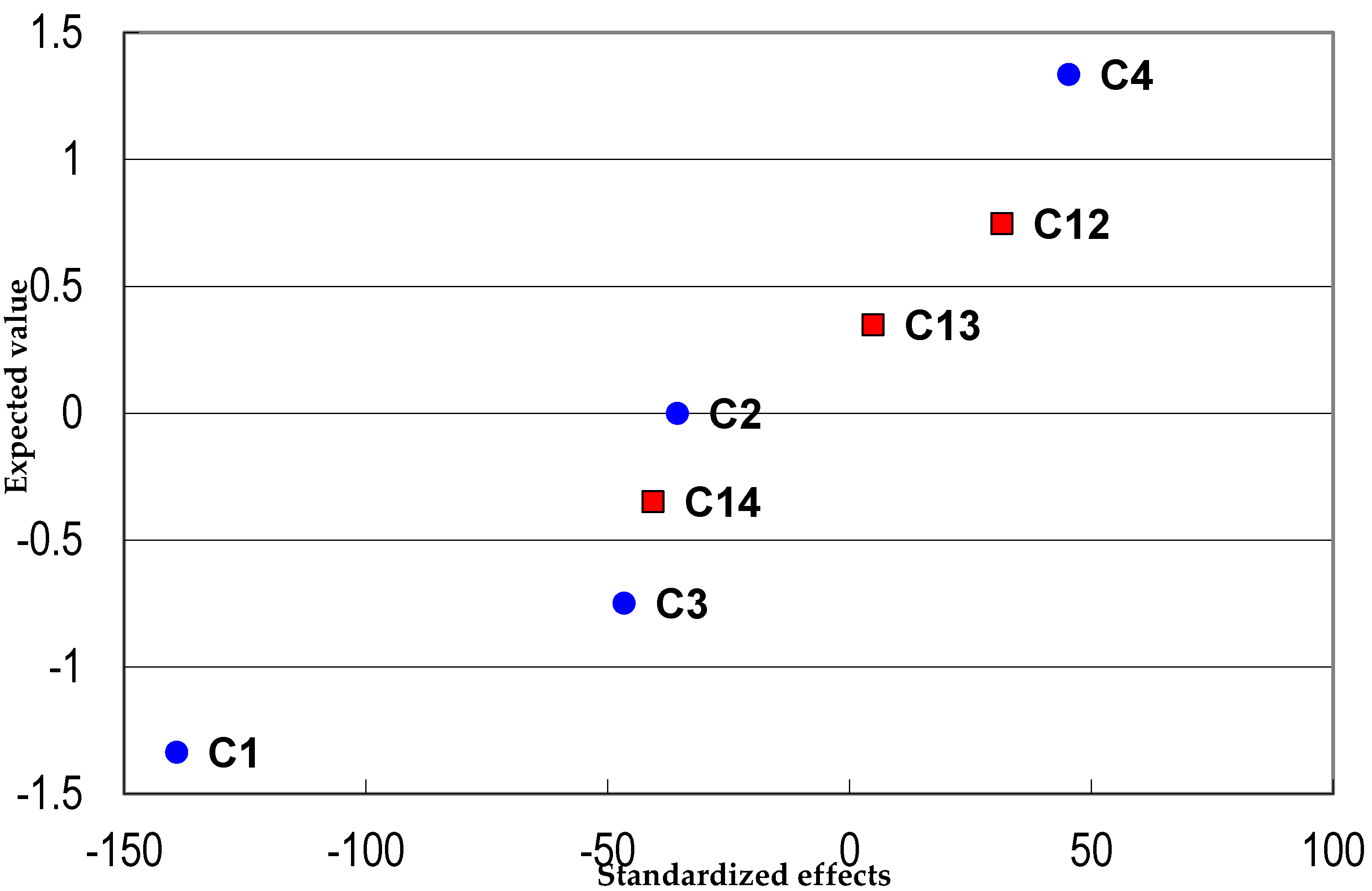
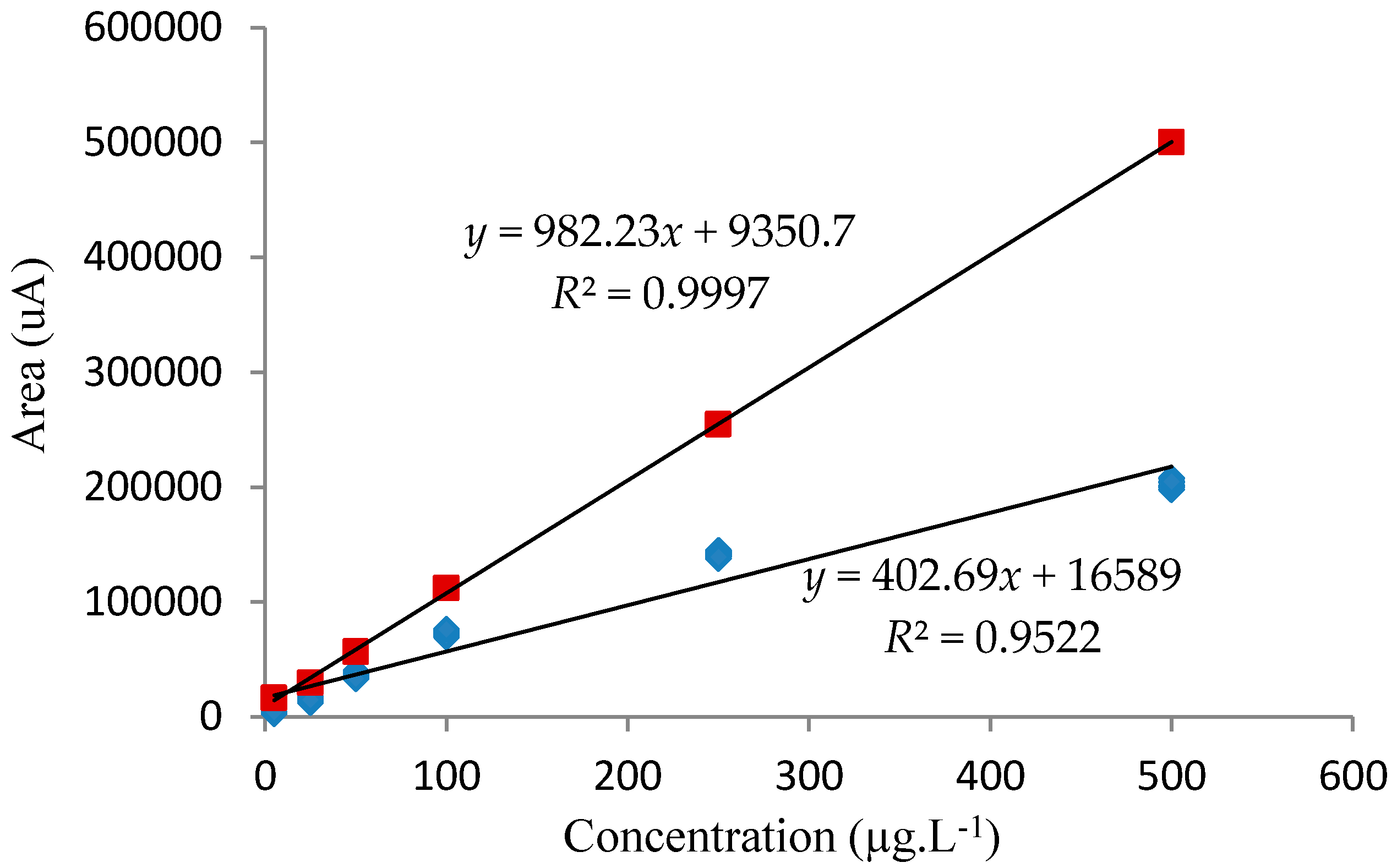
3.3.3. Analytical Curve Adjustment and Working Range
3.3.4. Limits of Detection and Quantification
3.3.5. Precision and Accuracy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Protano, C.; Andreoli, R.; Manini, P.; Guidotti, M.; Vitali, M. A tobacco-related carcinogen: Assessing the impact of smoking behaviours of cohabitants on benzene exposure in children. Tob. Control 2012, 21, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Guarieiro, L.L.N.; Vasconcellos, P.C.; Solci, M.C. Air Pollutants from the burning of fossil fuels and biofuels: A brief review. Rev. Virtual Quim. 2011, 3, 434–445. [Google Scholar] [CrossRef]
- Manini, P.; de Palma, G.; Andreoli, R.; Poli, D.; Mozzoni, P.; Folesani, G.; Mutti, A.; Apostoli, P. Environmental and biological monitoring of benzene exposure in a cohort of Italian taxi drivers. Toxicol. Lett. 2006, 167, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Kivistö, H.; Pekari, K.; Peltonen, K.; Svinhufvud, J.; Veidebaum, T.; Sorsa, M.; Aitio, A. Biological monitoring of exposure to benzene in the production of benzene and in a cokery. Sci. Total Environ. 1997, 199, 49–63. [Google Scholar] [CrossRef]
