Abstract
Chemotherapeutic agents are frequently used to treat various cancers, but the mechanisms mediating the cellular response to the drugs are still not fully understood. We previously reported that the nuclear translocation of serine/arginine protein kinases (SRPKs), triggered by the exposure of cells to DNA damage-inducers, plays a pivotal role in drug responsiveness. Here, we investigated the mechanism linking the nuclear accumulation of SRPK2 to the cisplatin treatment of HeLa cells. We present experimental evidence that nuclear SRPK2 acts downstream of Chk2 in the ATM/Chk2 cascade. The inhibition of ATM or Chk2 kinase activity by specific low-molecular-weight inhibitors restricted SRPK2 to the cytoplasm and conferred tolerance to cisplatin treatment. A similar effect was achieved by treating cells with SRPIN340, a selective SRPK1/2 inhibitor, thus confirming previous findings that kinase activity is indispensable for the nuclear import of SRPKs. These data add to previous findings that support a decisive role of SRPKs in coordinating cellular response to DNA damage.
1. Introduction
The SRPK family of protein kinases is highly conserved throughout eukaryotes [1]. SRPKs were initially considered to specifically phosphorylate the RS domains of SR splicing factors, thereby regulating pre-mRNA splicing [1]. However, several non-splicing functions of SRPKs have also been reported, and these could be ascribed to the phosphorylation of various substrates involved in multiple cellular processes [1,2].
Both SRPK1 and SRPK2 are predominantly localized in the cytoplasm of cancer cells, and they contain two conserved kinase domains that are interrupted by a unique spacer sequence. SRPK1 undergoes cytoplasmic-to-nuclear relocation by removing the spacer sequence, and this translocation may lead to effects including the alteration of splice site selection, the aggregation of splicing factors, and severe splicing defects [3,4]. The effects were found to be even more harmful in budding yeast, where the mis-localization of Sky1 (a yeast homologue of human SRPK1) to the nucleus, due to the removal of its spacer region, was toxic to cell growth [5]. In contrast to SRPK1, the deletion of the spacer domain did not seem to affect SRPK2 subcellular distribution [6].
Various signals induce the nuclear translocation of SRPKs. The epidermal growth factor (EGF) activates the phosphatidylinositol 3-kinase (PI3K)/Akt cascade, which, in turn, was shown to induce the nuclear translocation of SRPK1 and the reprogramming of alternative splicing [7]. A similar redistribution was also observed by osmotic shock [4], and we recently showed that the ATR/ATM signaling pathways were activated upon the treatment of HeLa and T24 cells with 5-FU, resulting in the phosphorylation of threonine 326 and serine 408 within the spacer region of SRPK1 and the subsequent re-localization of the kinase from the cytoplasm to the nucleus [8]. Most importantly, the nuclear accumulation of the kinase mediated 5-FU sensitivity. The phosphorylation of threonine 326 was also implicated in the PI3K/Akt-induced nuclear translocation of SRPK1 [7]. SRPK2 was shown to re-localize to the nucleus after genotoxic treatments of human neuroblastoma cells [6] and non-small cell lung cancer cells [9]. Edmond et al. showed that nuclear SRPK2 induced apoptosis [9], while in another report, Jang et al. demonstrated that the nuclear entry of SRPK2 in neurons resulted in cyclin D1 up-regulation, cell cycle re-entry, and neuronal apoptosis [10]. Moreover, the mTORC1 signaling pathway, when activated by insulin, promoted SRPK2 nuclear translocation and led to the splicing of lipogenic enzymes and de novo lipogenesis [11]. The phosphorylation of various residues in the C-terminal domain of SRPK2 seems to trigger the nuclear translocation of SRPK2 [6,10,11], but these phosphorylation events appear to be regulated by distinct molecular mechanisms. Acetylation/deacetylation has also been reported to be related to SRPK2 localization [9].
