Modeling of the Free Radical Copolymerization Kinetics of n-Butyl Acrylate, Methyl Methacrylate and 2-Ethylhexyl Acrylate Using PREDICI®



Abstract
:1. Introduction
2. Experimental Section
3. Model Development
3.1. Initiation
3.2. Propagation
3.3. Chain Transfer
3.4. Cross-Propagation and Cross-Chain Transfer
3.5. Backbiting of BA and Reactions with Mid-Chain Tertiary Radicals
3.6. Termination
3.7. Diffusion-Controlled Effects
3.8. Parameter Estimation Strategy
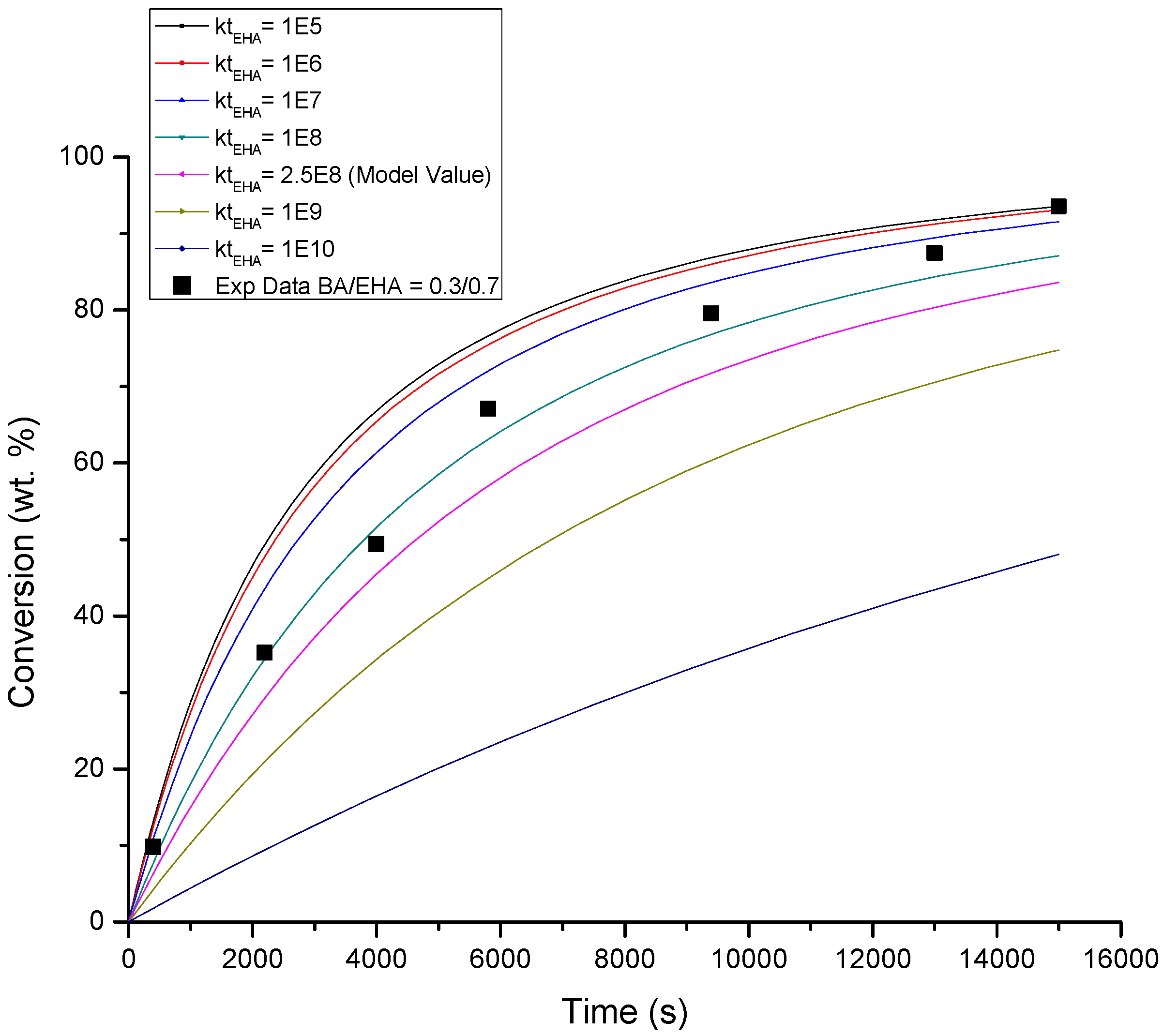
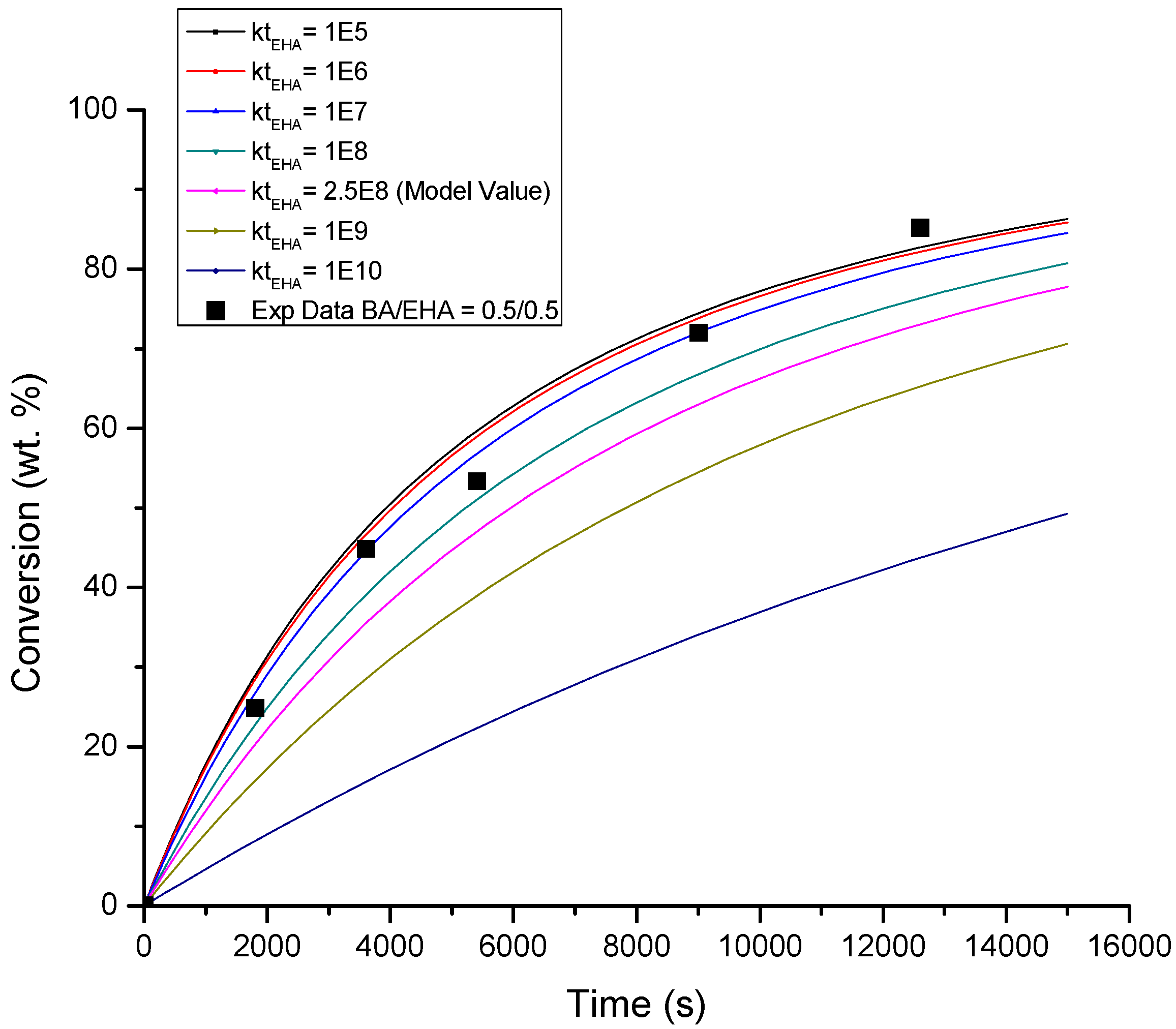
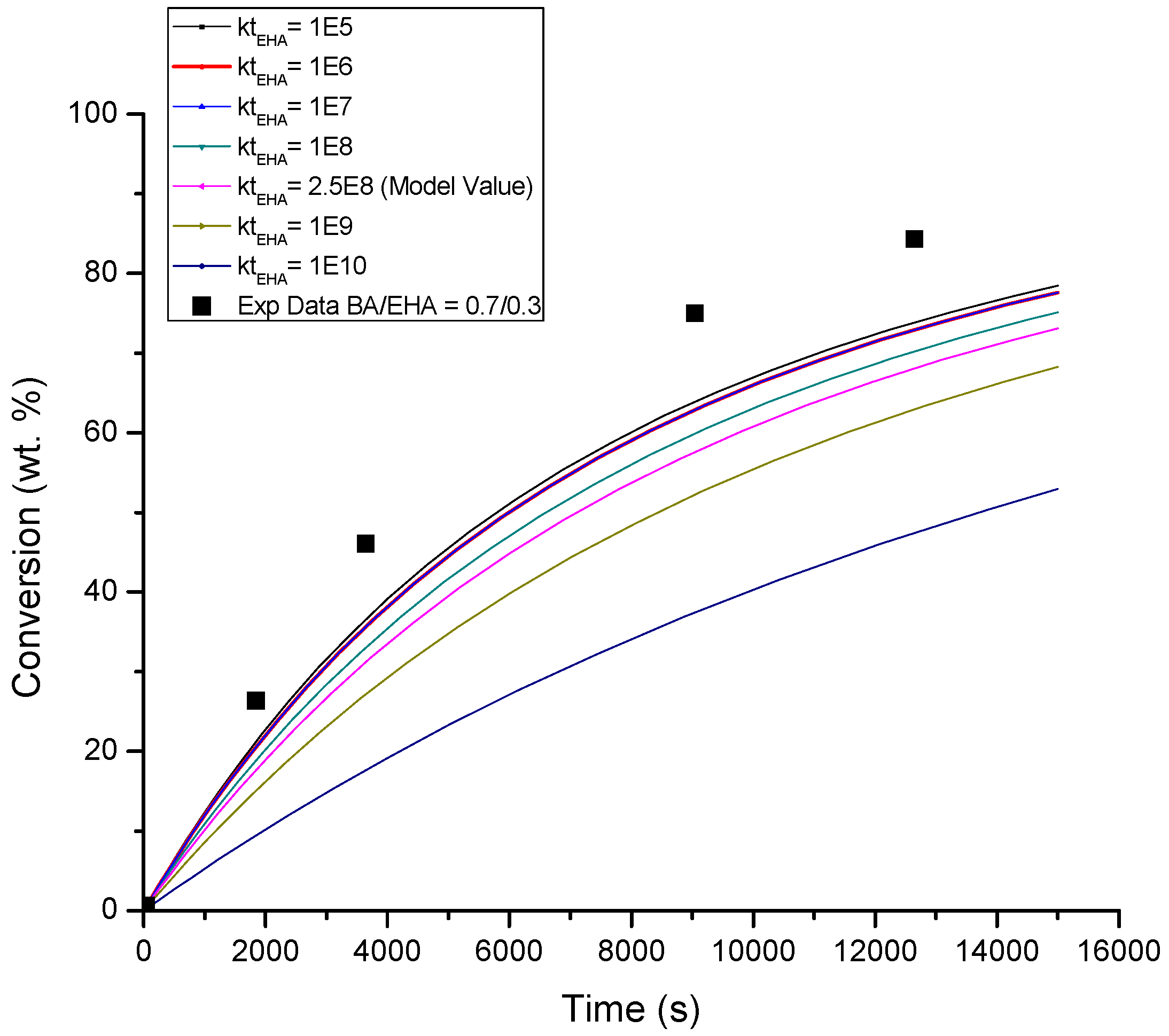
4. Results and Discussion
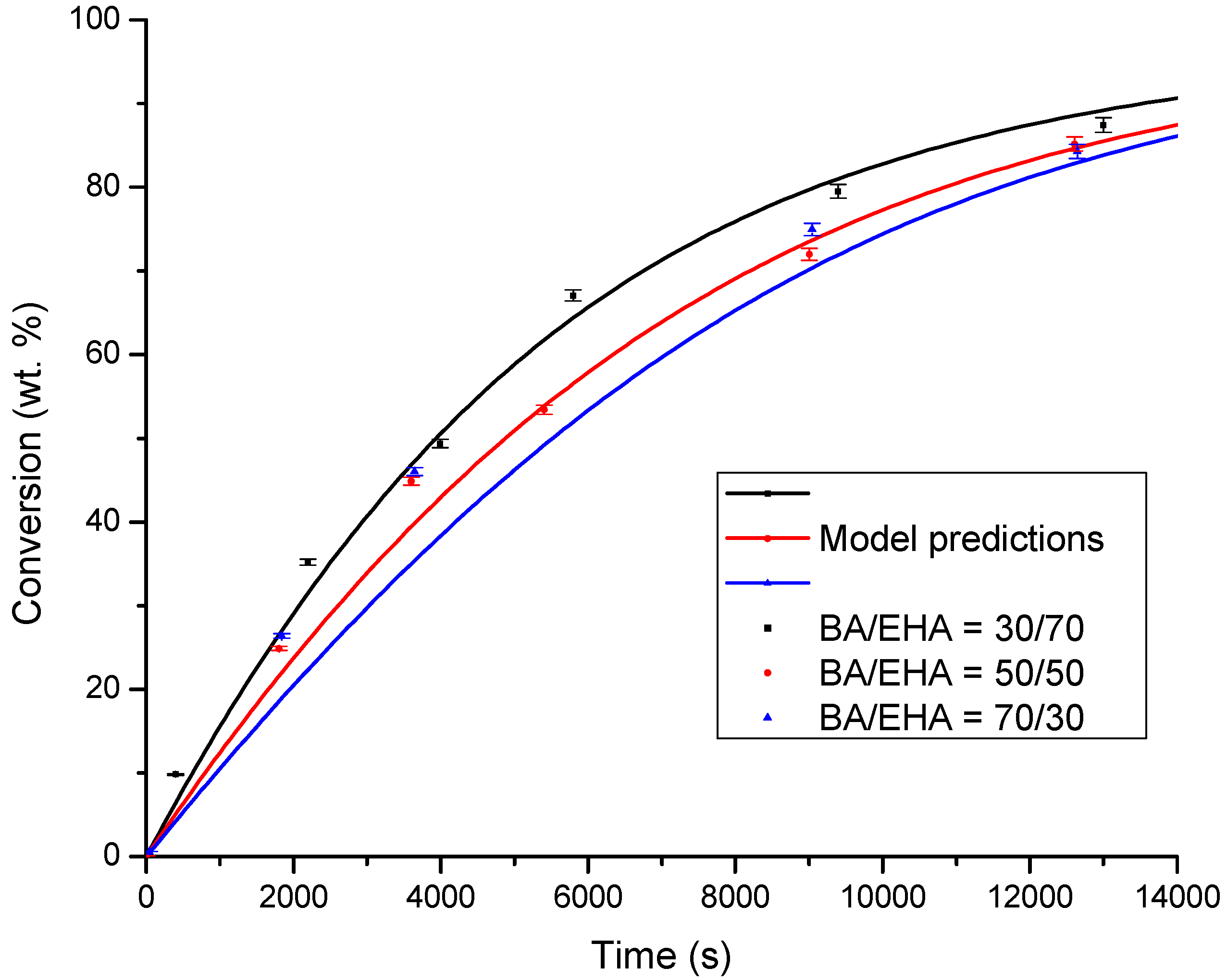
4.1. Conversion versus Time Profiles
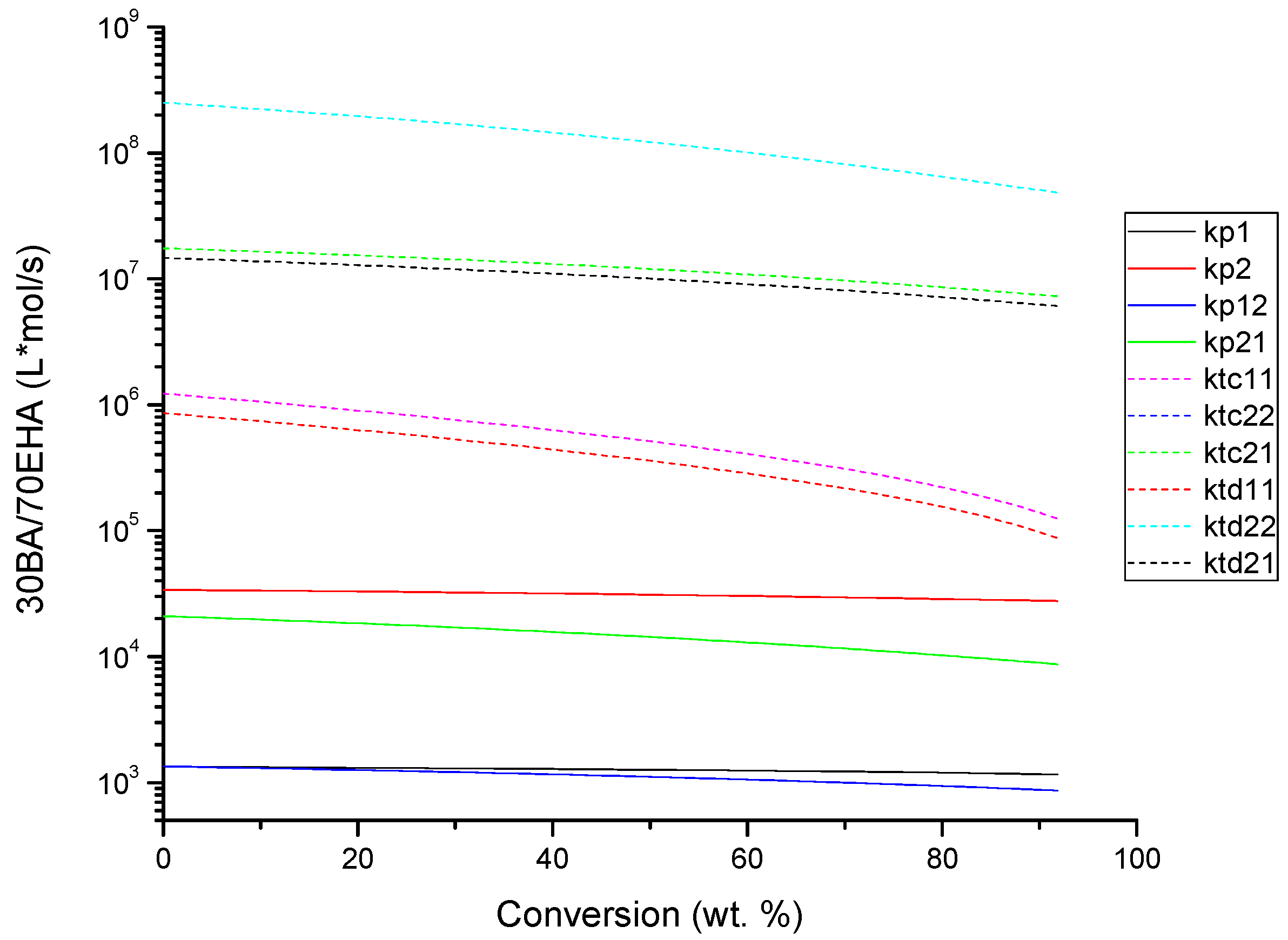
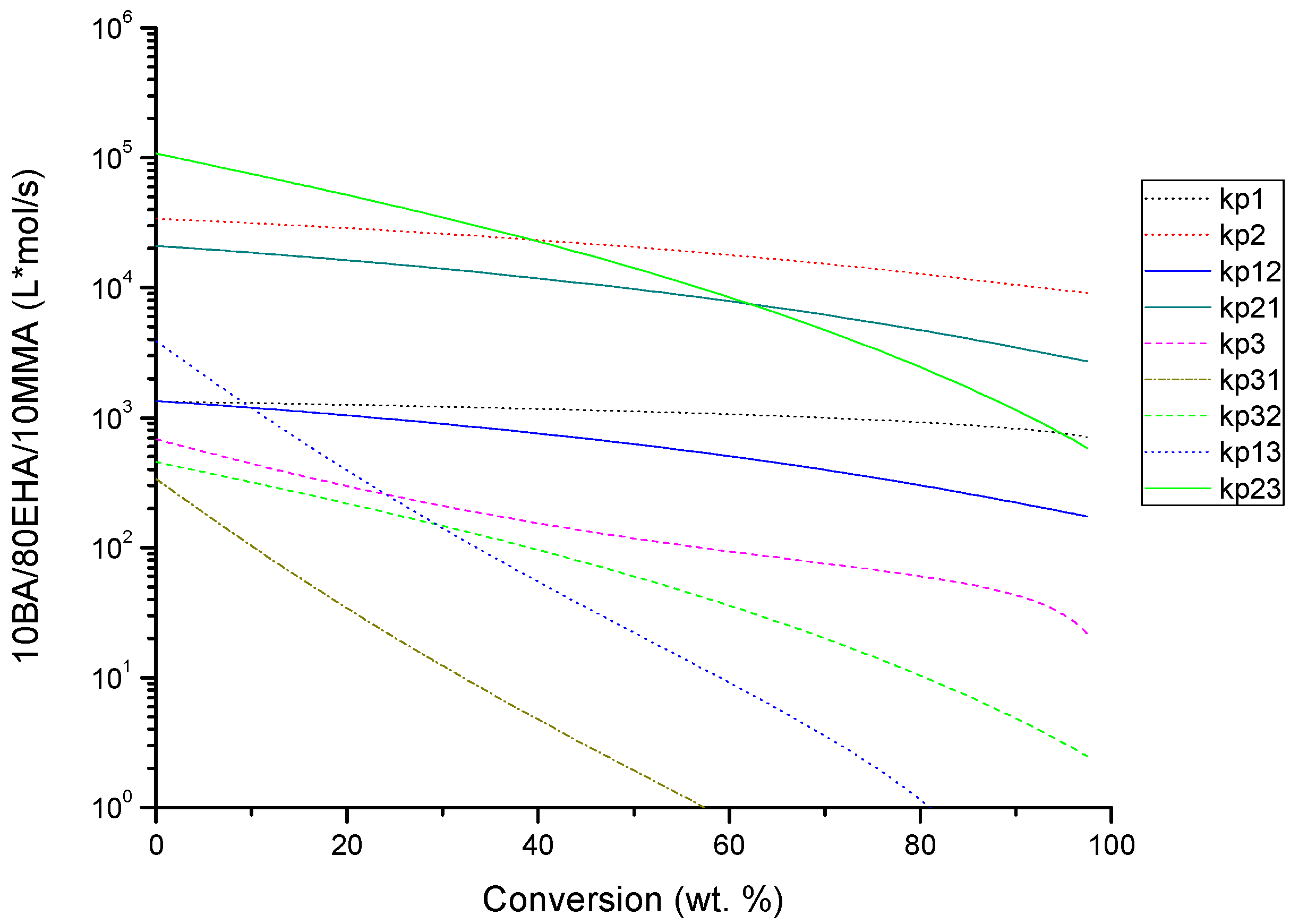
4.2. Evolution of Kinetic Rate Coefficients Due to DC Effects
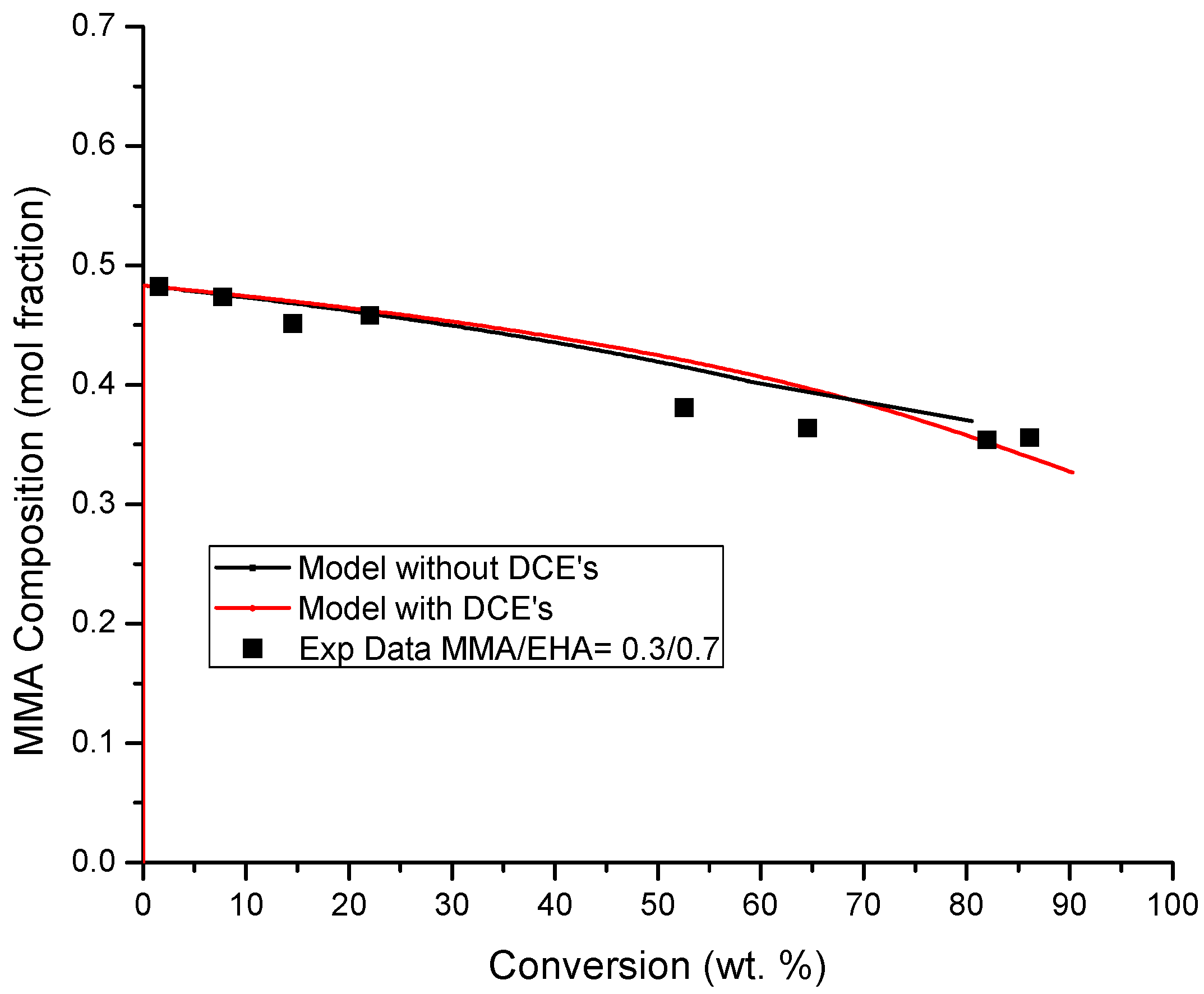
4.3. Copolymer Composition versus Conversion Profiles
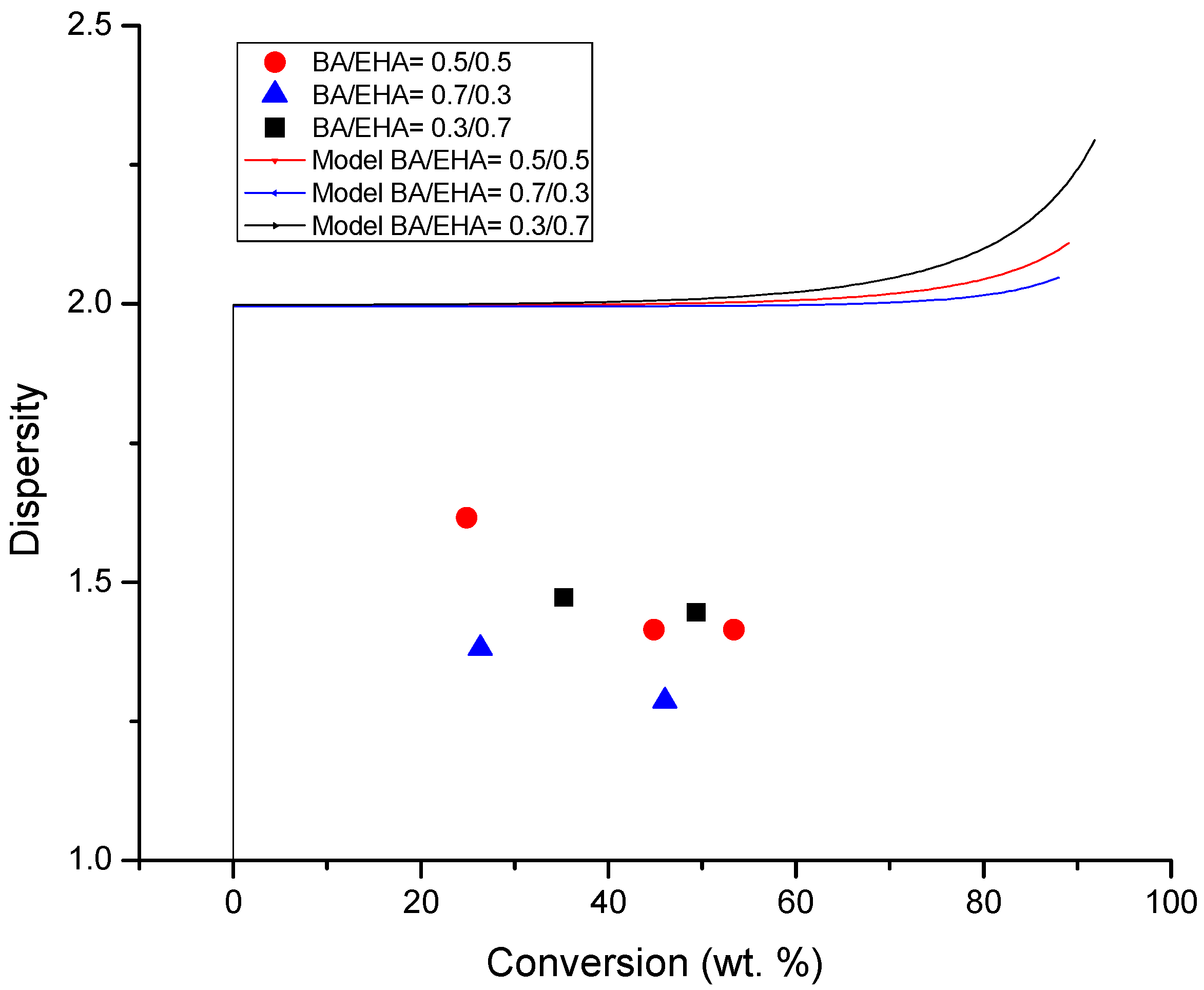
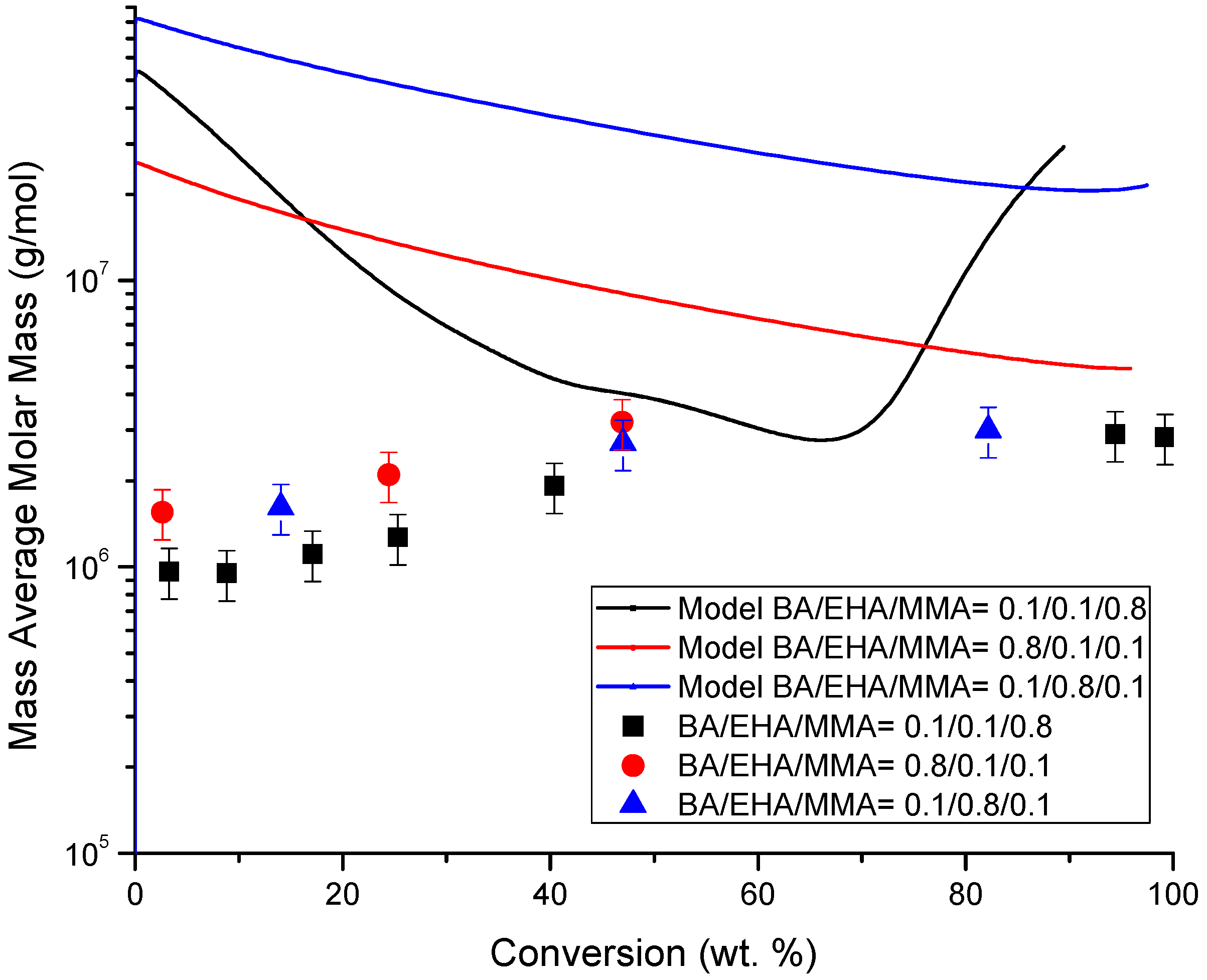
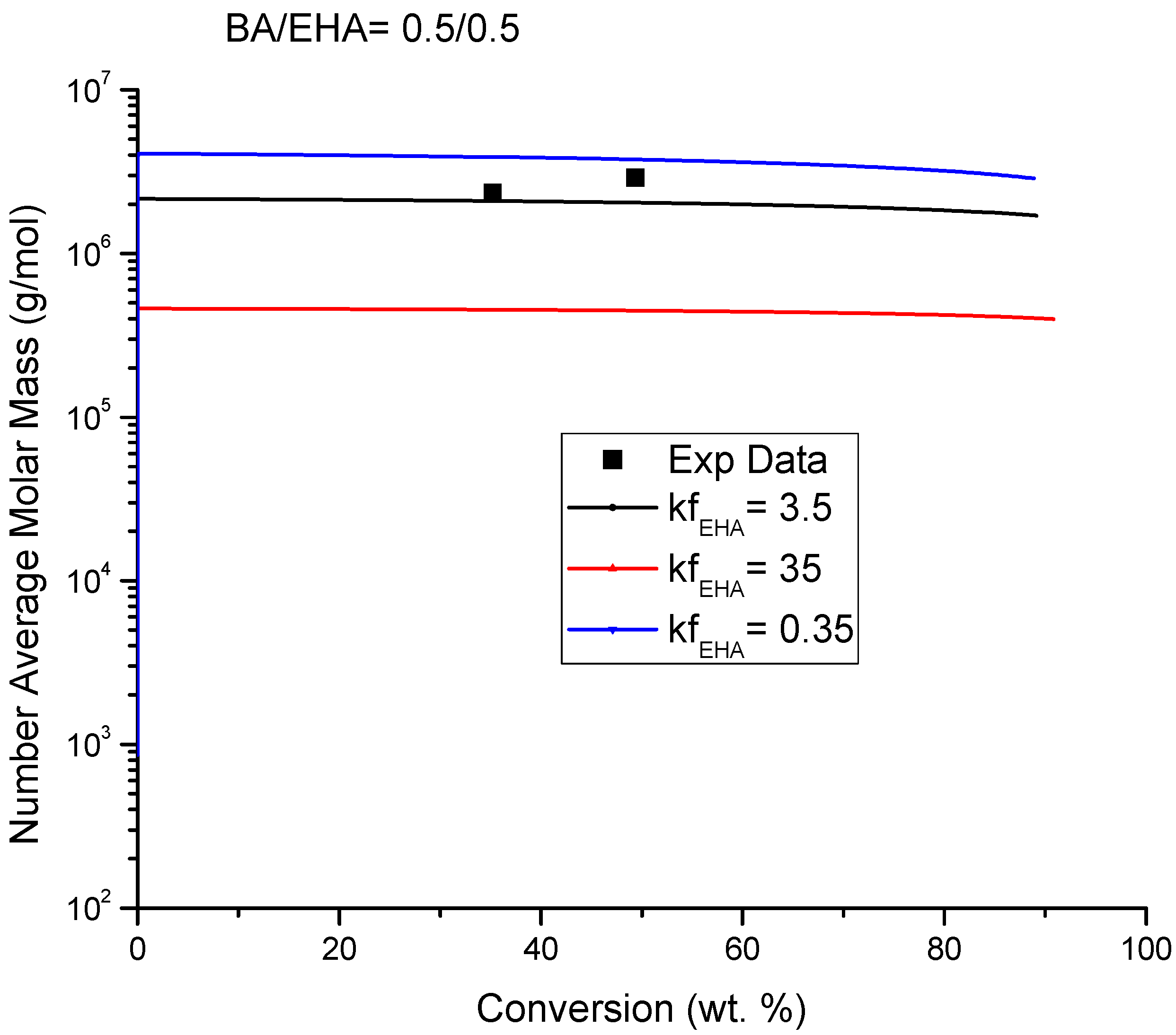
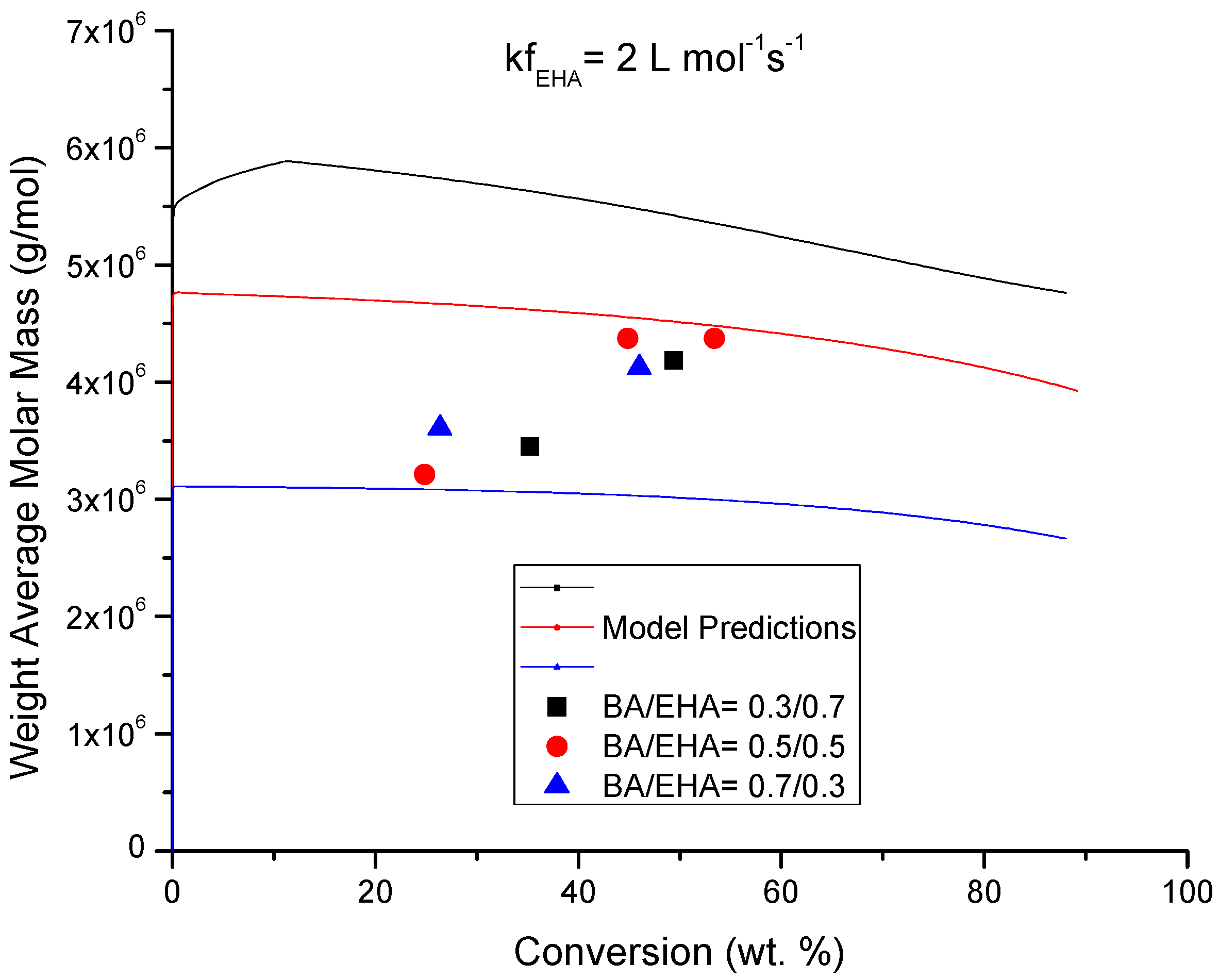
4.4. Molecular Weight versus Conversion Profiles
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Nomenclature
| BA | n-Butyl Acrylate |
| EHA | 2-Ethylhexyl Acrylate |
| MMA | Methyl Methacrylate |
| DC | Diffusion-Controlled (Effects) |
| PSA | Pressure Sensitive Adhesive |
| AIBN | Azobisisobutyronitrile |
| fi | Monomer Fraction for species i |
| I | Initiator |
| I• | Primary Radical |
| Mi | Monomer for species i |
| Pi(s)• | Secondary Radical for species i |
| Qi(s)• | Tertiary Radical for species i |
| Pi | Polymer for species i |
| Pi(s)• | Secondary Radical for species i |
| ki | Initiator Decomposition Kinetic Rate Coefficient |
| f | Initiator Efficiency Factor |
| kpi | Propagation Kinetic Rate Coefficient for species i |
| kpij | Cross-Propagation Kinetic Rate Coefficient for species iand j |
| kfi | Chain Transfer Kinetic Rate Coefficient for species i |
| kfij | Cross-Chain Transfer Kinetic Rate Coefficient for species i |
| kbb | Backbiting Kinetic Rate Coefficient for n-Butyl Acrylate |
| kti | Global Termination Kinetic Rate Coefficient for species i |
| ktci | Termination by Combination Kinetic Rate Coefficient for species i |
| ktdi | Termination by Disproportionation Kinetic Rate Coefficient for species i |
| kfitert | Tertiary Radical Chain Transfer Kinetic Rate Coefficient for species i |
| kpitert | Tertiary Radical Propagation Kinetic Rate Coefficient for species i |
| ktcij | Cross-Termination by Combination Kinetic Rate Coefficient for species i |
| ktdij | Cross-Termination by Disproportionation Kinetic Rate Coefficient for species i |
| rij | Reactivity Ratio beween species iand j |
| β | Attraction/Separation Overlap Factor in free-volume theory |
| Expansion Coefficient for Monomer | |
| Expansion Coefficient for Polymer | |
| vf | Free Volume at a given calculated time |
| vfo | Free volume at initial conditions |
| Vmi | Monomer Volume for species i |
| Vpi | Polymer Volume for species i |
| TgP | Polymer Glass Temperature |
| TgM | Monomer Glass Temperature |