- Choi, Y.; Shin, D.; Park, S.; Chung, Y.; Kim, M. Biological monitoring of benzene in residents living near petrochemical industrial areas in Korea. J. Occup. Health 2000, 42, 31–37. [Google Scholar] [CrossRef]
- Kirkeleit, J.; Riise, T.; Bråtveit, M.; Pekari, K.; Mikkola, J.; Moen, B.E. Biological monitoring of benzene exposure during maintenance work in crude oil cargo tanks. Chem.-Biol. Interact. 2006, 164, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Brugnone, F.; Perbellini, L.; Maranelli, G.; Romeo, L.; Guglielmi, G.; Lombardini, F. Reference values for blood benzene in the occupationally unexposed general population. Int. Arch. Occup. Environ. Health 1992, 64, 179–184. [Google Scholar] [CrossRef]
- Aprea, C.; Sciarra, G.; Bozzi, N.; Pagliantini, M.; Perico, A.; Bavazzano, P.; Leandri, A.; Carrieri, M.; Scapellato, M.; Bettinelli, M.; et al. Reference Values of Urinary trans,trans-muconic Acid: Italian Multicentric Study. Arch. Environ. Contam. Toxicol. 2008, 55, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Fustinoni, S.; Campo, L.; Mercadante, R.; Manini, P. Methodological issues in the biological monitoring of urinary benzene and S-phenylmercapturic acid at low exposure levels. J. Chromatogr. B 2010, 878, 2534–2540. [Google Scholar] [CrossRef] [PubMed]
- Waidyanatha, S.; Rothman, N.; Fustinoni, S.; Smith, M.T.; Hayes, R.B.; Bechtold, W.; Dosemeci, M.; Guilan, L.; Yin, S.; Rappaport, S.M. Urinary benzene as a biomarker of exposure among occupationally exposed and unexposed subjects. Carcinogenesis 2001, 22, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Coutrim, M.X.; de Carvalho, L.R.F.; Arcuri, A.S.A. Evaluation of the analytical methods to determinate benzene metabolites as potencial biomarkers for determining human exposure to benzene in air. Quím. Nova 2000, 23, 653–663. [Google Scholar] [CrossRef]
- Ministry of Labor and Employment of Brazil. Portaria No. 34, 20 December 2001, Attachment: Protocolo para a Utilização de Indicador Biológico da Exposição Ocupacional ao Benzeno. Available online: http://www.capecanaveral4045.com/legislacao/port_34_2001_benzeno_indicador_biologico.html (accessed on 12 September 2015).