Given that SRPK1 is a downstream target of ATR/ATM, we sought to investigate whether SRPK2 also functions as a downstream effector in the DNA damage response signaling pathway orchestrated by the ATM and ATR kinases. In this respect, we treated HeLa cells with cisplatin, which (as previously shown) induced a dramatic nuclear accumulation of SRPK2 but barely affected the cytoplasmic localization of SRPK1 [8]. We now provide evidence that nuclear SRPK2 acts as a downstream effector of Chk2 in the ATM/Chk2 cascade. Blocking the activity of ATM (but not ATR) or Chk2 by specific low-molecular-weight inhibitors prevented the nuclear import of SRPK2. Furthermore, cells co-treated with cisplatin and ATM/Chk2 inhibitors exhibited higher degrees of drug tolerance, indicating that the nuclear activity of SRPK2 mediates the cytotoxic effect of cisplatin. These findings, coupled with our previous findings [8] and data from the literature showing that SRPK1 is associated with a large number of increasingly phosphorylated proteins following the exposure of cells to DNA damage-inducers [12], further support an important role of SRPKs in the DNA damage response pathway.
2. Materials and Methods
2.1. Cell Culture and Drug Treatments
HeLa cells were provided by the ATCC (American Type Culture Collection) and maintained in a DMEM medium supplemented with 10% (v/v) fetal bovine serum (FBS) and antibiotics/antimycotics. Cells were incubated at 37 °C with 5% CO2 and treated with cisplatin (5–40 µM), SRPIN340 (SRPK1/2 inhibitor, 5–80 µM), KU-55933 (ATM inhibitor, 2.5–15 μM), VE-821 (ATR inhibitor, 2.5–15 μM), and C3742 (Chk2 Inhibitor II hydrate, 0.5–20 μM) either alone or in combination, as is indicated. SRPIN340 was a kind gift from M. Gammons (MRC Laboratory of Molecular Biology, University of Cambridge, Cambridge UK). All other chemicals were purchased from Sigma-Aldrich.
2.2. Immunofluorescence Microscopy
HeLa cells were plated in 24-well plates (4 × 104 cells per well) with glass coverslips and subjected to treatment with various chemicals for the indicated time periods. After incubation, the coverslips containing cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min at room temperature, and excess aldehyde was quenched with 100 mM Tris-HCl pH 7.5. Cells were then permeabilized with 0.2% Triton X-100 in PBS for 10 min and blocked with 0.5% fish skin gelatin (FSG) in PBS for 30 min. Probing with the primary antibody (anti-SRPK2 monoclonal antibody, BD Biosciences, diluted 1:200), secondary antibody (FITC-conjugated goat anti-mouse, diluted 1:400; Molecular Probes), and DNA staining (propidium iodide) were performed as previously described [13]. After three washes, the coverslips were mounted with a mounting medium (0.01% p-phenylenediamine and 50% glycerol in PBS) and visualized with a Nikon confocal microscope using the EZ-C1 3.20 software.
2.3. Cell Fractionation and Western Blotting
The subcellular fractionation of HeLa cells was performed using the REAP method [14], as previously described [8]. Loading was adjusted to give equal cell numbers/lane for nuclear and cytoplasmic fractions. SRPK2 was detected by Western blotting using the anti-SRPK2 monoclonal antibody (BD Biosciences, diluted 1:1000) and an alkaline phosphatase-coupled goat anti-mouse secondary antibody (Millipore). Alkaline phosphatase activity was detected with BCIP (5-bromo-4-chloro-3-indolyl phosphate)/NBT (nitro blue tetrazolium)-plus substrate (Sigma-Aldrich). The intensity of the bands corresponding to cytoplasmic and nuclear SRPK2 was quantified using ImageJ software. To assess the purity of each fraction, a mouse monoclonal α-GAPDH antibody (a kind gift from A. Bakopoulou, School of Dentistry, Aristotle University) and a polyclonal anti-lamin (α-L1) antibody (a kind gift from S. Georgatos, Medical School, University of Ioannina) were used.
2.4. MTT Assays
HeLa cells were seeded in 96-well plates (3 × 103 HeLa cells per well), and after 24 h, they were exposed to cisplatin and kinase inhibitors (SRPIN340, KU-55933, VE-821, C3742) either alone or in combination, as is indicated. Viable cells were quantified by an (3-(4,5-imethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) metabolic assay, as previously described [15]. The absorbance readings were normalized to the untreated cells (exposed to medium only), which was set as 100% viability. Values shown represent the means ± SE of three independent experiments run in triplicate.