| T | System Temperature |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BA βt/βp | EHA βt/βp | BA/EHA βt/βp | Figure | |
|---|---|---|---|---|
| Set 1 | 32 | 16 | 4 | A4 |
| Set 2 (Model) | 16 | 8 | 2 | A5 (2) |
| Set 3 | 8 | 4 | 1 | A6 |









References
- Dubé, A.M.; Zapata-González, I. Ch. 6: Copolymerization. In Handbook of Polymer Synthesis, Characterization, and Processing; Saldívar-Guerra, E., Vivaldo-Lima, E., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 105–125. [Google Scholar]
- Learner, T.J.S.; Krueger, J.W.; Schilling, M.R. Modern Paints Uncovered. In Proceedings of the Modern Paints Uncovered Symposium, London, UK, 16–19 May 2006; Learner, J.S., Smithen, P., Krueger, J.W., Schilling, M.R., Eds.; Getty Conservation Institute: London, UK, 2007. [Google Scholar]
- Moghbeli, M.R.; Zamir, S.M.; Molaee, B. Resultant Synergism in the Shear Resistance of AcrylicPressure-Sensitive Adhesives Prepared by EmulsionPolymerization of n-Butyl Acrylate/2-Ethyl HexylAcrylate/Acrylic Acid. J. Appl. Polym. Sci. 2008, 108, 606–613. [Google Scholar] [CrossRef]
- Srivastava, S. Co-polymerization of Acrylates. Des. Monomers Polym. 2009, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Benedek, I. Pressure-Sensitive Adhesives and Applications, 2nd ed.; Springer-Verlag: Leipzig, Germany, 2004; pp. 5–10. [Google Scholar]
- Gao, J.; Penlidis, A. A Comprehensive Simulator Database Package for Reviewing Free-Radical Copolymerizations. Macromol. React. Eng. 1998, 38, 651–780. [Google Scholar] [CrossRef]
- Beuermann, S.; Paquet, D.A.; McMinn, J.H.; Hutchinson, R.A. Determination of Free-Radical Propoagation Rate Coefficients of Butyl, 2-Ethylhexyl, and Dodecyl Acrylates by Pulsed-Laser Polymerization. Macromolecules 1996, 29, 4206–4215. [Google Scholar] [CrossRef]
- Agirre, A.; Nase, J.; Degrandi, E.; Creton, C.; Asua, J.M. MiniemulsionPolymerization of 2-Ethylhexyl Acrylate. Polymer Architecture Control and Adhesion Properties. Macromolecules 2010, 43, 8924–8932. [Google Scholar] [CrossRef]
- Capek, I.; Juranicova, V.; Barton, J.; Asua, J.M.; Ito, K. Microemulsion Radical Polymerization ofAlkyl Acrylates. Polym. Int. 1997, 43, 1–7. [Google Scholar] [CrossRef]
- Plessis, C.; Arzamendi, G.; Alberdi, J.M.; Agnely, M.; Leiza, J.R.; Asua, J.M. Intramolecular Chain Transfer to Polymer in the Emulsion Polymerization of 2-EthylhexylAcrylate. Macromolecules 2001, 34, 6138–6143. [Google Scholar] [CrossRef]
- Heatley, F.; Lovell, P.A.; Yamashita, T. Chain Transfer to Polymer in Free-Radical Solution Polymerization of2-Ethylhexyl Acrylate Studied by NMR Spectroscopy. Macromolecules 2001, 34, 7636–7641. [Google Scholar] [CrossRef]
- Couvreur, L.; Piteau, G.; Castignolles, P.; Tonge, M.; Coutin, B.; Charleux, B.; Vairon, J.P. Pulsed-Laser Radical Polymerization and Propagation Kinetic Parameters of Some Alkyl Acrylates. Macromol. Symp. 2001, 174, 197–207. [Google Scholar] [CrossRef]
- Junkers, T.; Schneider-Baumann, M.; Koo, S.S.; Castignolles, P.; Barner-Kowollik, C. Determination of propagation rate coefficients for methyl and 2-ethylhexyl acrylate via high frequency PLP-SEC under consideration of the impact of chain branching. Macromolecules 2010, 43, 10427–10434. [Google Scholar] [CrossRef]
