
Thermal Stabilization of Lipases Bound to Solid-Phase Triazine-Scaffolded Biomimetic Ligands: A Preliminary Assessment


1
Department of Bioengineering, Instituto Superior Técnico, Av. Rovisco Pais, 1049-001 Lisbon, Portugal
2
iBB-Institute for Bioengineering and Biosciences and i4HB-Institute for Health and Bioeconomy, Instituto Superior Técnico, Av. Rovisco Pais, 1049-001 Lisbon, Portugal
*
Author to whom correspondence should be addressed.
Processes 2024, 12(2), 371; https://doi.org/10.3390/pr12020371
Submission received: 1 January 2024
/
Revised: 2 February 2024
/
Accepted: 7 February 2024
/
Published: 11 February 2024
(This article belongs to the Special Issue Bioprocess Engineering: Sustainable Manufacturing for a Green Society)
Abstract
:Biomimetic ligands are synthetic compounds that mimic the structure and binding properties of natural biological ligands. The first uses of textile dyes as pseudo-affinity ligands paved the way for the rational design and de novo synthesis of low-cost, non-toxic and highly stable triazine-scaffolded affinity ligands. A novel method to assess and enhance protein stability, employing triazine-based biomimetic ligands and using cutinase from Fusarium solani pisi as a protein model, has been previously reported. This innovative approach combined the concepts of molecular modeling and solid-phase combinatorial chemistry to design, synthesize and screen biomimetic compounds able to bind cutinase through complementary affinity-like interactions while maintaining its biological functionality. The screening of a 36-member biased combinatorial library enabled the identification of promising lead ligands. The immobilization/adsorption of cutinase onto a particular lead (ligand 3′/11) led to a noteworthy enhancement in thermal stability within the temperature range of 60–80 °C. In the present study, similar triazine-based compounds, sourced from the same combinatorial library and mimicking dipeptides of diverse amino acids, were selected and studied to determine their effectiveness in binding and/or improving the thermal stability of several lipases, enzymes which are closely related in function to cutinases. Three ligands with different compositions were screened for their potential thermostabilizing effect on different lipolytic enzymes at 60 °C. An entirely distinct enzyme, invertase from Saccharomyces cerevisiae, was also assessed for binding to the same ligands and functioned as a ‘control’ for the experiments with lipases. The high binding yield of ligand 3′/11 [4-({4-chloro-6-[(2-methylbutyl)amino]-1,3,5-triazin-2-yl}amino)benzoic acid] to cutinase was confirmed, and the same ligand was tested for its ability to bind lipases from Aspergillus oryzae (AOL), Candida rugosa (CRL), Chromobacterium viscosum (CVL), Rhizomucor miehei (RML) and Rhizopus niveus (RNL). The enzymes CRL, CVL, RNL and invertase showed significant adsorption yields to ligand 3′/11—32, 29, 36 and 94%, respectively, and the thermal stability at 60 °C of free and adsorbed enzymes was studied. CVL and RNL were also stabilized by adsorption to ligand 3′/11. In the case of CRL and invertase, which bound but were not stabilized by ligand (3′/11), other ligands from the original combinatorial library were tested. Between the two alternative ligands, one was effective at stabilizing C. rugosa lipase, while none stabilized invertase.
1. Introduction
The protein surface serves as the interface by which a protein senses its external environment. Its composition of ionic, polar and hydrophobic residues is crucial for proteins’ interactions with other molecules and solvents [1]. Globular proteins exhibit marginal stabilities in solution, equivalent to only a few non-covalent interactions [2]. Enhancement of protein’s stability may arise from the presence of extrinsic factors, such as compatible solutes, co-factors or specific ligands (e.g., substrates, inhibitors, amino acids) [3,4]. The binding of a protein to a specific ligand or carrier involves the formation of intermolecular interactions that induce changes in the physical–chemical environment around the putative binding site. Such changes can lead to local rigidification of proteins’ tertiary structure with positive effects on stability. As such, immobilization in a multiplicity of supports and conditions has been scrutinized with different enzyme systems and varied experimental designs as one of the most effective approaches for proteins’ stabilization [5,6,7,8,9]. Additionally, it has been demonstrated by some authors that when immobilization takes place at specific protein surface sites (oriented immobilization) more effective stabilization is achieved, resulting from reduced mobility of the 3D structure and likely of the unfolding-prone regions [10,11,12,13]. In a particular study involving the stabilization of thermolysine-like protease from Baccillus stearothermophilus (TLP-ste), the authors evidenced that the stabilization effect due to immobilization is strongly dependent on the position of attachment being most effective if the protein is attached to the support at the region where unfolding is initiated [10,11].