3. Results
In a previous report, we showed that the nuclear import of SRPK1 was closely correlated with the appearance of γH2AX foci and mediated 5-FU sensitivity [8]. To determine whether there was a link between translocation to the nucleus and the phosphorylation of SRPK1, we showed that 5-FU treatment triggered the phosphorylation of two residues within the spacer domain of the kinase, Thr326 and Ser408, in an ATR/ATM-dependent manner. Blocking ATR/ATM kinase activities by selective low-molecular-weight inhibitors impeded the nuclear translocation of SRPK1 and partially reversed the cytotoxic effects of the drug [8]. Interestingly, while the treatment of HeLa cells with 20 μg/mL of 5-FU resulted in the approximately complete nuclear accumulation of SRPK1, the nuclear levels of the kinase were slightly induced by 20 μM cisplatin. In contrast, the treatment of HeLa cells with cisplatin caused a significant relocation of SRPK2 to the nucleus [8], implying that distinct signaling pathways are activated in response to 5-FU and cisplatin. These data prompted us to investigate the molecular mechanism underlying the cisplatin-induced nuclear translocation of SRPK2.
At first, we addressed the issue of whether the nuclear activity of SRPK2 mediates the cytotoxic effect of cisplatin. In this respect, we co-treated HeLa cells with cisplatin and SRPIN340, a selective inhibitor of both SRPK1/2 [16] and assessed cell viability in parallel with the localization of SRPK2. As shown in Figure 1, cisplatin exhibited a strong cytotoxic effect on HeLa cells and induced the nuclear accumulation of SRPK2. The addition of SRPIN340 alone did not have a significant effect on cell viability (~60% cell viability at a concentration of 80 μM), while SRPK2 predominantly localized to the cytoplasm, similarly to control cells. However, the co-treatment of HeLa cells with SRPIN340 and cisplatin impeded the nuclear translocation of the kinase; at the same time, the cells showed significantly more drug tolerance. To validate our immunofluorescence data, we performed subcellular fractionation experiments. As shown in Figure 1 (right panels), similar subcellular localizations of SRPK2 were observed in these experiments. The above findings confirm the critical role of the kinase activity for the nuclear import of SRPK2 and indicate that the presence of SRPK2 in the nucleus is essential for the cytotoxicity caused by cisplatin [3,8,13].
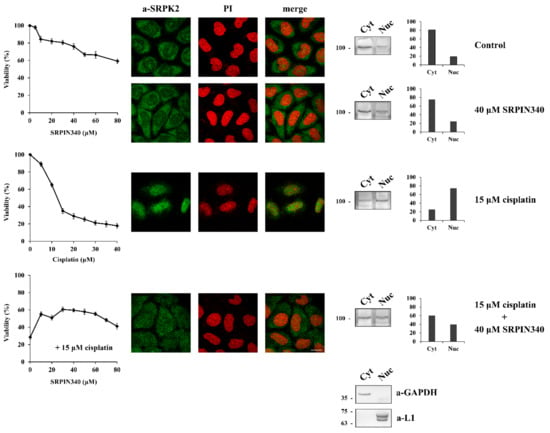
Figure 1.
SRPIN340 treatment prevents the nuclear import of SRPK2 in cisplatin-treated HeLa cells and protects the cells from the cytotoxic effects of the drug. HeLa cells were treated with SRPIN340 (5–80 µM) alone, cisplatin (5–40 µM) alone, and cisplatin (15 µM) combined with increasing concentrations of SRPIN340 (10–80 µM) for 24 h. The number of viable cells was measured with an MTT assay (left panel). Viable cells were quantified by normalizing the absorbance readings to the untreated control cells, which was set as 100% viability. Correspondingly, the figure shows fluorescent patterns of SRPK2 in HeLa cells treated for 24 h with SRPIN340 (40 µM) alone, cisplatin (15 µM) alone, and cisplatin (15 µM) combined with SRPIN340 (40 µM) (middle panel). SRPK2 was detected using the anti-SRPK2 monoclonal antibody, while nuclei were stained with propidium iodide (PI). Scale bar: 10 µm. A cell fractionation procedure was also employed to determine the distribution of SRPK2 (right panel). SRPK2 was detected in cytoplasmic (Cyt) and nuclear (Nuc) extracts by Western blotting using the anti-SRPK2 monoclonal antibody. The nuclear-to-cytoplasmic SRPK2 ratios in the Western blots shown were quantified using ImageJ software. Glyceraldehyde-3-phosphate dehydrogenase (a-GAPDH) and lamins (a-L1) were used as cytosolic and nuclear markers, respectively.