- Gabriel, V.A.; Dubé, M.A. Bulk Free-Radical Co- and Terpolymerization of n-Butyl Acrylate/2-Ehylhexyl Acrylate/MethylMethacrylate. Macromol. React. Eng. 2018. [Google Scholar] [CrossRef]
- Bicak, N.; Ozlem, M. Graft Copolymerization of Butyl Acrylate and 2-Ethyl HexylAcrylate from Labile Chlorines of Poly(vinyl chloride) byAtom Transfer Radical Polymerization. J. Polym. Sci. 2003, 41, 3457–3462. [Google Scholar] [CrossRef]
- Percec, V.; Popov, A.V.; Ramirez-Castillo, E.; Hinojosa-Falcon, L.A. Synthesis of Poly(vinyl chloride)-b-Poly(2-ethylhexylacrylate)-b-Poly(vinyl chloride) by the Competitive Single-Electron-Transfer/Degenerative-Chain-Transfer MediatedLiving Radical Polymerization of Vinyl Chloride Initiatedfromα,ω-Di(iodo)poly(2-ethylhexyl acrylate) and Catalyzedwith Sodium Dithionite in Water. J. Polym. Sci. 2005, 43, 2276–2280. [Google Scholar]
- Vidts, K.R.M.; Dervaux, B.; Prez, F.E.D. Block, Blocky Gradient and Random Copolymers of 2-Ethylhexyl Acrylate and Acrylic Acid by Atom Transfer Radical Polymerization. Polymer 2006, 47, 6028–6037. [Google Scholar] [CrossRef]
- Haloi, D.J.; Singha, N.K. Synthesis of Poly(2-ethylhexyl acrylate)/Clay Nanocompositeby In Situ Living Radical Polymerization. J. Polym. Sci. 2011, 49, 1564–1571. [Google Scholar] [CrossRef]
- Achilias, D.S. Review of Modeling of Diffusion Controlled Polymerization Reactions. Macromol. Theory Simul. 2007, 16, 319–347. [Google Scholar] [CrossRef]
- Buback, M.; Huckestein, B.; Russell, G.T. Modeling of termination in intermediate and high conversion free radical polymerizations. Macromol. Chem. Phys. 1994, 195, 539–554. [Google Scholar] [CrossRef]
- Stickler, M.; Panke, D.; Hamielec, A.E. Polymerization of Methyl Methacrylate up to High Degrees of Conversion: Experimental Investigation of the Diffusion-controlled Polymerization. J. Polym. Sci. Polym. Chem. 1984, 22, 2243–2253. [Google Scholar] [CrossRef]
- Arzamendi, G.; Plessis, C.; Leiza, J.R.; Azua, J.M. Effect of the Intramolecular Chain Transfer to Polymer on PLP/SEC Experiments of Alkyl Acrylates. Macromol. Theory Simul. 2003, 12, 315–324. [Google Scholar] [CrossRef]
- Lena, J.B.; Deschamps, M.; Sciortino, N.F.; Masters, S.L.; Squire, M.A.; Russell, G.T. Effects of Chain Transfer Agent and Temperature on Banching and β-Scission in Radical Polymerization of 2-Ethylhexyl Acrylate. Macromol. Chem. Phys. 2018, 219, 1700579. [Google Scholar] [CrossRef]
- Plessis, C.; Arzamendi, G.; Alberdi, J.M. Evidence of Branching in Poly(butyl acrylate) Produced in Pulsed Laser Polymerization Experiments. Macromol. Rapid Commun. 2003, 24, 173–177. [Google Scholar] [CrossRef]
- Ren, S.; Vivaldo-Lima, E.; Dubé, M.A. Modeling of the Copolymerization Kinetics of n-Butyl Acrylate and D-Limonene Using PREDICI. Processes 2015, 4, 1. [Google Scholar] [CrossRef]
- Zhu, S.; Hamielec, A.E. Heat effects for free-radical polymerization in glass ampoules reactors. Polymer 1991, 32, 3021–3025. [Google Scholar] [CrossRef]
- Zoller, A.; Gigmes, D.; Guillaneuf, Y. Simulation of radical polymerization of methyl methacylate at room temperaturre using a tertiary amine/BPO initiating system. Polym. Chem. 2015, 6, 1–3. [Google Scholar] [CrossRef]