- Menezes, M.; Balbão, M.S.; Siqueira, M.E.P.B.; Martins, I. Influence of tobacco smoking on urinary excretion of trans,trans-muconic acid. Braz. J. Pharm. Sci. 2008, 44, 459–464. [Google Scholar]
- Roma-Torres, J.; Teixeira, J.P.; Silva, S.; Laffon, B.; Cunha, L.; Mendez, J.; Mayan, O. Evaluation of genotoxicity in a group of workers from a petroleum refinery aromatics plant. Mutat. Res. 2006, 604, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Violante, F.; Sanguinetti, G.; Barbieri, A.; Accorsi, A.; Mattioli, S.; Cesari, R.; Fimognari, C.; Hrelia, P. Lack of correlation between environmental or biological indicators of benzene exposure at parts per billion levels and micronuclei induction. Environ. Res. 2003, 91, 135–142. [Google Scholar] [CrossRef]
- Kouniali, A.; Cicolella, A.; Gonzalez-Flesca, N.; Dujardin, R.; Gehanno, J.-F.; Bois, F.Y. Environmental benzene exposure assessment for parent-child pairs in Rouen. France. Sci. Total Environ. 2003, 308, 73–82. [Google Scholar] [CrossRef]
- Cocco, P.; Tocco, M.G.; Ibba, A.; Scano, L.; Ennas, M.G.; Flore, C.; Randaccio, F. trans,trans-Muconic acid excretion in relation to environmental exposure to benzene. Int. Arch. Occup. Environ. Health 2003, 76, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Marrubini, G.; Coccini, T.; Manzo, L. Direct analysis of urinary trans,trans-muconic acid by coupled column liquid chromatography and spectrophotometric ultraviolet detection: Method applicability to human urine. J. Chromatogr. B 2001, 758, 295–303. [Google Scholar] [CrossRef]
- Waidyanatha, S.; Rothman, N.; Li, G.; Smith, M.T.; Yin, S.; Rappaport, S.M. Rapid determination of six urinary benzene metabolites in occupationally exposed and unexposed subjects. Anal. Biochem. 2004, 327, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Vermeulen, R.; Waidyanatha, S.; Johnson, B.A.; Lan, Q.; Rothman, N.; Smith, M.T.; Zhang, L.; Li, G.; Shen, M.; et al. Using urinary biomarkers to elucidate dose-related patterns of human benzene metabolism. Carcinogenesis 2006, 27, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Mudiam, M.K.R.; Chauhan, A.; Singh, K.P.; Gupta, S.K.; Jain, R.; Ch, R.; Murthy, R.C. Determination of t,t-muconic acid in urine samples using a molecular imprinted polymer combined with simultaneous ethyl chloroformate derivatization and pre-concentration by dispersive liquid-liquid microextraction. Anal. Bioanal. Chem. 2013, 405, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.; Skov, H.; Autrup, H.; Hertel, O.; Loft, S. Urban benzene exposure and oxidative DNA damage: Influence of genetic polymorphisms in metabolism genes. Sci. Total Environ. 2003, 309, 69–80. [Google Scholar] [CrossRef]
- Wiwanitkit, V.; Suwansaksri, J.; Soogarun, S. A note on urine trans, trans muconic acid level among a sample of Thai police: Implication for an occupational health issue. Yale J. Biol. Med. 2003, 76, 103–108. [Google Scholar] [PubMed]
- Ruppert, T.; Scherer, G.; Tricker, A.R.; Rauscher, D.; Adlkofer, F. Determination of urinary trans,trans-muconic acid by gas chromatography-mass spectrometry. J. Chromatogr. B 1995, 666, 71–76. [Google Scholar] [CrossRef]
- Franqui, L.S.; Vieira, A.C.; Maia, P.P.; Figueiredo, E.C. Molecularly imprinted solid-phase extraction of urinary trans,trans-muconic acid and analysis by gas chromatography-mass spectrometry. Quím. Nova 2012, 35, 1577–1581. [Google Scholar] [CrossRef]