We next sought to investigate the signaling pathway underlying the nuclear import of SRPK2 after cisplatin treatment. Considering that SRPK1 was shown to be a target of the DNA damage response kinases of ATR (mainly) and ATM (to a lesser extent) [8], we sought to investigate whether ATR and/or ATM are also responsible for phosphorylating SRPK2. In this respect, there is only one serine residue in the SRPK2 sequence, Ser448, which is found within the consensus motif (S/TQ) targeted by ATM/ATR, while PhosphoNET, a predictor for identifying phosphorylation sites in human protein, suggests that ATR could be a candidate kinase that may target Ser448. As shown in Figure 2, blocking ATM by the specific inhibitor KU-55933 impeded the nuclear accumulation of SRPK2 in cisplatin-treated cells, whereas the co-treatment of HeLa cells with cisplatin and VE-821, a specific inhibitor of the ATR kinase, marginally affected the nuclear translocation of SRPK2. In line with the immunofluorescence data, only KU-55933, out of the two inhibitors, was able to confer a 1.6–1.8-fold resistance to the drug, thus suggesting that SRPK2 is not a direct target of ATR. Furthermore, the total lack of predicted ATM sites by PhosphoNET implies that SRPK2 is also not a direct target of ATM but rather a downstream effector of the ATM signaling pathway.
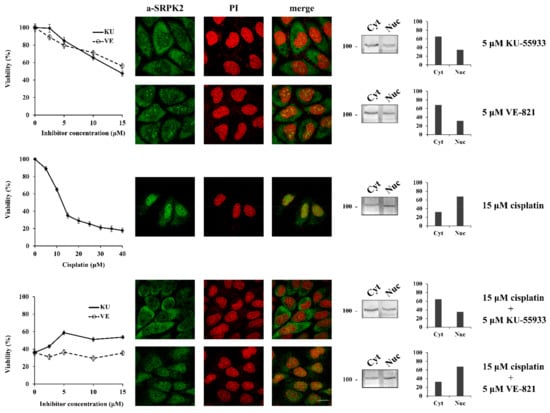
Figure 2.
ATM inhibition prevents the nuclear translocation of SRPK2 in cisplatin-treated cells and partially protects the cells from drug cytotoxicity. HeLa cells were treated with increasing concentrations of KU-55933 (2.5–15 µM) alone, VE-821 (2.5–15 µM) alone, cisplatin (5–40 µM) alone, and cisplatin (15 µM) combined with increasing concentrations of KU-55933 (2.5–15 µM) or VE-821 (2.5–15 µM) for 24 h. The number of viable cells was measured with an MTT assay (left panel). Viable cells were quantified by normalizing the absorbance readings to the untreated control cells, which was set as 100% viability. Correspondingly, the figure shows fluorescent patterns of SRPK2 in HeLa cells treated for 24 h with KU-55933 (5 µM) alone, VE-821 (5 µM) alone, cisplatin (15 µM) alone, and cisplatin (15 µM) combined with KU-55933 (5 µM) or VE-821 (5 µM) (middle panel). SRPK2 was detected using the anti-SRPK2 monoclonal antibody, while nuclei were stained with PI. Scale bar: 10 µm. The distribution of SRPK2 after each treatment was also determined by a cell fractionation procedure (right panel). SRPK2 was detected in cytoplasmic (Cyt) and nuclear (Nuc) extracts by Western blotting using the anti-SRPK2 monoclonal antibody. The nuclear-to-cytoplasmic SRPK2 ratios in the Western blots shown were quantified using ImageJ software.