- Wulkow, M. Computeir Aided Modeling of Polymer Reaction Engineering-The Status of Predici, 1-Simulation. Macromol. React. Eng. 2008, 2, 461–494. [Google Scholar] [CrossRef]
- Peikert, P.; Pflug, K.M.; Busch, M. Modeling of High-Pressure Ethene Homo- and Copolymerization. Chem. Ing. Tech. 2019, 91, 1–6. [Google Scholar]
- Louie, B.M.; Carratt, G.M.; Soong, D.S. Modeling the free radical solution and bulk polymerization of methyl methacrylate. J. Appl. Polym. Sci. 1985, 30, 3985–4012. [Google Scholar] [CrossRef]
- Penlidis, A. Watpoly Database; University of Waterloo: Waterloo, ON, Canada, 1992. [Google Scholar]
- Rantow, E.S.; Soroush, M.; Grady, M.C.; Kalfas, G.A. Spontaneous polymerization and chain microstructure evolution in high-temperature solution polymerization of n-butyl acrylate. Polymer 2006, 47, 1423–1435. [Google Scholar] [CrossRef]
- Dorschner, D. Multicomponent Free Radical Polymerization Model Refinements and Extensions with Depropagations. Master’s Thesis, University of Waterloo, Department of Chemical Engineering, Waterloo, ON, Canada, 2010. [Google Scholar]
- McManus, N.T.; Dubé, M.A. PeNLIDIS. Polym. React. Eng. 1999, 7, 131. [Google Scholar] [CrossRef]
- Hungenberg, K.D.; Wulkow, M. Modeling and Simulation in Polymer Reaction Engineering (A Modular Approach); Wiley-VCH: Weinheim, Germany, 2018; pp. 92–256. [Google Scholar]
- Scott, A.J.; Penlidis, A. Ready-to-Use Computattional Package for Copolymerzation Reactivity Ratio Estimation: Improved Acces to the Error-in-Variables-Model. Process 2017, 5, 1–41. [Google Scholar]
- Peck, A.N.F.; Hutchinson, R.A. Secondaryreactions in thehigh-temperature free radical polymerizationofbutyl acrylate. Macromolecules 2004, 37, 5944–5951. [Google Scholar] [CrossRef]
- Ahmad, N.M.; Heatley, F.; Lovell, P.A. Chain Transfer Copolymer in Free-radical Solution Polymerizationof n-Butyl Acrylatestudiedby NMR spectroscopy. Macromolecules 1998, 31, 2822–2827. [Google Scholar] [CrossRef]
- Theis, A.; Feldermann, A.; Charton, N.; Davies, T.P.; Stenzel, M.H.; Kowollik, C.B. Living free radical polymerization (RAFT) of dodecyl acrylate: Chain length dependent termination, mid-chain radicals and monomer reaction order. Polymer. 2005, 46, 6797–6809. [Google Scholar] [CrossRef]
- Hamielec, A.E. Recent Developments in Free Radical Polymerization at High Conversion Diffusion Controlled Termination and Propagation. Chem. Eng. Commun. 1983, 24, 1–19. [Google Scholar] [CrossRef]
- D’hooge, R.D.; Reyniers, M.F.; Marin, G.B. The Crucial Roles of Diffusional Limitantion in Controlled Radical Polymerization. Macromol. React. Eng. 2013, 7, 362–379. [Google Scholar]
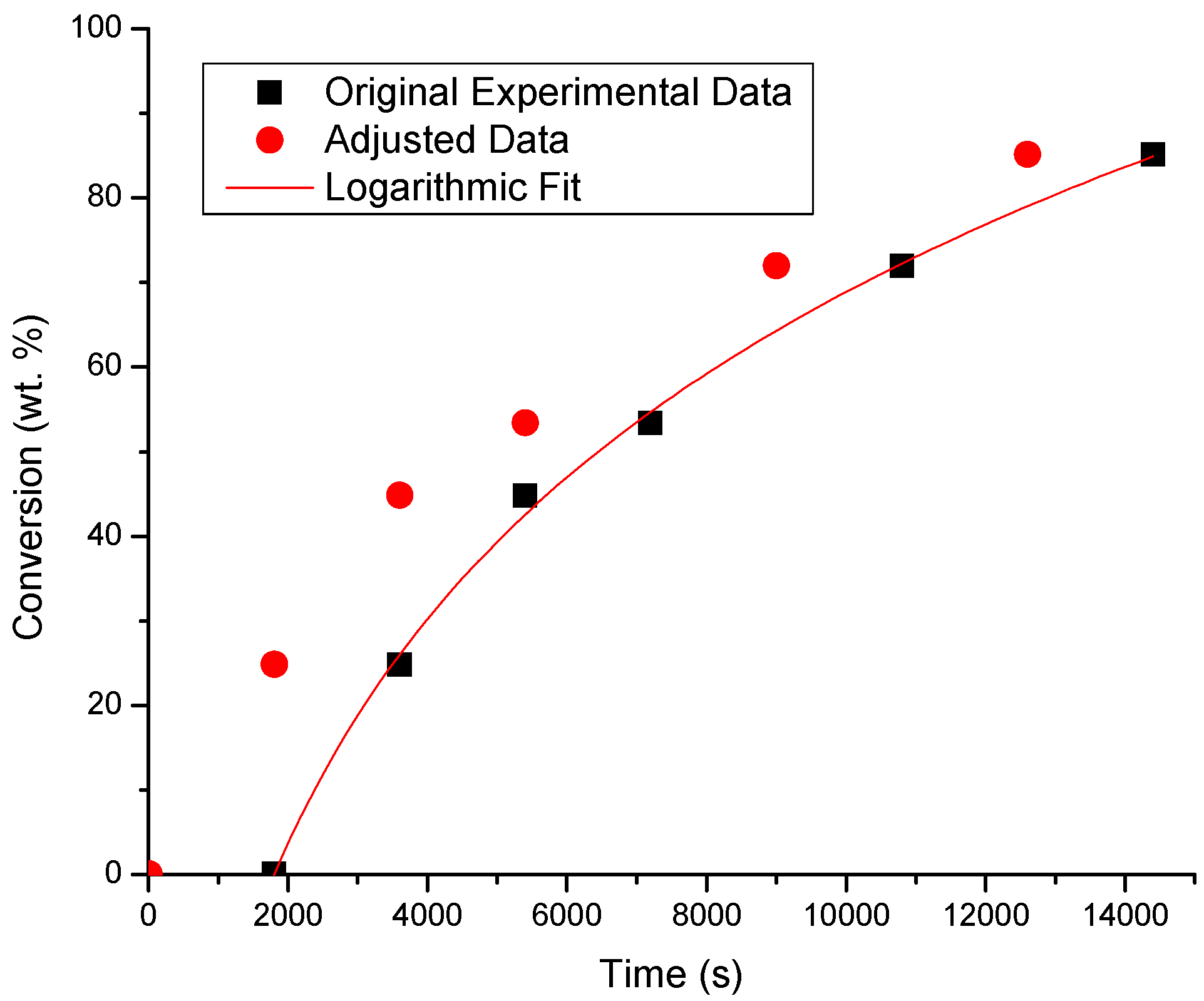
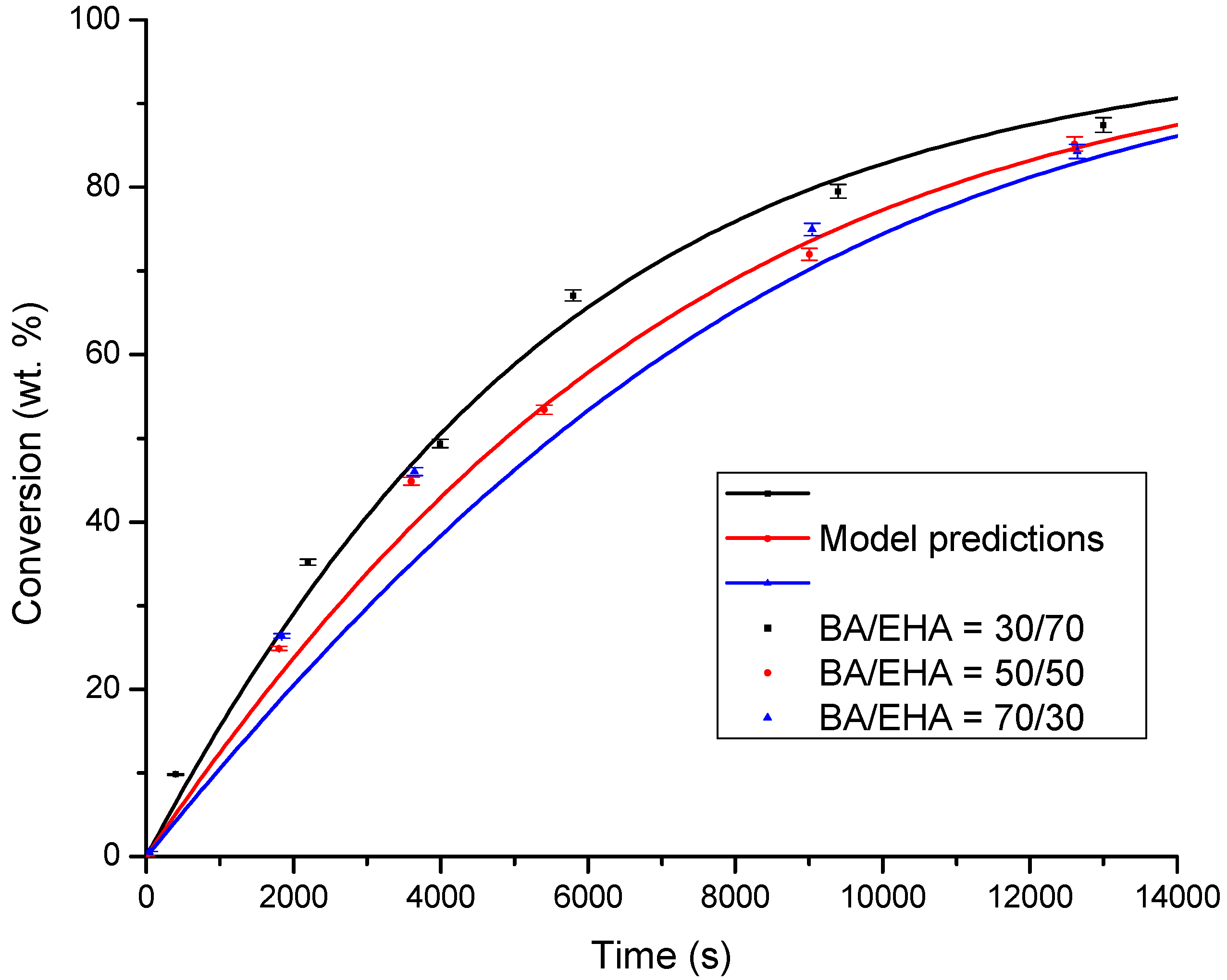

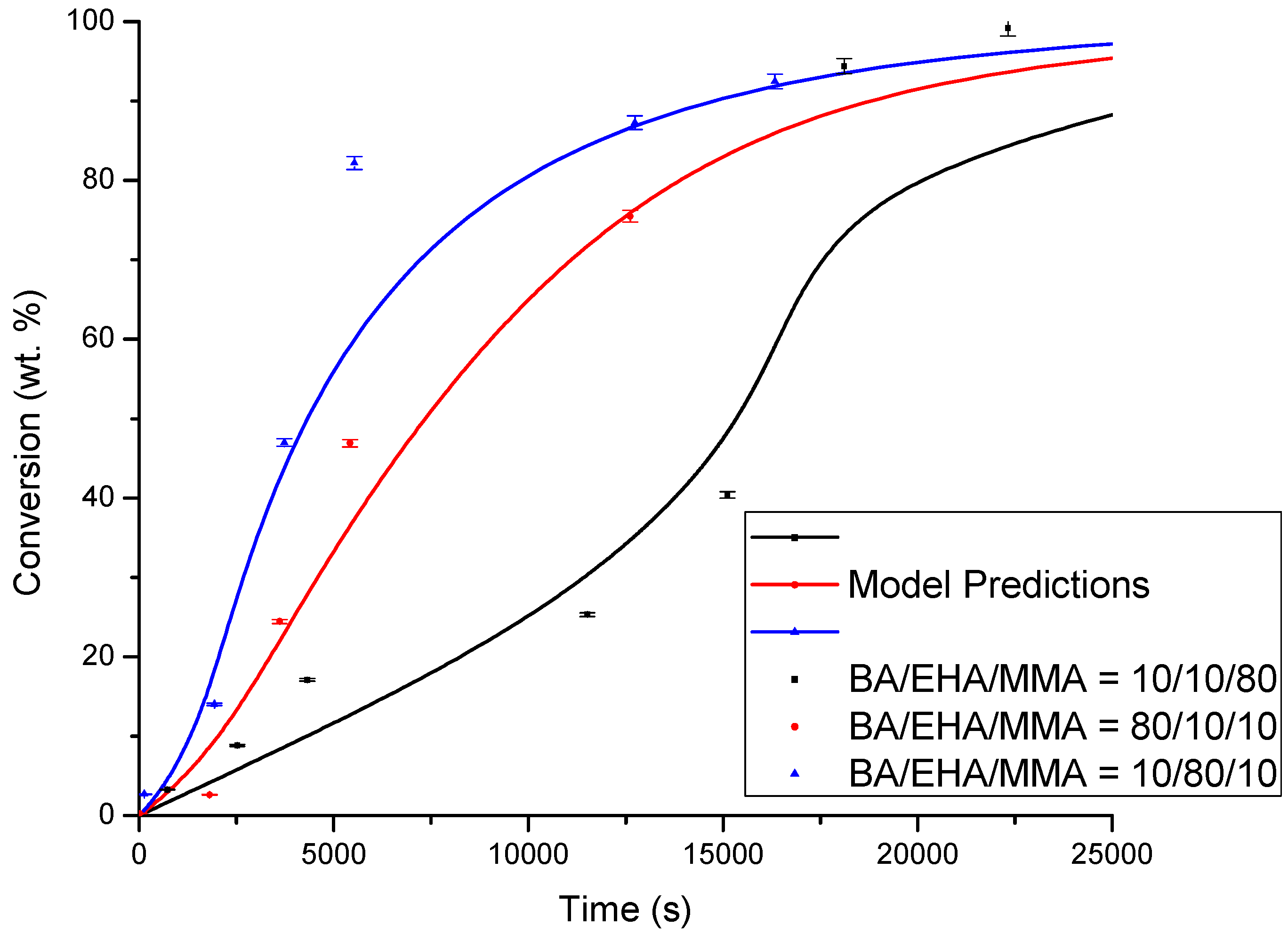

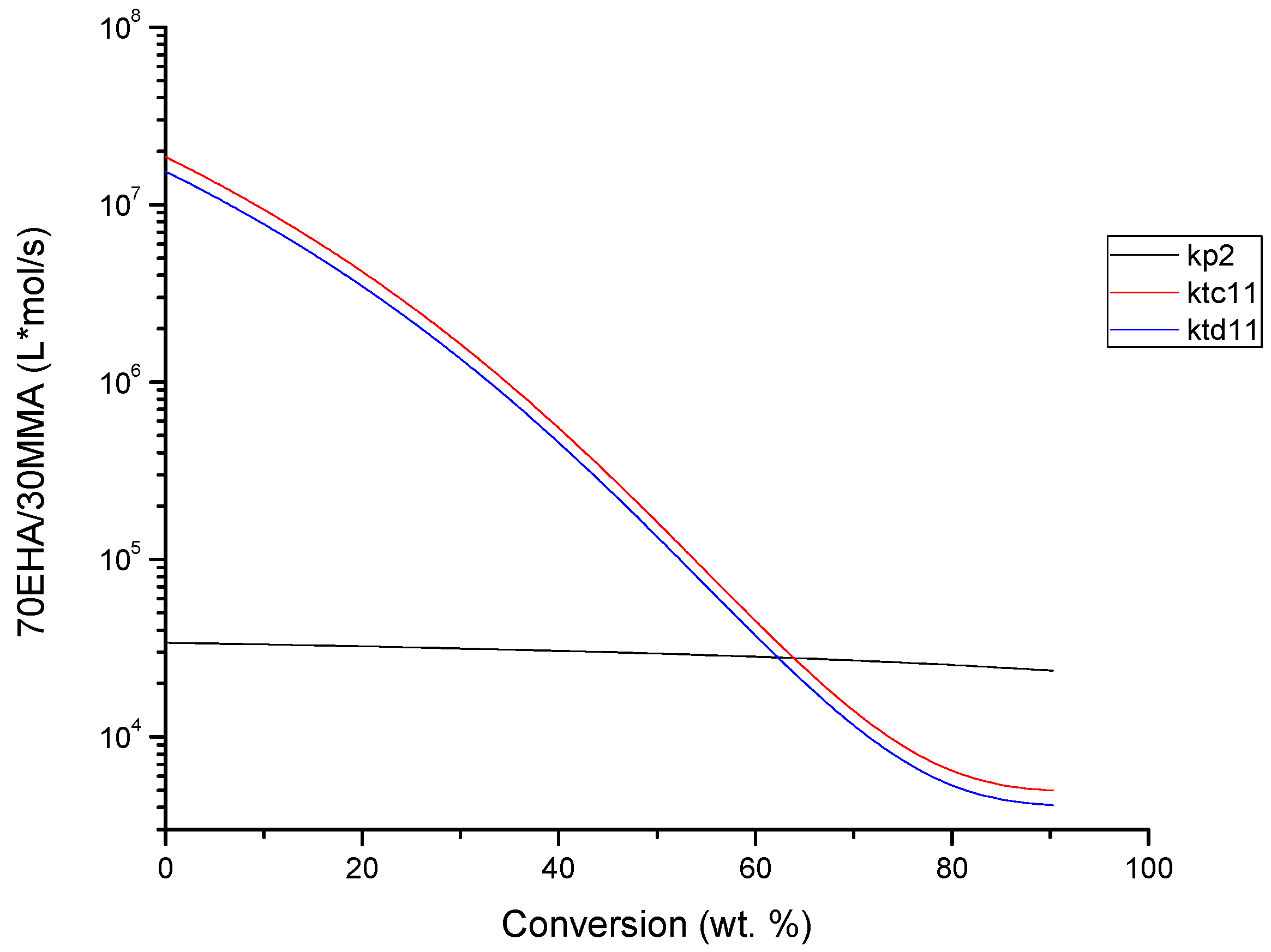

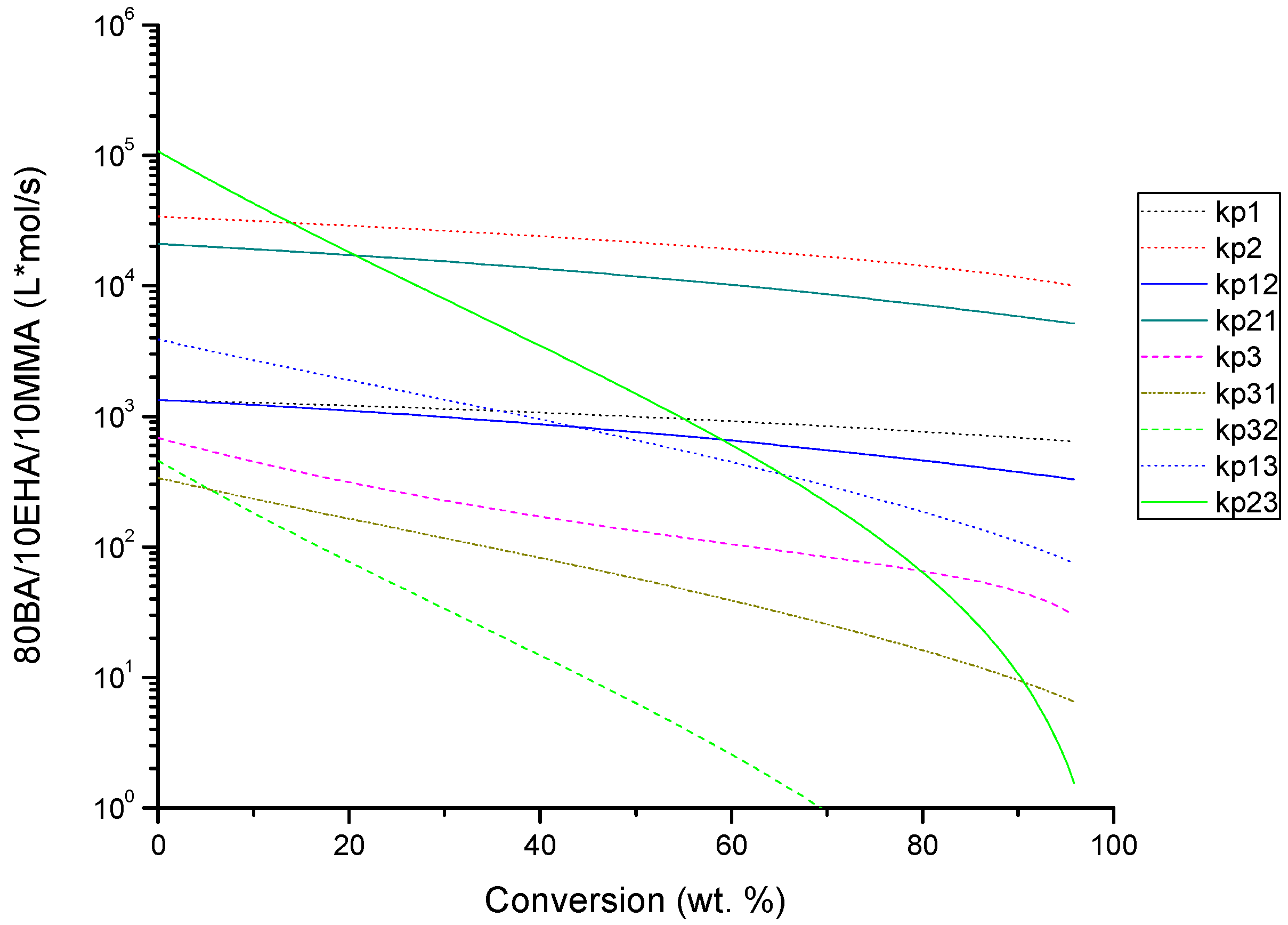
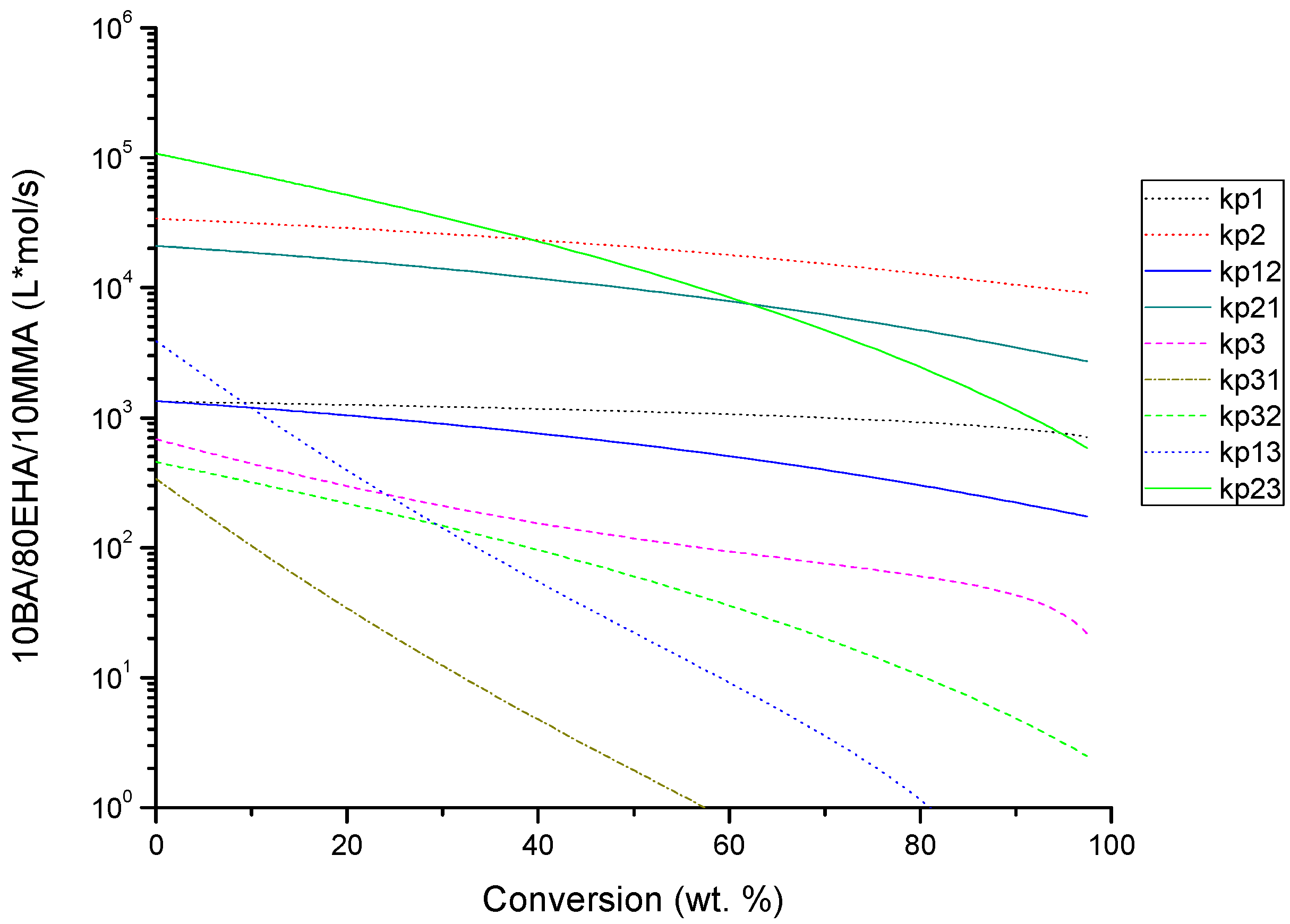
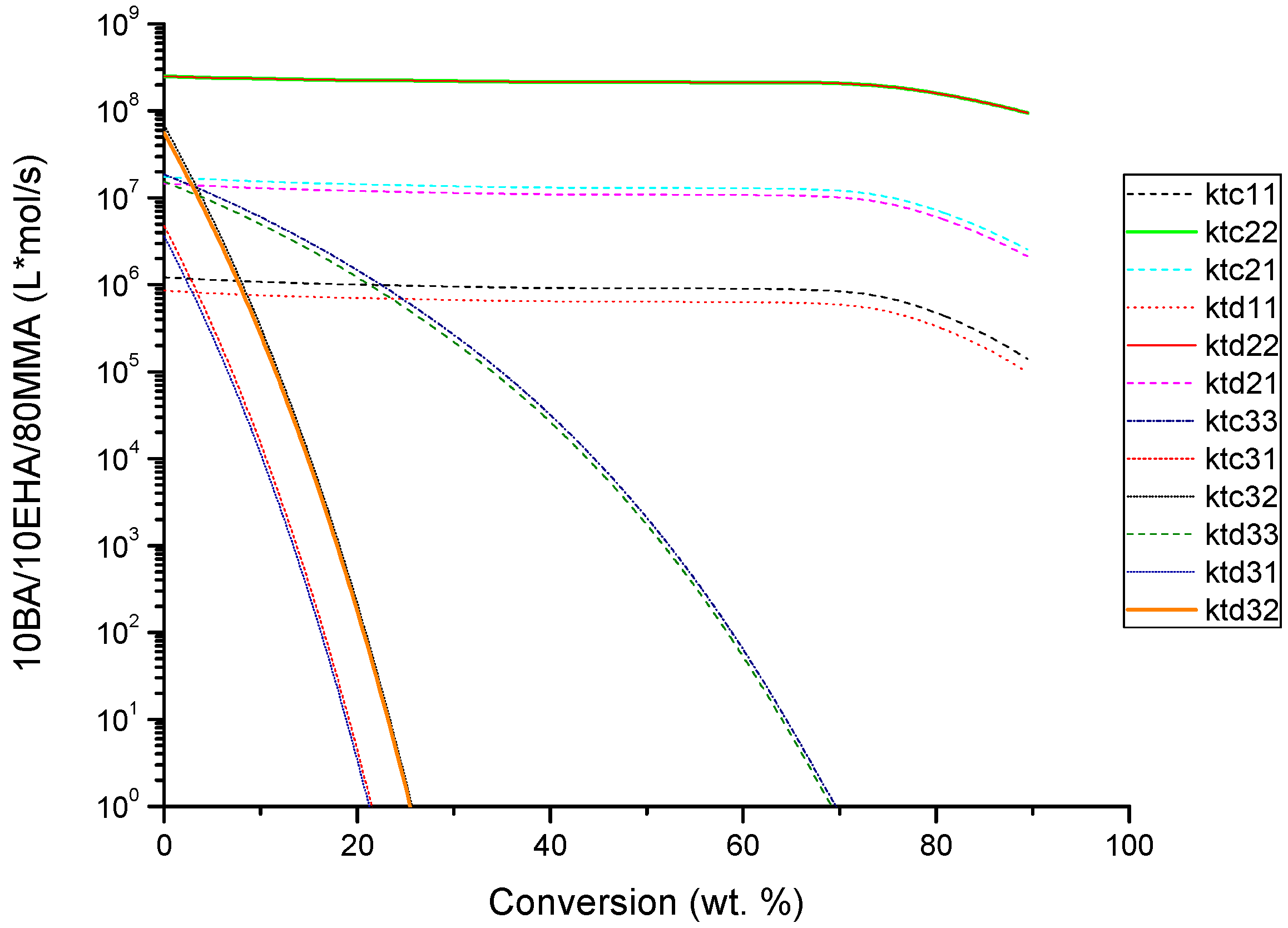


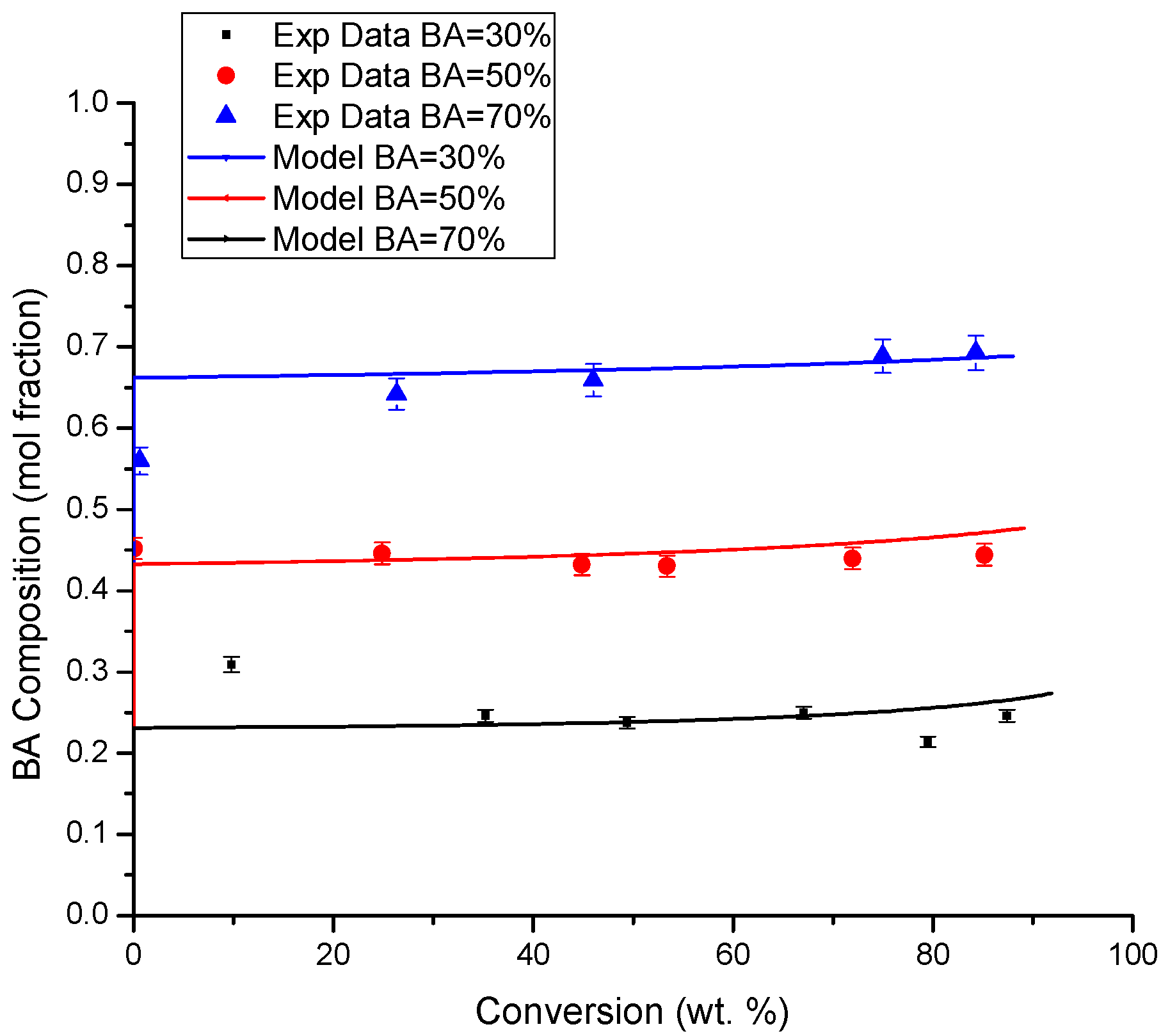
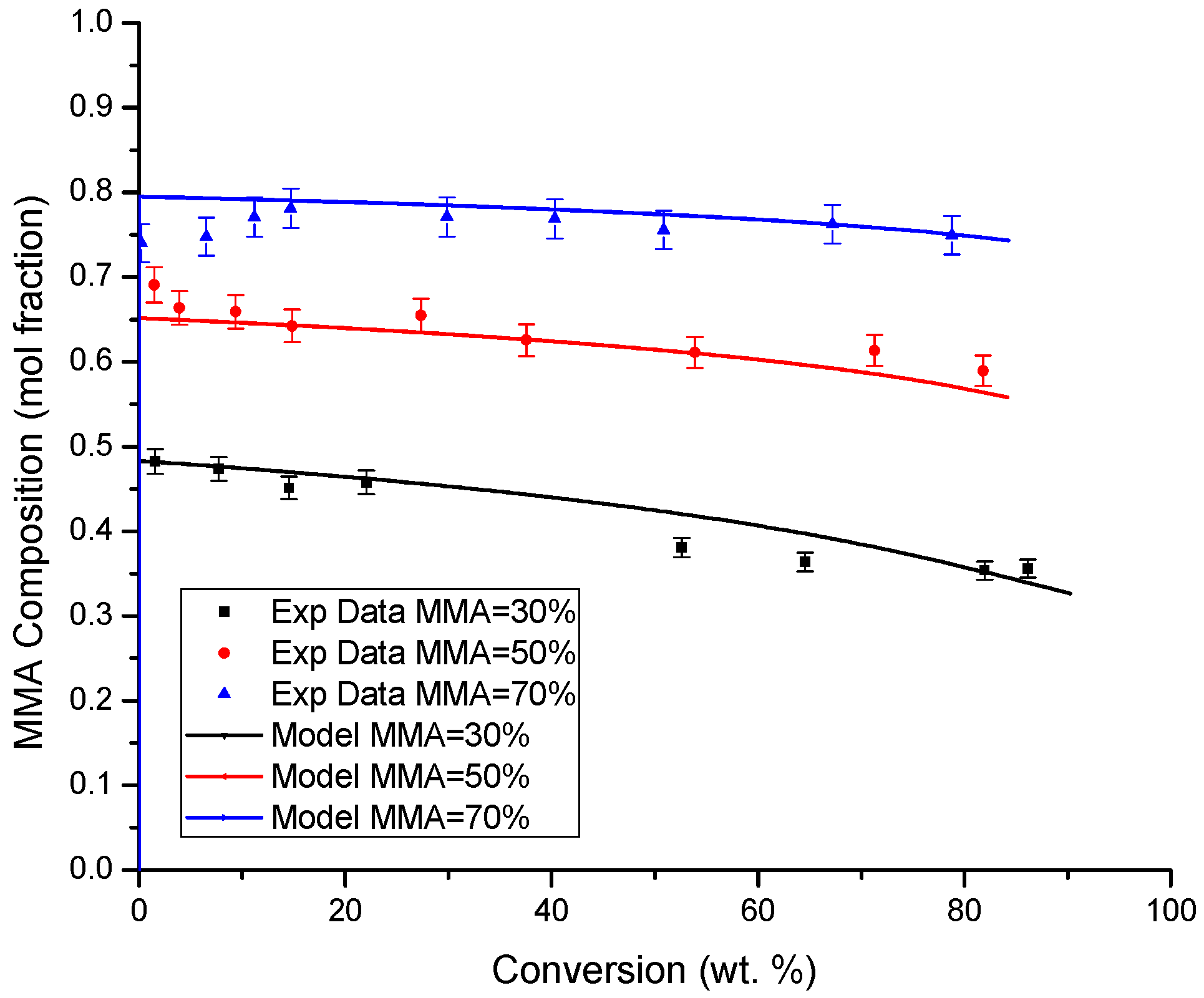
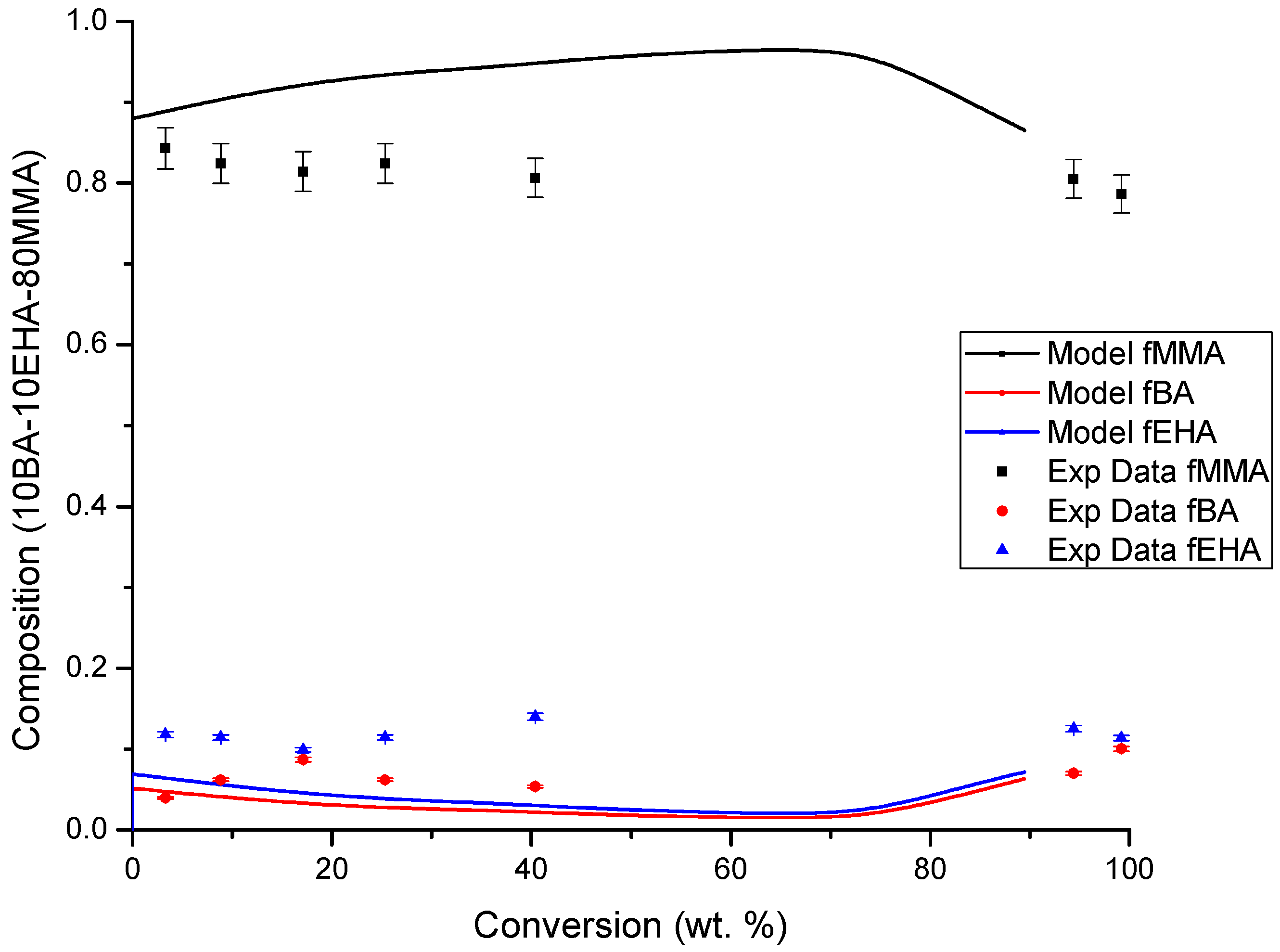