- Johnson, E.S.; Langård, S.; Lin, Y.-S. A critique of benzene exposure in the general population. Sci. Total Environ. 2007, 374, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Ducos, P.; Gaudin, R.; Robert, A.; Francin, J.; Maire, C. Improvement in HPLC analysis of urinary trans,trans-muconic acid, a promising substitute for phenol in the assessment of benzene exposure. Int. Arch. Occup. Environ. Health 1990, 62, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Bono, R.; Maestri, L.; Ghittori, S.; Imbriani, M. High-pressure liquid chromatographic-mass spectrometric determination of sorbic acid in urine: Verification of formation of trans,trans-muconic acid. Chem.-Biol. Interact. 2005, 153, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Schroijen, C.; Baeyens, W.; Schoeters, G.; den Hond, E.; Koppen, G.; Bruckers, L.; Nelen, V.; van de Mieroop, E.; Bilau, M.; Covaci, A.; et al. Internal exposure to pollutants measured in blood and urine of Flemish adolescents in function of area of residence. Chemosphere 2008, 71, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Serena, P.; Tapparo, A.; Bombi, G. Direct determination of t,t-muconic acid in human urine by two-dimensional liquid chromatography. Analyst 2000, 125, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Melikian, A.A.; Connor, R.; Prahalad, A.; Hu, P.; Li, H.; Kagan, M.; Thompson, S. Determination of the urinary benzene metabolites S-phenylmercapturic acid and trans,trans-muconic acid by liquid chromatography-tandem mass spectrometry. Carcinogenesis 1999, 20, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Senzolo, C.; Frignani, S.; Pavoni, B. Environmental and biological monitoring of occupational exposure to organic micropollutants in gasoline. Chemosphere 2001, 44, 67–82. [Google Scholar] [CrossRef]
- Barbieri, A.; Sabatini, L.; Accorsi, A.; Roda, A.; Violante, F. Simultaneous determination of t,t-muconic, S-phenylmercapturic and S-benzylmercapturic acids in urine by a rapid and sensitive liquid chromatography/electrospray tandem mass spectrometry method. Rapid Commun. Mass Spectrom. 2004, 18, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Manini, P.; Andreoli, R.; Niessen, W.M.A. Liquid chromatography-mass spectrometry in occupational toxicology: A novel approach to the study of biotransformation of industrial chemicals. J. Chromatogr. A 2004, 1058, 21–37. [Google Scholar] [CrossRef]
- Fustinoni, S.; Buratti, M.; Campo, L.; Colombi, A.; Consonni, D.; Pesatori, A.C.; Bonzini, M.; Farmer, P.; Garte, S.; Valerio, F.; et al. Urinary t,t-muconic acid, S-phenylmercapturic acid and benzene as biomarkers of low benzene exposure. Chem.-Biol. Interact. 2005, 153, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Ong, H.; Ong, Y.; Ong, C. A sensitive liquid chromatographic method for the spectrophotometric determination of urinary trans,trans-muconic acid. J. Chromatogr. B 2005, 818, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Navasumrit, P.; Chanvaivit, S.; Intarasunanont, P.; Arayasiri, M.; Lauhareungpanya, N.; Parnlob, V.; Settachan, D.; Ruchirawat, M. Environmental and occupational exposure to benzene in Thailand. Chem.-Biol. Interact. 2005, 153, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, M.; Bonfiglio, E.; Scapellato, M.L.; Maccà, I.; Tranfo, G.; Faranda, P.; Paci, E.; Bartolucci, G.B. Comparison of exposure assessment methods in occupational exposure to benzene in gasoline filling-station attendants. Toxicol. Lett. 2006, 162, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Scherer, G.; Engl, J.; Urban, M.; Gilch, G.; Janket, D.; Riedel, K. Relationship between machine-derived smoke yields and biomarkers in cigarette smokers in Germany. Regul. Toxicol. Pharm. 