The DNA damage response has traditionally been divided into two major kinase branches, the ATM/Chk2 and the ATR/Chk1 pathways [17]. The fact that specifically blocking ATM but not ATR prevented the nuclear import of SRPK2 in cisplatin-treated cells and partially reversed the cytotoxic effects of the drug, combined with the lack of ATM phosphorylation sites in the SRPK2 molecule, positions SRPK2 nuclear activity at a downstream point of the ATM/Chk2 cascade. To further confirm this suggestion, we co-treated HeLa cells with cisplatin and C3742 (Chk2 Inhibitor II hydrate), a specific inhibitor of Chk2. The addition of C3742 impeded the nuclear translocation of SRPK2 in cisplatin-treated cells and significantly protected the cells from drug cytotoxicity (more than 2-fold increase in cell viability at a concentration of 5 μM), a d the treatment of HeLa cells with the inhibitor alone did not affect the localization of SRPK2 and exhibited a mild effect on cell viability (Figure 3).
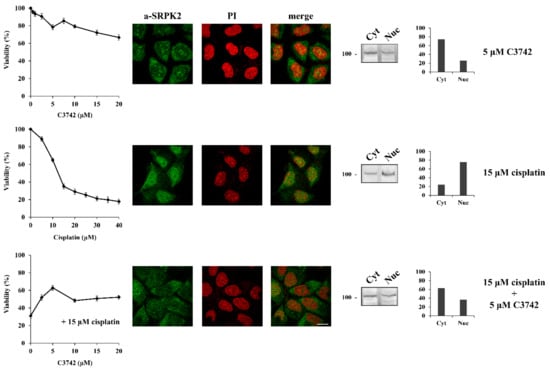
Figure 3.
Chk2 inhibition prevents the nuclear translocation of SRPK2 in cisplatin-treated cells and significantly protects the cells from drug cytotoxicity. HeLa cells were treated with increasing concentrations of C3742 (0.5–20 μM) alone, cisplatin (5–40 µM) alone, and cisplatin (15 µM) combined with increasing concentrations of C3742 for 24 h. The number of viable cells was measured with an MTT assay (left panel). Viable cells were quantified by normalizing the absorbance readings to the untreated control cells, which was set as 100% viability. Correspondingly, HeLa cells were treated with C3742 (5 µM) alone, cisplatin (15 µM) alone, and cisplatin (15 µM) combined with C3742 (5 µM) for 24 h and subjected to immunofluorescence staining with the anti-SRPK2 monoclonal antibody (middle panel). Nuclei were stained with PI. Scale bar: 10 µm. The distribution of SRPK2 after each treatment was also determined by a cell fractionation procedure (right panel). SRPK2 was detected in cytoplasmic (Cyt) and nuclear (Nuc) extracts by Western blotting using the anti-SRPK2 monoclonal antibody. The nuclear-to-cytoplasmic SRPK2 ratios in the Western blots shown were quantified using ImageJ software.
4. Discussion
Genotoxic agents exert their biological activity by activating downstream effectors. Of great importance are the identification of these effectors and the elucidation of the molecular mechanisms responsible for conveying the respective signals into the nucleus of cancer cells. In a previous report, we showed that SRPK1 translocated to the nucleus following the treatment of HeLa and T24 cells with 5-FU and mediated drug cytotoxicity. Mechanistically, this was achieved via the phosphorylation of two residues within the spacer domain of the kinase, Thr326 and Ser408, in an ATR/ATM-dependent manner [8]. However, treatment with cisplatin barely affected the localization of SRPK1 in the cytoplasm but resulted in the nuclear accumulation of SRPK2, implying that distinct signaling pathways are activated in response to these two chemotherapeutic agents. In this report, we provide evidence that in cisplatin-treated cells, while blocking the activity of ATM by a specific chemical inhibitor prevented the nuclear accumulation of SRPK2, blocking the activity of ATR had only a marginal impact on the translocation of the kinase to the nucleus. Interestingly, blocking the activity of Chk2, the main kinase that functions as a downstream effector of ATM, also hindered the nuclear translocation of SRPK2 in cisplatin-treated cells, though not as effectively as ATM inhibition. These data suggest that nuclear SRPK2 may act downstream of Chk2 in the ATM/Chk2 cascade. However, we cannot exclude the possibility that ATM either directly (by phosphorylating an unknown residue within SRPK2) or indirectly (through the activation of another pathway, apart from CHK2) also contributes to the nuclear translocation of SRPK2. In this respect, previous studies have shown that, in the presence of wild-type p53, the suppression of ATM or its downstream effector Chk2 actually protects tumor cells from genotoxic agents [18].