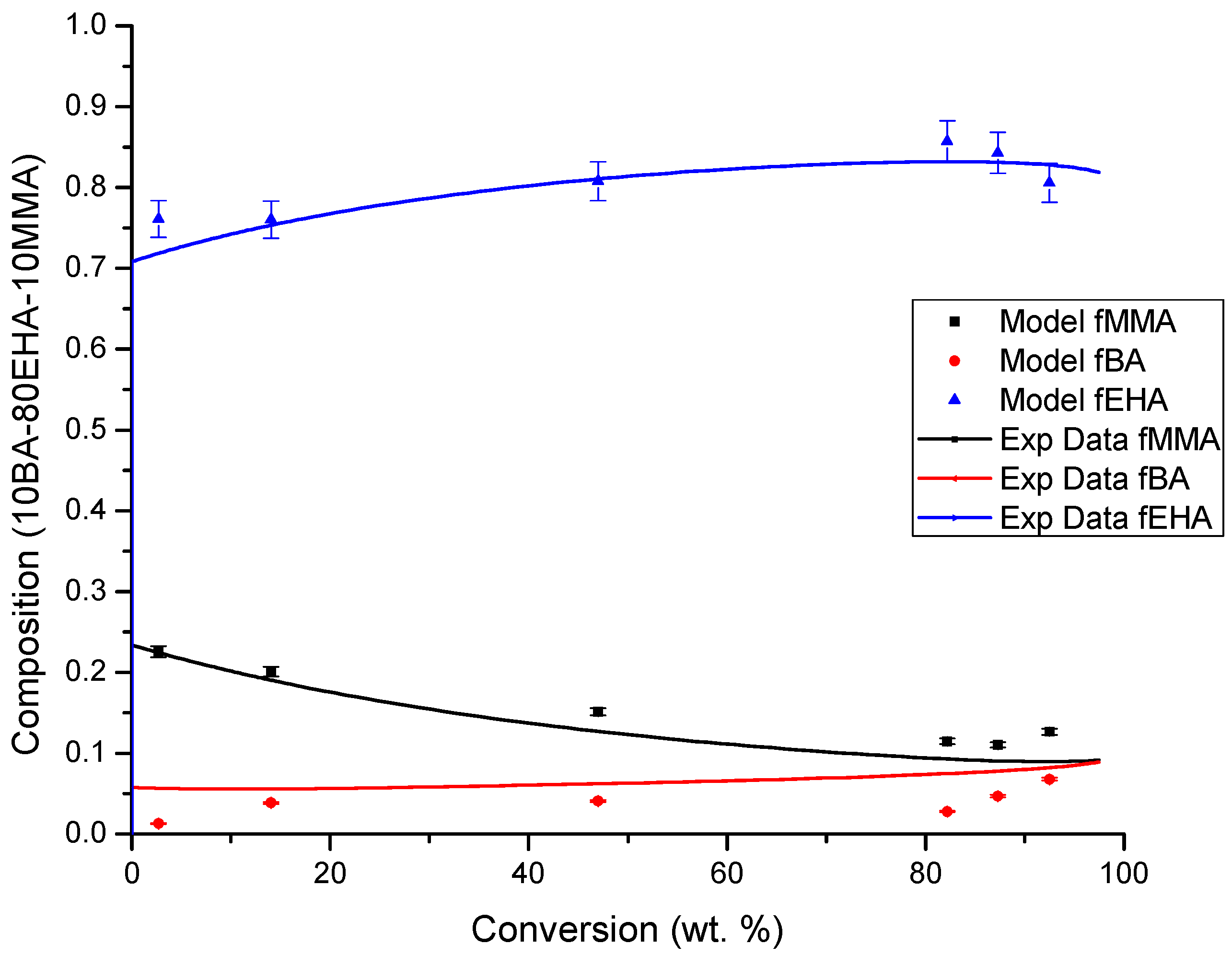
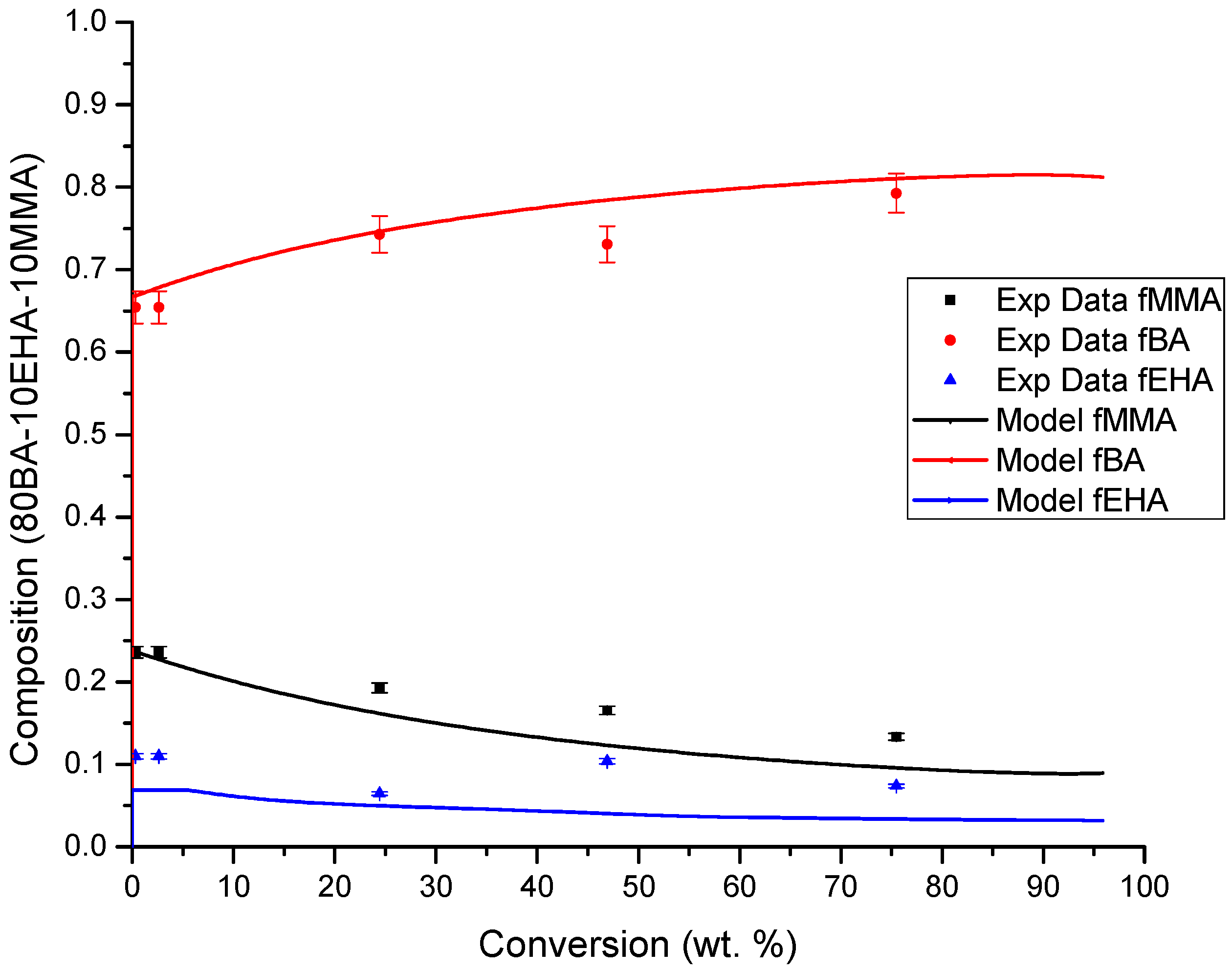
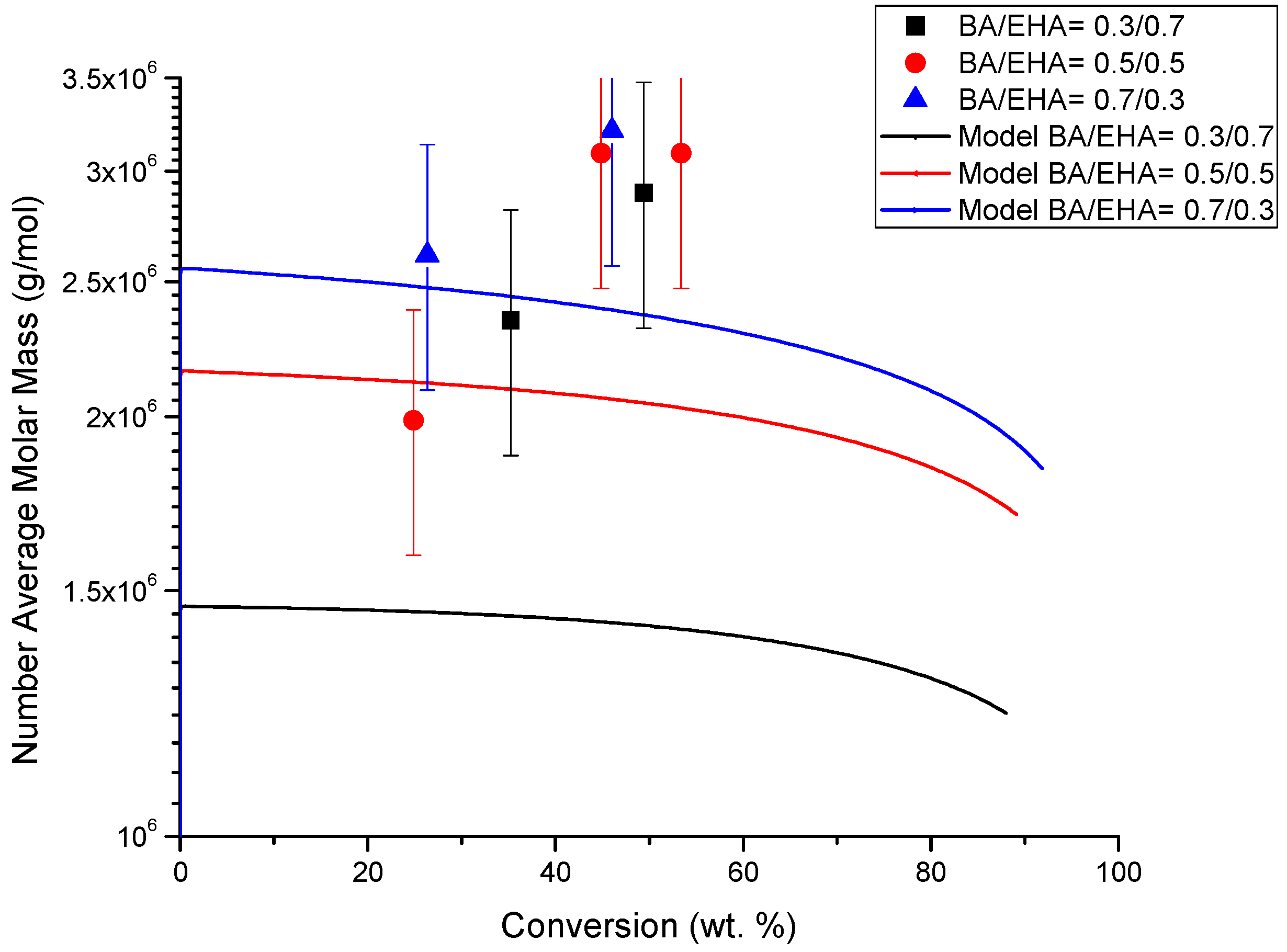



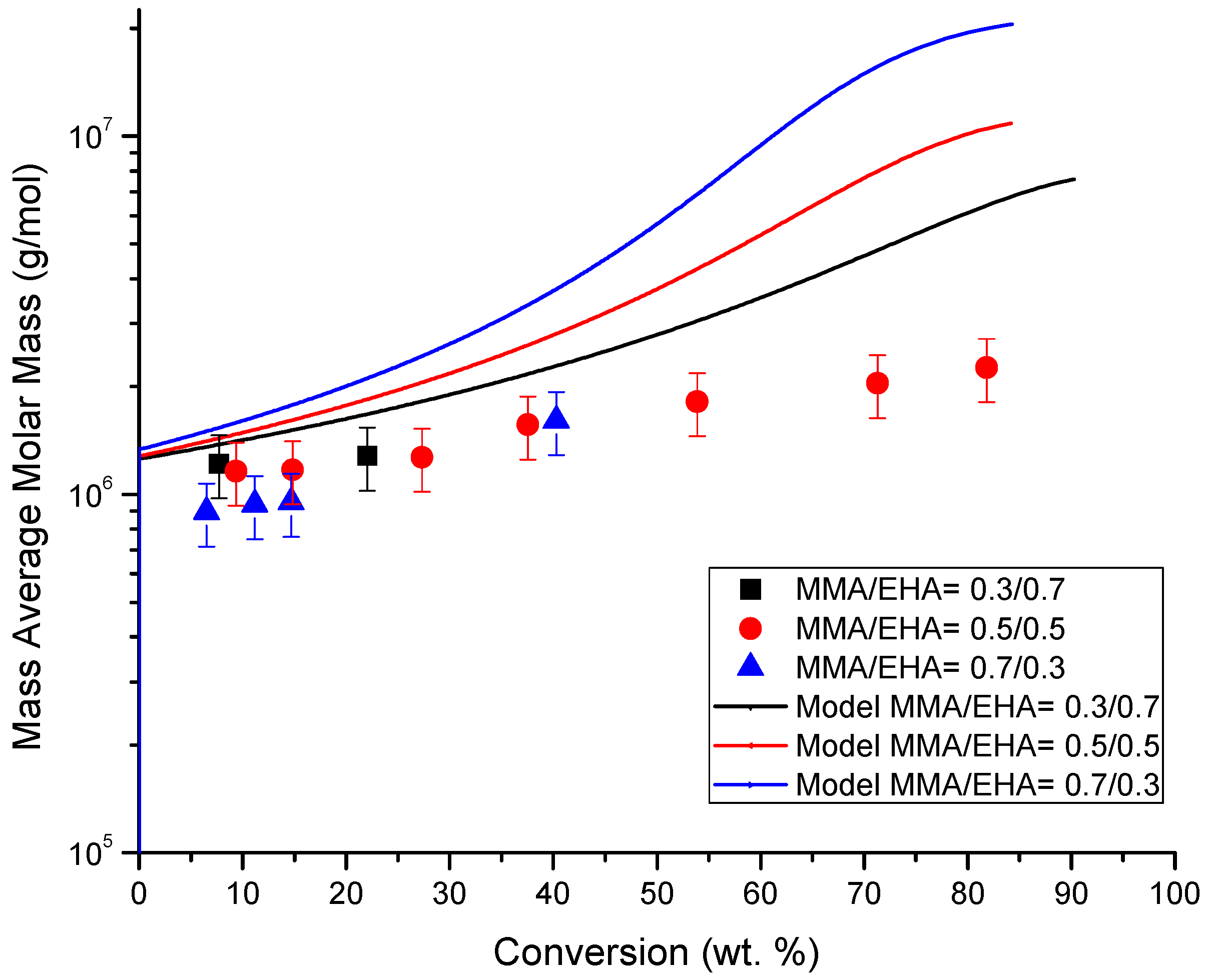

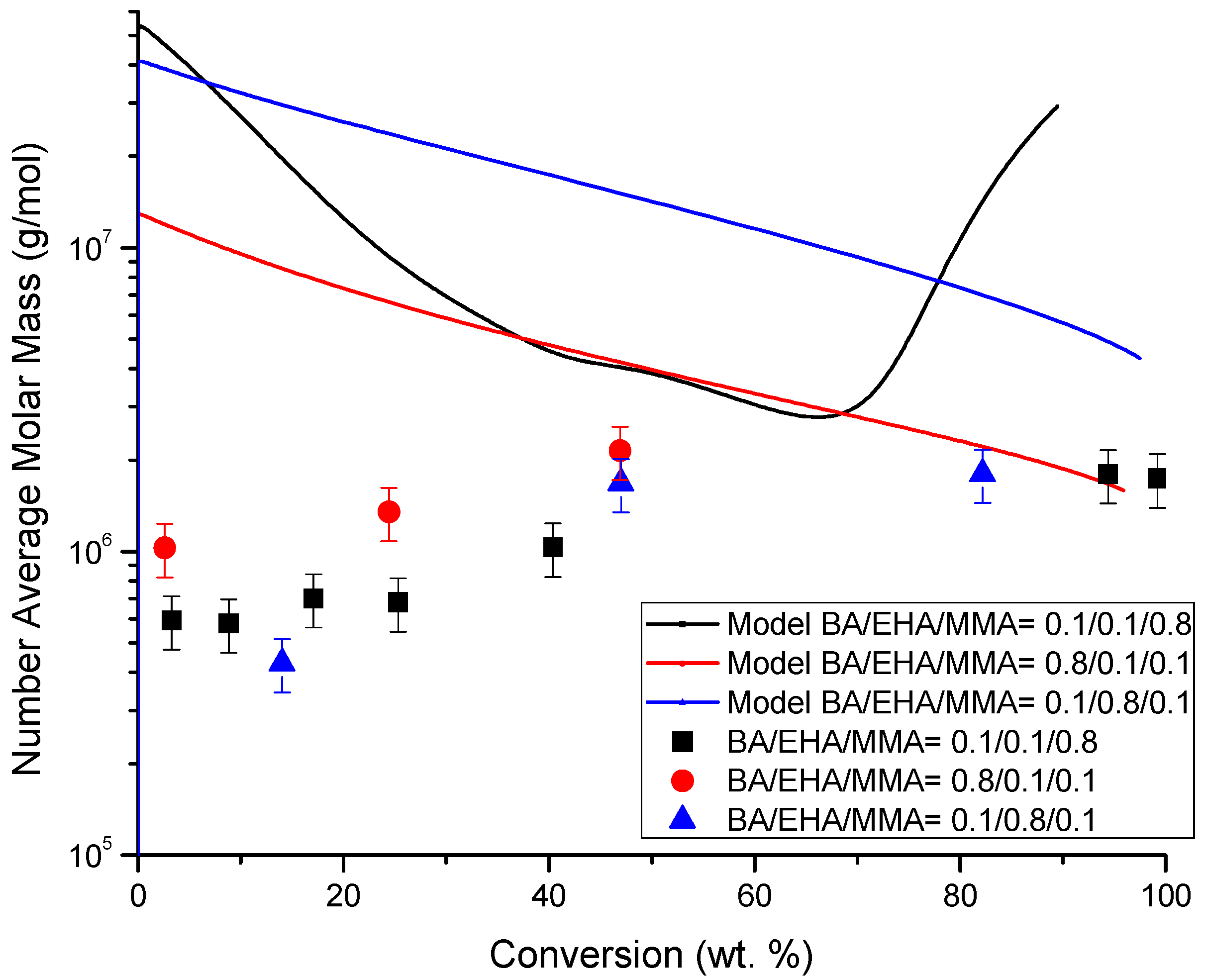
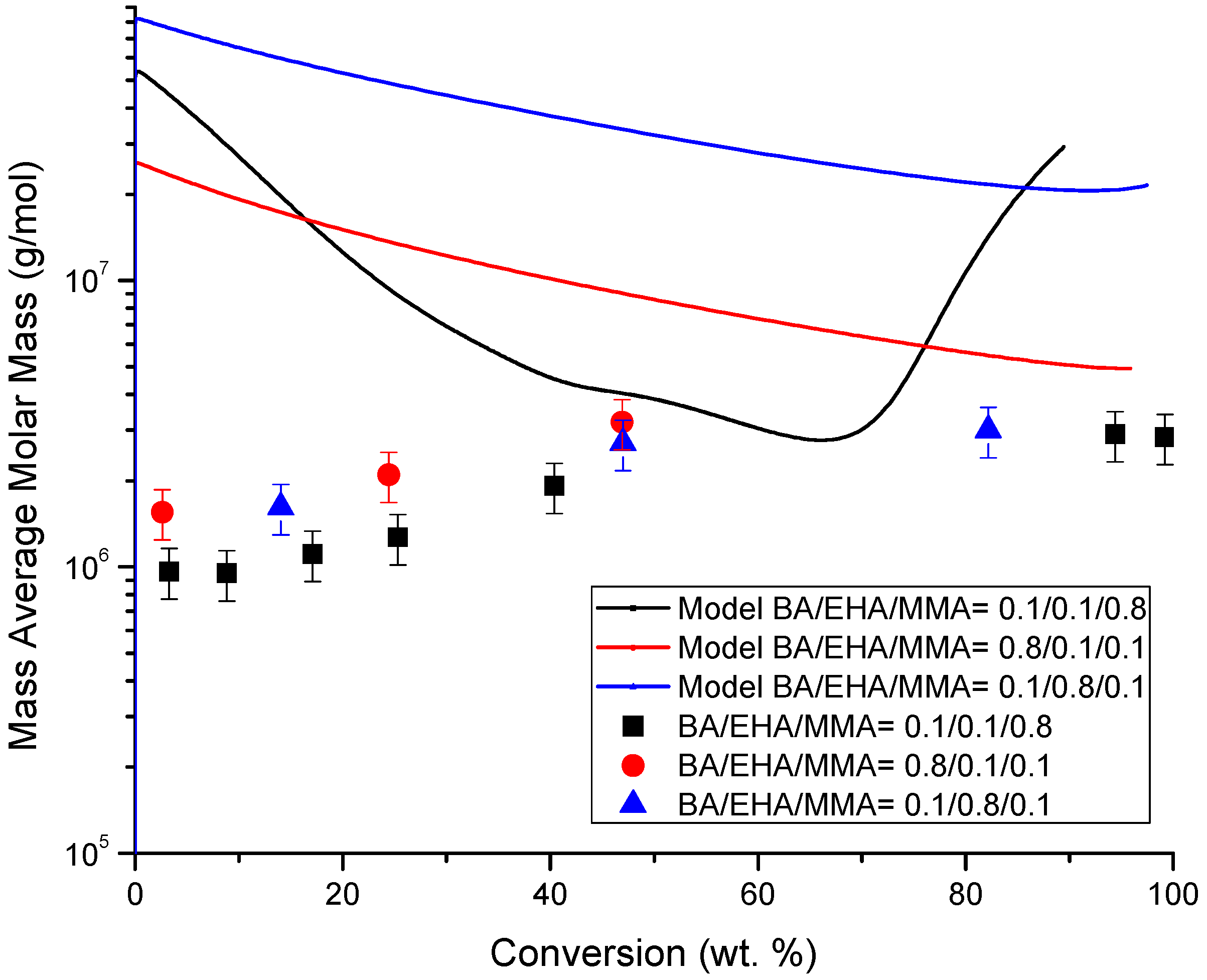
| Monomer Feed (BA Molar Fraction) | Monomer Feed (EHA Molar Fraction) | BA (mol L−1) | EHA (mol L−1) | AIBN (mol L−1) |
|---|---|---|---|---|
| fBA = 0.3 | fEHA = 0.7 | 1.596 | 3.699 | 0.00414 |
| fBA = 0.5 | fEHA = 0.5 | 2.844 | 2.836 | 0.00414 |
| fBA = 0.7 | fEHA = 0.3 | 4.284 | 1.837 | 0.00414 |
| Monomer Feed (MMA Molar Fraction) | Monomer Feed (EHA Molar Fraction) | MMA (mol L−1) | EHA (mol L−1) | AIBN (mol L−1) |
|---|---|---|---|---|
| fMMA = 0.3 | fEHA = 0.7 | 1.6847 | 3.9407 | 0.00414 |
| fMMA = 0.5 | fEHA = 0.5 | 3.1794 | 3.1762 | 0.00438 |
| fMMA = 0.7 | fEHA = 0.3 | 5.1093 | 2.1897 | 0.00438 |
| Monomer Feed (BA Molar Fraction) | Monomer Feed (EHA Molar Fraction) | Monomer Feed (MMA Molar Fraction) | BA (mol L−1) | EHA (mol L−1) | MMA (mol L−1) | AIBN (mol L−1) |
|---|---|---|---|---|---|---|
| fBA = 0.8 | fEHA = 0.1 | fMMA = 0.1 | 5.456 | 0.683 | 0.677 | 0.00414 |
| fBA = 0.1 | fEHA = 0.8 | fMMA = 0.1 | 0.512 | 4.207 | 0.509 | 0.00419 |
| fBA = 0.1 | fEHA = 0.1 | fMMA = 0.8 | 0.835 | 0.834 | 6.689 | 0.00450 |
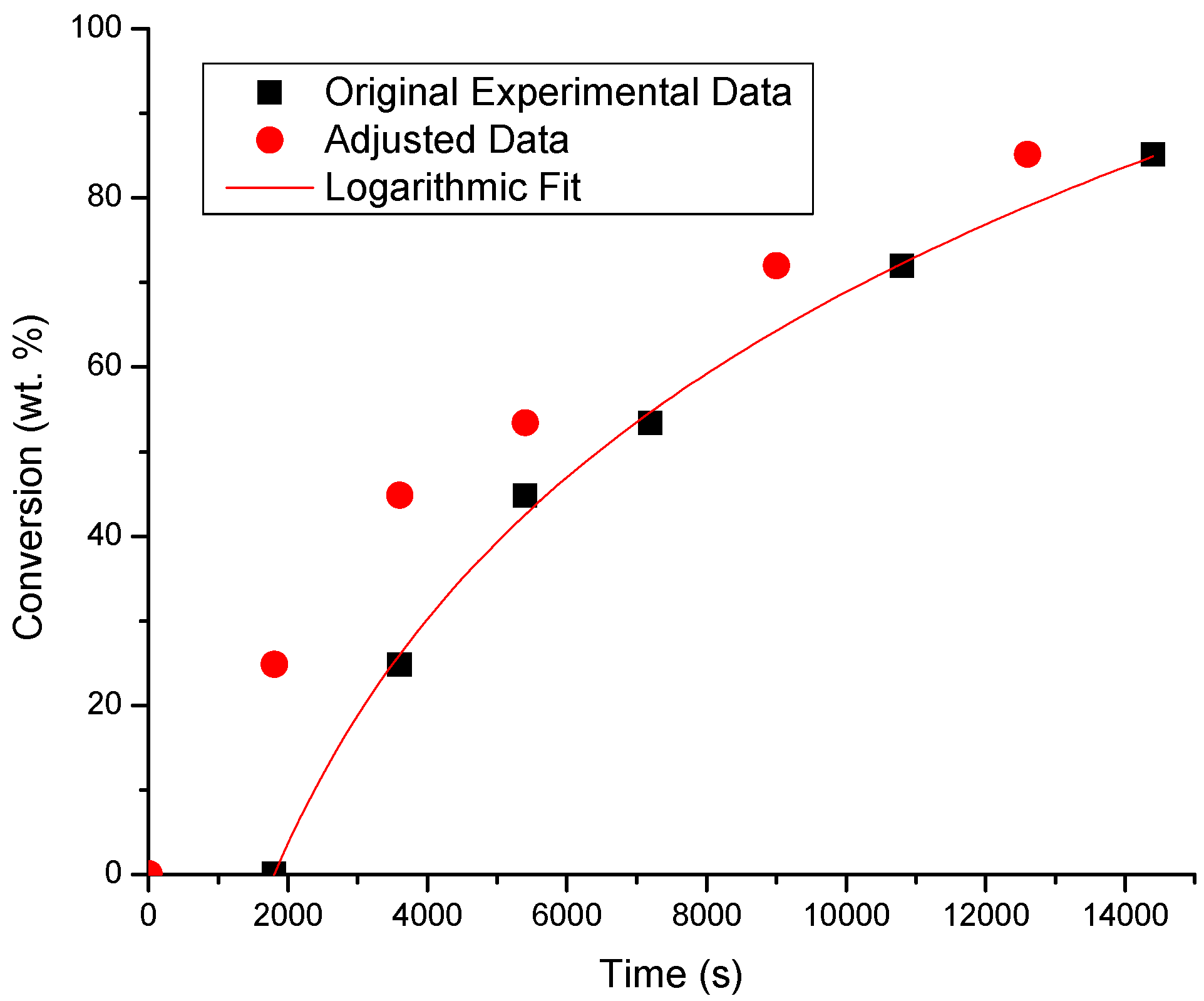
| System | r2 Value | Fitting Equation (x: Conversion; t: Time) | Induction Time (s) |
|---|---|---|---|
| BA/50EHA | 0.997 | x = 38.457ln(t) − 288.17 | 1796 |
| 30BA/70EHA | 0.993 | x = 38.464ln(t) − 278.76 | 1404 |
| 70BA/30EHA | 0.998 | x = 40.656ln(t) − 303.78 | 1758 |
| 50MMA/50EHA | 0.996 | x = 7E−08t2+ 0.002t− 3.0858 | 1468 |
| 30MMA/70EHA | 0.996 | x = 4E−08t2 + 0.0034t − 4.8554 | 1405 |
| 70MMA/30EHA | 0.996 | x = 4E−08t2 + 0.0025t − 4.1244 | 1608 |
| 10BA/10MMA/80EHA | 0.969 | x = 39.607ln(t) − 293.78 | 1665 |
| 10BA/80MMA/10EHA | 0.987 | x = 0.0038t − 10.949 | 2881 |
| 80BA/10MMA/10EHA | 0.899 | x = 33.614ln(t) − 251.67 | 1785 |
| Description | Step in PREDICI® | Parameter | Value (L mol−1 s−1 Unless Otherwise Stated) | Reference |
|---|---|---|---|---|
| Initiation | ||||
| Initiator decomposition | I→2fI• | ki, f | 7.4609E−6, 0.58 (s−1) | [30] |
| First Propagation for BA | I• + M1→P1(1) | kp1 | 1333.8 | [31] |
| First Propagation for EHA | I• + M2→P2(1) | kp2 | 33,395 | [7] |
| Propagation | ||||
| Self-propagation for BA | P1(s)• + M1→P1(s+1)• | kp1 | 1333.8 | [31] |
| Self-propagation forEHA | P2(s)• + M2→P2(s+1)• | kp2 | 33,395 | [7] |
| Cross-Propagation | P1(s)• + M2→P2(s+1)• | kp12 | 1333.8/r12 | [14,31] |
| Cross-Propagation | P2(s)• + M1→P1(s+1)• | kp21 | 33,395/r21 | [7,14] |
| Chain Transfer | ||||
| Chain Transfer to BA | P1(s)• + M1→P(s) + P1(1)• | kf1 | 0.1492 | [31] |
| Chain Transfer to BA | P2(s)• + M1→P(s) + P1(1)• | kf21 | 3.5/r21 | This work/[14] |
| Chain Transfer to EHA | P1(s)• + M2→P(s) + P2(1)• | kf12 | 0.1492/r12 | [14,31] |
| Chain Transfer to EHA | P2(s)• + M2→P(s) + P2(1)• | kf2 | 3.5 | This work |
| Intramolecular chain transfer to BA | ||||
| Backbitingfor BA | P1(s)•→Q1(s)• | kbb | 553 | [32] |
| Short-chainbranching BA | Q1(s)• + M1→P1(s+1)• | kp1tert | 49.401 | [33] |