2007, 47, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.; Roychoudhury, S.; Mukherjee, S.; Lahiri, T. Occupational benzene exposure from vehicular sources in India and its effect on hematology, lymphocyte subsets and platelet P-selectin expression. Toxicol. Ind. Health 2007, 23, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Chakroun, R.; Faidi, F.; Hedhili, A.; Charbaji, K.; Nouaigui, H.; Laiba, M.B. Inhalant Abuse Detection and Evaluation in Young Tunisians. J. Forensic Sci. 2008, 53, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Weaver, V.; Buckley, T.; Groopman, J.D. Lack of specificity of trans,trans-muconic acid as a benzene biomarker after ingestion of sorbic acid-preserved foods. Cancer Epidemiol. Biomarkers Prev. 2000, 9, 749–755. [Google Scholar] [PubMed]
- Fritz, J.S. Review: Principles and applications of ion-exclusion chromatography. J. Chromatogr. 1991, 546, 111–118. [Google Scholar] [CrossRef]
- Kelebek, H.; Selli, S. Determination of volatile, phenolic, organic acid and sugar components in a Turkish cv. Dortyol (Citrus sinensis L. Osbeck) orange juice. J. Agric. Food Chem. 2011, 91, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Eyeghe-Bickong, H.; Alexandersson, E.O.; Gouws, L.; Young, P.; Vivier, M.A. Optimisation of an HPLC method for the simultaneous quantification of the major sugars and organic acids in grapevine berries. J. Chromatogr. B 2012, 885, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Brigham, C.J.; Kim, Y.; Kim, H.; Yi, D.; Kim, H.; Rha, C.; Sinskey, A.; Yang, Y. Biosynthesis of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (P(HB-co-HHx)) from butyrate using engineered Ralstonia eutropha. Appl. Microbiol. Biotech. 2014, 98, 5461–5469. [Google Scholar] [CrossRef] [PubMed]
- Teófilo, R.F.; Ferreira, M.M.C. Chemometrics II: Spreadsheets for experimental design calculations, a tutorial. Quím. Nova 2006, 29, 338–350. [Google Scholar] [CrossRef]
- Brazil, National Institute of Metrology (INMETRO). Orientação Sobre Validação de Métodos Analíticos, 3rd Review, February 2010. Available online: http://www.inmetro.gov.br/sidoq/arquivos/cgcre/doq/doq-cgcre-8_03.pdf (accessed on 14 September 2015).
- Ribani, M.; Bottoli, C.B.G.; Collins, C.H.; Jardim, I.C.S.F.; Melo, L.F.C. Validation for chromatographic and electrophoretic methods. Quím. Nova 2004, 27, 771–780. [Google Scholar] [CrossRef]
- Martins, I.; Siqueira, M.E.P.B. Determination of trans,trans-muconic acid in urine: Validation of a high performance liquid chromatographic method. Braz. J. Pharmac. Sci. 2002, 38, 205–213. [Google Scholar]
- Tranfo, G.; Paci, E.; Sisto, R.; Pigini, D. Validation of an HPLC/MS/MS method with isotopic dilution for quantitative determination of trans,trans-muconic acid in urine samples of workers exposed to low benzene concentrations. J. Chromatogr. B 2008, 867, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Brazilian National Health Surveillance Agency (ANVISA). Resolution RE No. 899, 29 May 2003, Attachment: Guia para Validação de Métodos Analíticos e Bioanalíticos, pp. 56–59. Available online: http://pesquisa.in.gov.br/imprensa/jsp/visualiza/index.jsp?data=02/06/2003&jornal=1&pagina=56&totalArquivos=176 (accessed on 14 September 2015).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Level | ||
|---|---|---|---|
| −1 | 0 | 1 | |
| Solution buffering | No buffering | pH 8.0 | pH 10.0 |