A critical question is whether SRPK2 is directly targeted by Chk2. The study of natural and artificial Chk2 substrates revealed a rather loose consensus site [19]. The leucine residue at the −5 position has been identified as the most critical residue for substrate recognition. An arginine residue at position −3 also seems to be required, but there are well-known Chk2 substrates with a glutamic acid residue at this position, such as p53 that is phosphorylated by Chk2 at Ser20 (ETFS20) [20]. In this respect, SRPK2 harbors an amino acid sequence that can be considered a ChK2 consensus motif, with Ser588 being the putative phosphorylatable amino acid and positions -5 and -3 occupied by Leu and Glu residues, respectively (583LFEPHS588). PhosphoNET suggests that CHK2 is a candidate kinase for targeting Ser588, and there is convincing evidence from the literature that the phosphorylation of this residue is required for the nuclear translocation of SRPK2 upon genotoxic treatments that induce DNA damage. More specifically, Vivarelli et al. showed that the treatment of neuroblastoma cells with genotoxic agents such as paraquat (a highly toxic pesticide that causes oxidative stress by inducing the overproduction of reactive oxygen species (ROS)), cisplatin, or irradiation induced the phosphorylation and subsequent nuclear translocation of SRPK2 [6]. The phosphorylation of Ser588 (Ser581 in this report since we used a construct expressing mouse SRPK2) was shown to be both necessary and sufficient for the nuclear accumulation of SRPK2 since a mutated SRPK2 in which Ser581 was substituted to Ala (S581A) was unable to re-localize to the nucleus upon paraquat treatment. On the other hand, the respective phosphomimetic mutation, S581D, was nuclear even in the absence of paraquat [6]. Ser581 (Ser588 in human SRPK2) was considered by Vivarelli et al. to be a casein kinase 2 (CK2) site on the basis of the previous mapping and analysis of phosphorylation sites in SRPK1 [21]; however, they clearly demonstrated that caffeine, an efficacious inhibitor of ATM/ATR kinases that does not inhibit the kinase activity of CK2, prevented SRPK2 nuclear translocation [6].
Other phosphorylation events have also been implicated in the nuclear translocation of SRPK2, with different functional outcomes. Jang et al. reported that Akt phosphorylates SRPK2 on Thr492, thus promoting its nuclear import [10]. Moreover, Lee et al. showed that mTORC1-activated S6K1 phosphorylated SRPK2 at Ser494, which then primed Ser497 phosphorylation by casein kinase 1 (CK1) [11]. Even though it is not known at present whether these phosphorylation events are related (and to what extent) to the genotoxic treatment of cells, an appealing idea is that the nuclear entry of SRPK2 may be mediated by different signaling pathways in distinct cellular environments. In each case, the balance between the nuclear and cytoplasmic SRPK2 levels, which is tightly regulated by the type and extent of phosphorylation, may dictate the final functional outcome. The translocation of a strictly regulated number of SRPK2 molecules may be beneficial for the cell by promoting specific biological responses, such as fine-tuning protein expression through the regulation of pre-mRNA splicing. On the other hand, the massive nuclear accumulation of SRPK2 induced by genotoxic/stress signals (chemotherapeutics, irradiation, oxidative stress) may result in toxic effects. In this respect, blocking the nuclear entry of SRPK2, either by inhibiting its activity—since kinase activity was shown to be required for the nuclear translocation of SRPKs [3,8,13]—or blocking the activity of the kinases that mediate SRPK2 phosphorylation can confer tolerance to cells. However, there is a threshold concentration of cisplatin (at the beginning of viability curve flattening: 25–35% viability) above which the cytotoxicity of the drug cannot be reversed.