| Short-chainbranching EHA | Q1(s)• + M2→P2(s+1)• | kp12tert | 49.401/r12 | [14,33] |
| Degradativechain transfer | Q1(s)• + M1→P(s) + P1(1)• | kf1tert | 6 | This work |
| Degradativechain transfer | Q1(s)• + M2→P(s) + P2(1)• | kf12tert | 6/r12 | This work/[14] |
| Termination | ||||
| Bycombination | P1(s)• + P1(r)•→P(s+r) | ktc11 | 1.2259E6 | [31] |
| - | P1(s)• + P2(r)•→P(s+r) | ktc21 | √ktc11·ktc22 | [31]/This work |
| - | P2(s)• + P2(r)•→P(s+r) | ktc22 | 2.5E8 | This work |
| Bydisproportionation | P1(s)• + P1(r)•→P(s) + P(r) | ktd11 | 8.5815E5 | [31] |
| - | P1(s)• + P2(r)•→P(s) + P(r) | ktd21 | √ktd11·ktd22 | [31]/This work |
| - | P2(s)• + P2(r)•→P(s) + P(r) | ktd22 | 2.5E8 | This work |
| Termination of BA tertiary radicals | ||||
| Bycombination | Q1(s)• + P1(r)•→P(s+r) | ktc11 | 1.2259E6 | [31] |
| - | Q1(s)• + P2(r)•→P(s+r) | ktc21 | √ktc11·ktc22 | [31]/This work |
| - | Q1(s)• + Q1(r)•→P(s+r) | ktc11 | 1.2259E6 | [31] |
| Bydisproportionation | Q1(s)• + P1(r)•→P(s) + P(r) | ktd11 | 8.5815E5 | [31] |
| - | Q1(s)• + P2(r)•→P(s) + P(r) | ktd21 | √ktd11·ktd22 | [31]/This work |
| - | Q1(s)• + Q1(r)•→P(s) + P(r) | ktd11 | 8.5815E5 | [31] |
| Description | Step in PREDICI® | Parameters | Value (L mol−1 s−1, Unless Otherwise Stated) | Reference |
|---|---|---|---|---|
| Initiation | ||||
| Initiator decomposition | I→2fI• | ki, f | 7.4609E−6, 5.8E−1 (s−1) | [30] |
| First Propagation for MMA | I• + M1→P1(1) | kp1 | 683.24 | [31] |
| First Propagation for EHA | I• + M2→P2(1) | kp2 | 33,395 | [7] |
| Propagation | ||||
| Self-propagation for MMA | P1(s)• + M1→P1(s+1)• | kp1 | 683.24 | [31] |
| Self-propagation for EHA | P2(s)• + M2→P2(s+1)• | kp2 | 33,395 | [7] |
| Cross-Propagation | P1(s)• + M2→P2(s+1)• | kp12 | 683.24/r12 | [14,31] |
| Cross-Propagation | P2(s)• + M1→P1(s+1)• | kp21 | 33,395/r21 | [7,14] |
| Chain Transfer | ||||
| Chain Transfer to MMA | P1(s)• + M1→P(s) + P1(1)• | kf1 | 1.9321E−2 | [31] |
| Chain Transfer to MMA | P2(s)• + M1→P(s) + P1(1)• | kf21 | 3.5/r21 | This work/[14] |
| Chain Transfer to EHA | P1(s)• + M2→P(s) + P2(1)• | kf12 | 1.9321E−2/r12 | [14,31] |
| Chain Transfer to EHA | P2(s)• + M2→P(s) + P2(1)• | kf2 | 3.5 | This work |
| Termination | ||||
| By combination | P1(s)• + P1(r)•→P(s+r) | ktc11 | 1.859E7 | [31] |
| - | P1(s)• + P2(r)•→P(s+r) | ktc21 | √ktc11·ktc22 | [31]/This work |
| - | P2(s)• + P2(r)•→P(s+r) | ktc22 | 2.5E8 | This work |
| By disproportionation | P1(s)• + P1(r)•→P(s) + P(r) | ktd11 | 1.5382E7 | This work |
| - | P1(s)• + P2(r)•→P(s) + P(r) | ktd21 | √ktd11·ktd22 | [31]/This work |
| - | P2(s)• + P2(r)•→P(s) + P(r) | ktd22 | 2.5E8 | This work |
| Description | Step in PREDICI® | Parameters | Value (L mol−1 s−1, Unless Otherwise Stated) | Reference |
|---|---|---|---|---|
| Initiation | ||||
| Initiator decomposition | I→2fI• | ki, f | 7.4609E−6, 0.58 (s−1) | [30] |
| First Propagation for BA | I• + M1→P1(1) | kp1 | 1333.8 | [31] |
| First Propagation for EHA | I• + M2→P2(1) | kp2 | 33,395 | [7] |
| First Propagation for MMA | I• + M3→P3(1) | kp3 | 683.24 | [31] |
| Propagation | ||||