| Removal of interferents (washing) | No addition of water | 1.5 mL of water | 3.0 mL of water |
| Final volume | 3.0 mL | 4.0 mL | 5.0 mL |
| Sample volume | 1.0 mL | 2.0 mL | 3.0 mL |
| Variable | Buffering of the Solution | Washing | Final Volume | Sample Volume | |
|---|---|---|---|---|---|
| Tests | |||||
| Test 1 | no buffering (−1) | no water addition (−1) | 3.0 mL (−1) | 1.0 mL (−1) | |
| Test 2 | pH 10.0 (1) | no water addition (−1) | 3.0 mL (−1) | 3.0 mL (1) | |
| Test 3 | no buffering (−1) | 3.0 mL of water (1) | 3.0 mL (−1) | 3.0 mL (1) | |
| Test 4 | pH 10.0 (1) | 3.0 mL of water (1) | 3.0 mL (−1) | 1.0 mL (−1) | |
| Test 5 | no buffering (−1) | no water addition (−1) | 5.0 mL (1) | 3.0 mL (1) | |
| Test 6 | pH 10.0 (1) | no water addition (−1) | 5.0 mL (1) | 1.0 mL (−1) | |
| Test 7 | no buffering (−1) | 3.0 mL of water (1) | 5.0 mL (1) | 1.0 mL (−1) | |
| Test 8 | pH 10.0 (1) | 3.0 mL of water (1) | 5.0 mL (1) | 3.0 mL (1) | |
| CP 1 | pH 8.0 (0) | 1.5 mL of water (0) | 4.0 mL (0) | 2.0 mL (0) | |
| CP 2 | pH 8.0 (0) | 1.5 mL of water (0) | 4.0 mL (0) | 2.0 mL (0) | |
| CP 3 | pH 8.0 (0) | 1.5 mL of water (0) | 4.0 mL (0) | 2.0 mL (0) | |
| Test | X1 | X2 | X3 | X4 | y |
|---|---|---|---|---|---|
| 1 | −1 | −1 | −1 | −1 | 106,519 |
| 2 | 1 | −1 | −1 | 1 | 38,378 |
| 3 | −1 | 1 | −1 | 1 | 116,419 |
| 4 | 1 | 1 | −1 | −1 | 33,805 |
| 5 | −1 | −1 | 1 | 1 | 124,593 |
| 6 | 1 | −1 | 1 | −1 | 14,086 |
| 7 | −1 | 1 | 1 | −1 | 44,460 |
| 8 | 1 | 1 | 1 | 1 | 14,293 |
| CP1 | 0 | 0 | 0 | 0 | 38,802 |
| CP2 | 0 | 0 | 0 | 0 | 37,527 |
| CP3 | 0 | 0 | 0 | 0 | 37,513 |
| CP (mean) | 37,947 | ||||
| CP (standard deviation) | 740.2 | ||||
| CP (RSD, %) | 1.95 | ||||
| Parameter | Condition |
|---|---|
| Buffering | None |
| Sample volume | 3.0 mL |
| Sample washing | 5.0 mL of acetic acid (1%) |
| Final volume | 5.0 mL of acetic acid (10%) |
| Chromatographic Conditions | Description |
|---|---|
| Column | Aminex HPX-87H (300 mm × 7.8 mm) ion exclusion |
| Mobile phase flow rate | 0.6 mL·min−1 |
| Injection volume | 20 μL (automatic injection system) |
| Column temperature | 55 °C |
| Run time | 60 min |
| Urine Sample | Average Peak Area (µA) | CV (%) |
|---|---|---|
| Pool of 10 samples | 96,461 | 1.22 |
| Pool with addition of 5.0 µg·L−1 | 99,892 | 0.53 |
| Pool with addition of 50.0 µg·L−1 | 133,092 | 1.47 |
| Pool with addition of 500 µg·L−1 | 299,546 | 1.16 |
| Concentration of tt-MA Added (µg·L−1) | Concentration of tt-MA Found (µg·L−1) | Recovery (%) |
|---|---|---|
| 5.0 | 4.4 | 87.20 |
| 50.0 | 50.9 | 101.84 |
| 500.0 | 510.6 | 102.12 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, R.D.P.; Sanson, A.L.; Lobo, F.A.; Afonso, R.J.d.C.F.; Coutrim, M.X. Method for the Determination of Benzene Metabolite t,t-Muconic Acid in Urine by HPLC-UV with an Ion Exclusion Column. Separations 2016, 3, 14. https://doi.org/10.3390/separations3020014
Gomes RDP, Sanson AL, Lobo FA, Afonso RJdCF, Coutrim MX. Method for the Determination of Benzene Metabolite t,t-Muconic Acid in Urine by HPLC-UV with an Ion Exclusion Column. Separations. 2016; 3(2):14. https://doi.org/10.3390/separations3020014
Chicago/Turabian StyleGomes, Rafaela De Paiva, Ananda Lima Sanson, Fabiana Aparecida Lobo, Robson José de Cássia Franco Afonso, and Mauricio Xavier Coutrim. 2016. "Method for the Determination of Benzene Metabolite t,t-Muconic Acid in Urine by HPLC-UV with an Ion Exclusion Column" Separations 3, no. 2: 14. https://doi.org/10.3390/separations3020014