There is increasing evidence that SRPKs may play a decisive role in various nuclear events via the phosphorylation of not only SR splicing factors but also other proteins not directly related to splicing. In an attempt to identify factors and delineate pathways that mediate the cellular response to DNA damage, Boeing et al. found that SRPK1 might influence transcription rates after UV irradiation, and SRPK1 was also found to be closely related to 20 proteins presenting UV-induced phosphorylation [12]. The most highly phosphorylated SRPK1-interacting proteins were found to be BCLAF1, THRAP3, and apoptotic chromatin condensation inducer 1 (ACIN1), all of which contain extended RS domains and were also shown to interact with RNA polymerase II. Furthermore, BCLAF1 and THRAP3 are considered critical players in maintaining DNA genomic stability by regulating the transcription, mRNA splicing, and export of a number of DNA-repair proteins in response to DNA damage [22]. Based on these data, Boeing et al. proposed a new role for SRPK1 and its interactors in transcription-related DNA damage response. It is not yet clear whether these proteins are phosphorylated and to what extent solely by SRPK1 or by both SRPK1 and SRPK2. In any case, the findings presented in this report and our previous report [8] strongly suggest that SRPKs play an essential role in the DNA damage response.
5. Conclusions
Based on our data, we propose that the subcellular localization of SRPK2 plays an important role in its biological functions (Figure 4). In untreated cells, SRPK2 mainly localizes in the cytoplasm and promotes cell growth. Treatment with cisplatin activates the ATM/CHK2 signaling pathway that mediates the phosphorylation and the subsequent nuclear translocation of SRPK2. Nuclear SRPK2 mediates drug cytotoxicity and reduces cell viability. Blocking SRPK2 activity by SRPIN340 or blocking ATM/Chk2 activity by specific chemical inhibitors results in the retention of SRPK2 in the cytoplasm and renders HeLa cells significantly more resistant to cisplatin.
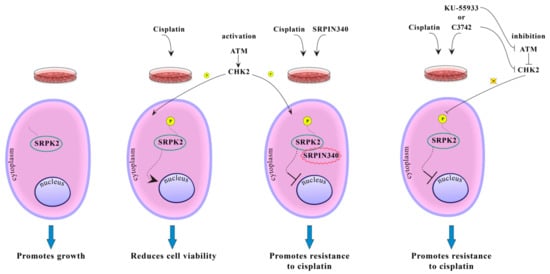
Figure 4.
Putative model showing the effect of phosphorylation on SRPK2 subcellular localization and function.
Author Contributions
Conceptualization, T.G. and E.N.; formal analysis, I.S. and A.K.; funding acquisition, T.G. and E.N.; investigation, I.S., A.K., A.M. and P.L.; methodology, I.S., A.K., and E.N.; resources, T.G. and E.N.; supervision, T.G. and E.N.; validation, I.S., A.K., A.M., P.L., T.G. and E.N.; visualization, I.S., A.K., A.M., P.L., T.G., and E.N.; writing—original draft, T.G. and E.N.; writing—review and editing, T.G. and E.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the EΔBM103 Program (MIS 5047934) co-financed by the European Union (European Social Fund-ESF) and Greek national funds through the Operational Program “Human Resources Development, Education and Lifelong Learning 2014–2020”.
Acknowledgments
We are grateful to Anastasia Tsingotjidou (Faculty of Veterinary Medicine, Aristotle University of Thessaloniki, Thessaloniki, Greece) for generously providing access to the Nikon d-Eclipse C1 CLSM.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Giannakouros, T.; Nikolakaki, E.; Mylonis, I.; Georgatsou, E. Serine-arginine protein kinases: A small protein kinase family with a large cellular presence. FEBS J. 2011, 278, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Bullock, N.; Oltean, S. The many faces of SRPK1. J. Pathol. 2017, 241, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.H.; Zhong, X.Y.; Hagopian, J.C.; Cruz, M.M.; Ghosh, G.; Feramisco, J.; Adams, J.A.; Fu, X.D. Regulated cellular partitioning of SR protein-specific kinases in mammalian cells. Mol. Biol. Cell 2006, 17, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.Y.; Ding, J.H.; Adams, J.A.; Ghosh, G.; Fu, X.D. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 2009, 23, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Siebel, C.W.; Feng, L.; Guthrie, C.; Fu, X.D. Conservation in budding yeast of a kinase specific for SR splicing factors. Proc. Natl. Acad. Sci. USA 1999, 96, 5440–5445. [Google Scholar] [CrossRef] [PubMed]