| Self-propagation for BA | P1(s)• + M1→P1(s+1)• | kp1 | 1333.8 | [31] |
| Self-propagation for EHA | P2(s)• + M2→P2(s+1)• | kp2 | 33,395 | [7] |
| Self-propagation forMMA | P3(s)• + M3→P3(s+1)• | kp3 | 683.24 | [31] |
| Cross-Propagation | P1(s)• + M2→P2(s+1)• | kp12 | 1333.8/r12 | [14,31] |
| Cross-Propagation | P1(s)• + M3→P3(s+1)• | kp13 | 1333.8/r13 | [31,34] |
| Cross-Propagation | P2(s)• + M1→P1(s+1)• | kp21 | 33,395/r21 | [7,14] |
| Cross-Propagation | P2(s)• + M3→P3(s+1)• | kp23 | 33,395/r23 | [7,14] |
| Cross-Propagation | P3(s)• + M1→P1(s+1)• | kp31 | 683.24/r31 | [31,34] |
| Cross-Propagation | P3(s)• + M2→P2(s+1)• | kp32 | 683.24/r32 | [31,34] |
| Chain Transfer | ||||
| Chain Transfer to BA | P1(s)• + M1→P(s) + P1(1)• | kf1 | 0.1492 | [31] |
| Chain Transfer to BA | P2(s)• + M1→P(s) + P1(1)• | kf21 | 3.5/r21 | This work/[14] |
| Chain Transfer to BA | P3(s)• + M1→P(s) + P1(1)• | kf31 | 1.9321E−2/r31 | [31,34] |
| Chain Transfer to EHA | P1(s)• + M2→P(s) + P2(1)• | kf12 | 0.1492/r12 | [14,31] |
| Chain Transfer to EHA | P2(s)• + M2→P(s) + P2(1)• | kf2 | 4.5 | This work |
| Chain Transfer to EHA | P3(s)• + M2→P(s) + P2(1)• | kf32 | 1.9321E−2/r32 | [14,31] |
| Chain Transfer to MMA | P1(s)• + M3→P(s) + P3(1)• | kf13 | 0.1492/r13 | [31,34] |
| Chain Transfer to MMA | P2(s)• + M3→P(s) + P3(1)• | kf23 | 3.5/r23 | This work/[31] |
| Chain Transfer to MMA | P3(s)• + M3→P(s) + P3(1)• | kf3 | 1.9321E−2 | [31] |
| Intramolecular chain transfer of BA | ||||
| Backbiting for BA | P1(s)•→Q1(s)• | kbb | 553 | [32] |
| Short-chain branching BA | Q1(s)• + M1→P1(s+1)• | kp1tert | 49.401 | [33] |
| Short-chain branching EHA | Q1(s)• + M2→P2(s+1)• | kp12tert | 49.401/r12 | [14,33] |
| Short-chain branching EHA | Q1(s)• + M3→P3(s+1)• | kp13tert | 49.401/r13 | [33,34] |
| Degradative chain transfer | Q1(s)• + M1→P(s) + P1(1)• | kf1tert | 6 | This work |
| Degradative chain transfer | Q1(s)• + M2→P(s) + P2(1)• | kf12tert | 6/r12 | This work/[14] |
| Degradative chain transfer | Q1(s)• + M3→P(s) + P3(1)• | kf13tert | 6/r13 | This work/[34] |
| Termination | ||||
| By combination | P1(s)• + P1(r)•→P(s+r) | ktc11 | 1.2259E6 | [31] |
| - | P1(s)• + P2(r)•→P(s+r) | ktc21 | √ktc11·ktc22 | [31]/This work |
| - | P1(s)• + P3(r)•→P(s+r) | ktc31 | √ktc11·ktc33 | [31] |
| - | P2(s)• + P2(r)•→P(s+r) | ktc22 | 2.5E8 | This work |
| - | P2(s)• + P3(r)•→P(s+r) | ktc32 | √ktc22·ktc33 | This work/[31] |
| - | P3(s)• + P3(r)•→P(s+r) | ktc33 | 1.859E7 | [31] |
| By disproportionation | P1(s)• + P1(r)•→P(s) + P(r) | ktd11 | 8.5815E5 | [31] |
| - | P1(s)• + P2(r)•→P(s) + P(r) | ktd21 | √ktd11·ktd22 | [31]/This work |
| - | P1(s)• + P3(r)•→P(s) + P(r) | ktd31 | √ktd11·ktd33 | [31] |
| - | P2(s)• + P2(r)•→P(s) + P(r) | ktd22 | 2.5E8 | This work |
| - | P2(s)• + P3(r)•→P(s) + P(r) | ktd32 | √ktd22·ktd33 | This work/[31] |
| - | P3(s)• + P3(r)•→P(s) + P(r) | ktd33 | 1.5382E7 | [31] |
| Termination of BA tertiary radicals | ||||
| By combination | Q1(s)• + P1(r)•→P(s+r) | ktc11 | 1.2259E6 | [31] |
| - | Q1(s)• + P2(r)•→P(s+r) | ktc21 | √ktc11·ktc22 | This work |
| - | Q1(s)• + P3(r)•→P(s+r) | ktc31 | √ktc11·ktc33 | [31] |
| - | Q2(s)• + Q2(r)•→P(s+r) | ktc22 | 2.5E8 | This work |