- Vivarelli, S.; Lenzken, S.C.; Ruepp, M.D.; Ranzini, F.; Maffioletti, A.; Alvarez, R.; Mühlemann, O.; Barabino, S.M. Paraquat modulates alternative pre-mRNA splicing by modifying the intracellular distribution of SRPK2. PLoS ONE 2013, 8, e61980. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Qiu, J.; Liu, W.; Zhou, Y.; Plocinik, R.M.; Li, H.; Hu, Q.; Ghosh, G.; Adams, J.A.; Rosenfeld, M.G.; et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol. Cell 2012, 47, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Sigala, I.; Koutroumani, M.; Koukiali, A.; Giannakouros, T.; Nikolakaki, E. Nuclear translocation of SRPKs is associated with 5-FU and cisplatin sensitivity in HeLa and T24 cells. Cells 2021, 10, 759. [Google Scholar] [CrossRef] [PubMed]
- Edmond, V.; Moysan, E.; Khochbin, S.; Matthias, P.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO J. 2011, 30, 510–523. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Fu, H.; Rees, H.; Yepes, M.; Levey, A.; Ye, K. Interaction of Akt-phosphorylated SRPK2 with 14-3-3 mediates cell cycle and cell death in neurons. J. Biol. Chem. 2009, 284, 24512–24525. [Google Scholar] [CrossRef]
- Lee, G.; Zheng, Y.; Cho, S.; Jang, C.; England, C.; Dempsey, J.M.; Yu, Y.; Liu, X.; He, L.; Cavaliere, P.M.; et al. Post-transcriptional regulation of de novo lipogenesis by mTORC1-S6K1-SRPK2 signaling. Cell 2017, 171, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Boeing, S.; Williamson, L.; Encheva, V.; Gori, I.; Saunders, R.E.; Instrell, R.; Aygün, O.; Rodriguez-Martinez, M.; Weems, J.C.; Kelly, G.P.; et al. Multiomic analysis of the UV-induced DNA damage response. Cell Rep. 2016, 15, 1597–1610. [Google Scholar] [CrossRef]
- Koutroumani, M.; Papadopoulos, G.E.; Vlassi, M.; Nikolakaki, E.; Giannakouros, T. Evidence for disulfide bonds in SR Protein Kinase 1 (SRPK1) that are required for activity and nuclear localization. PLoS ONE 2017, 12, e0171328. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Bose, P.; Leong-Quong, R.Y.; Fujita, D.J.; Riabowol, K. REAP: A two minute cell fractionation method. BMC Res. Notes 2010, 3, 294. [Google Scholar] [CrossRef] [PubMed]
- Sigala, I.; Tsamis, K.I.; Gousia, A.; Alexiou, G.; Voulgaris, S.; Giannakouros, T.; Kyritsis, A.P.; Nikolakaki, E. Expression of SRPK1 in gliomas and its role in glioma cell lines viability. Tumour Biol. 2016, 37, 8699–8707. [Google Scholar] [CrossRef]
- Fukuhara, T.; Hosoya, T.; Shimizu, S.; Sumi, K.; Oshiro, T.; Yoshinaka, Y.; Suzuki, M.; Yamamoto, N.; Herzenberg, L.A.; Hagiwara, M. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. USA 2006, 103, 11329–11333. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Yaffe, M.B. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 2009, 21, 245–255. [Google Scholar] [CrossRef]
- Jiang, H.; Reinhardt, H.C.; Bartkova, J.; Tommiska, J.; Blomqvist, C.; Nevanlinna, H.; Bartek, J.; Yaffe, M.B.; Hemann, M.T. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009, 23, 1895–1909. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.L.; Hupp, T.R. The regulation of CHK2 in human cancer. Oncogene 2004, 23, 8411–8418. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seo, G.J.; Kim, S.E.; Lee, Y.M.; Lee, J.W.; Lee, J.R.; Hahn, M.J.; Kim, S.T. Determination of substrate specificity and putative substrates of Chk2 kinase. Biochem. Biophys. Res. Commun. 2003, 304, 339–343. [Google Scholar] [CrossRef]
- Mylonis, I.; Giannakouros, T. Protein kinase CK2 phosphorylates and activates the SR protein-specific kinase 1. Biochem. Biophys. Res. Commun. 2003, 301, 650–656. [Google Scholar] [CrossRef]
- Meng, X.; Yang, S.; Camp, V.J.A. The interplay between the DNA Damage Response, RNA processing and extracellular vesicles. Front. Oncol. 2020, 9, 1538. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).