| - | Q2(s)• + Q3(r)•→P(s+r) | ktc32 | √ktc22·ktc33 | This work/[31] |
| - | Q3(s)• + Q3(r)•→P(s+r) | ktc33 | 1.859E7 | [31] |
| By disproportionation | Q1(s)• + P1(r)•→P(s) + P(r) | ktd11 | 8.5815E5 | [31] |
| - | Q1(s)• + P2(r)•→P(s) + P(r) | ktd21 | √ktd11·ktd22 | [31]/ This work |
| - | Q1(s)• + P3(r)•→P(s) + P(r) | ktd31 | √ktd11·ktd33 | [31] |
| - | Q2(s)• + P2(r)•→P(s) + P(r) | ktd22 | 2.5E8 | This work |
| - | Q2(s)• + P3(r)•→P(s) + P(r) | ktd32 | √ktd22·ktd33 | This work/[31] |
| - | Q3(s)• + Q3(r)•→P(s) + P(r) | ktd33 | 1.5382E7 | [31] |
| Species | TgM(K) | TgP(K) | ||
|---|---|---|---|---|
| BA | 185 | 218 | 0.001 | 0.00048 |
| EHA | 167 | 223 | 0.001 | 0.00048 |
| MMA | 167 | 387 | 0.001 | 0.00048 |
| System | Rate Coefficients Where DC Effects Were Considered | β Value | Rate Coefficients Where DC Effects Were Neglected (β = 0) |
|---|---|---|---|
| BA-EHA | kp1 | 0.25 | ki |
| kp2 | 0.25 | f | |
| kp12 | 0.25 | kf1 | |
| kp21 | 0.5 | kf21 | |
| kbb | 5 | kf12 | |
| ktc11 | 4 | kf2 | |
| ktc21 | 0.5 | kp1tert | |
| ktc22 | 2 | kp12tert | |
| ktd11 | 4 | kf1tert | |
| ktd21 | 0.5 | kf12tert | |
| ktd22 | 2 | ---- | |
| MMA-EHA | kp2 | 0.5 | ki |
| kf1 | 1 | f | |
| kf12 | 1 | kp1, kp12 | |
| kf2 | 1 | kp2, kp21 | |
| kf21 | 1 | ktc22, ktd22 | |
| ktc11 | 4 | ktc21, ktd21 | |
| ktd11 | 4 | ---- | |
| BA-EHA-MMA | kp1 | 1 | ki |
| kp12 | 1 | f | |
| kp2 | 1.5 | kf1 | |
| kp21 | 1 | kf21 | |
| kp23 | 2 | kf31 | |
| kp13 | 2 | kf12 | |
| kp3 | 2 | kf2 | |
| kp31 | 2 | kf32 | |
| kp32 | 2 | kf13 | |
| kbb | 2.5 | kf23 | |
| kf3 | 2 | kbb | |
| kf13tert | 2 | kp1tert | |
| ktc11 | 6 | kp12tert | |
| ktc21 | 2 | kp13tert | |
| ktc22 | 2 | kf1tert | |
| ktc31 | 10 | kf12tert | |
| ktc32 | 10 | ---- | |
| ktc33 | 10 | ---- | |
| ktd11 | 6 | ---- | |
| ktd21 | 2 | ---- | |
| ktd22 | 2 | ---- | |
| ktd31 | 10 | ---- | |
| ktd32 | 10 | ---- | |
| ktd33 | 10 | ---- |
| Parameter | Value | Description |
|---|---|---|
| ktcEHA | 2.5 × 108 | EHA termination by combination |
| ktdEHA | 2.5 × 108 | EHA termination by disproportionation |
| kfEHA | 45 | Chain transfer to EHA |
| kftertEHA | 60 | EHA intramolecular chain transfer to BA tertiary radicals |
| kftertMMA | 35 | MMA intramolecular chain transfer to BA tertiary radicals |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Reguera, J.A.; Vivaldo-Lima, E.; Gabriel, V.A.; Dubé, M.A. Modeling of the Free Radical Copolymerization Kinetics of n-Butyl Acrylate, Methyl Methacrylate and 2-Ethylhexyl Acrylate Using PREDICI®. Processes 2019, 7, 395. https://doi.org/10.3390/pr7070395
Gómez-Reguera JA, Vivaldo-Lima E, Gabriel VA, Dubé MA. Modeling of the Free Radical Copolymerization Kinetics of n-Butyl Acrylate, Methyl Methacrylate and 2-Ethylhexyl Acrylate Using PREDICI®. Processes. 2019; 7(7):395. https://doi.org/10.3390/pr7070395
Chicago/Turabian StyleGómez-Reguera, Javier A., Eduardo Vivaldo-Lima, Vida A. Gabriel, and Marc A. Dubé. 2019. "Modeling of the Free Radical Copolymerization Kinetics of n-Butyl Acrylate, Methyl Methacrylate and 2-Ethylhexyl Acrylate Using PREDICI®" Processes 7, no. 7: 395. https://doi.org/10.3390/pr7070395