Biomimetic triazine-scaffolded ligands have been extensively studied and proven to be stable, non-toxic and non-immunogenic synthetic analogues of proteins that can replace advantageously naturally occurring biological ligands, namely in protein purification processes [14]. Over the last decades the triazine scaffold has garnered interest for its ease of manipulation and structural rigidity, making it an attractive candidate to generate new bioactive compounds through solid-phase combinatorial methodologies [15,16]. My research team has proposed a novel approach for accessing and enhancing protein stability based upon binding to specific triazine-based compounds. This approach relies on the selection of biomimetic ligands capable of stabilizing an enzyme against thermal inactivation, through binding to its surface by affinity-like interactions [17,18,19]. Cutinase from Fusarium solani pisi was used as the protein model. Cutinases are small carboxylic ester hydrolases that bridge the properties between esterases and lipases, being able to hydrolyze a wide range of substrates, such as esters, polyesters, and a large variety of short- and long-chain triacylglycerols. By its reverse reaction, cutinases can also catalyze esterification, polymerization and transesterification reactions for the production of esters, polyesters and novel triacylglycerols, among other compounds [20]. As opposed to lipases, cutinases do not exhibit interfacial activation, i.e., they do not present a hydrophobic lid covering the active site [21]. This structural property explains the high catalytic versatility of cutinases and their easy adaptation to multiple substrates.
In previous studies with cutinase from F. solani pisi, a 169-membered solid-phase combinatorial ligand library containing in its composition substituents mimicking the side-chains of twelve essential amino acids was randomly screened for binding cutinase while maintaining its biological activity [18]. The results were combined with those obtained from the screening of a de novo rationally designed ligand library as described in [17]. The primary goal of designing this rational library was to stabilize specific surface regions (designated as ‘weak regions’), where unfolding of cutinase is likely initiated [22]. Based on the findings from the comprehensive screening approach, a semi-rational, second-generation solid-phase biased combinatorial library consisting of 36-members was synthesized. This library was then evaluated for its ability to bind and improve the thermal stability of adsorbed cutinase. Amongst the tested ligands, one conducting to the highest activity retention of bound cutinase (ligand 3′/11, [4-({4-chloro-6-[(2-methylbutyl)amino]-1,3,5-triazin-2-yl}amino)benzoic acid]) was chosen as the lead for further studies. Solid-phase synthesized ligand 3′/11 enhanced considerably the activity retention of adsorbed cutinase at high temperatures (60–80 °C) relative to the free enzyme. At 60 °C and pH 8.0, immobilized cutinase presented a relative specific activity above 90% upon 2 h of incubation [18].
The thermal stabilization observed with cutinase adsorbed to a biomimetic affinity support was the groundwork for the current study, the main goal of which was to further explore triazine-based lead compounds to enhance the stability of enzymes functionally related with cutinase through affinity-like immobilization. Three solid-phase synthetic ligands, sourced from the 36-member biased combinatorial library and mimicking dipeptides of diverse amino acids, were screened for binding and stabilizing five different microbial lipases. The biomimetic adsorbents were synthesized directly in agarose by a solid-phase method well described in [23]. Agarose beads are highly porous, mechanically resistant, chemically and physically inert and sharply hydrophilic. These features render them particularly suitable for enzyme immobilization with a wide range of derivatization methods [24,25]. Commercially available microbial lipolytic preparations from Aspergillus oryzae (AOL), Candida rugosa (CRL), Rhizomucor miehei (RML), Rhizopus niveus (RNL) and Chromabacterium viscosum (CVL) were adsorbed to selected solid-phase synthesized ligands and assessed for improved stability at 60 °C. Additionally, adsorption and stabilization of invertase (β-fructofuranosidase) from Saccharomyces cerevisiae, an enzyme entirely distinct from cutinase and lipases, was also evaluated as a control.
2. Materials and Methods
2.1. Materials
All chemicals and biochemicals were from Sigma-Aldrich (Darmstadt, Germany). Sepharose CL-6B was purchased from GE Healthcare (Uppsala, Sweeden).
Enzymes
Recombinant cutinase, from Fusarium solani pisi, cloned in pMa/c5-CUF plasmid, was produced and purified in iBB as described in [26]. Lipases from Aspergillus oryzae (AOL), Candida rugosa (CRL), Rhizomucor miehei (RML) and Rhizopus niveus (RNL) were from Sigma-Aldrich. Lipase from Chromobacterium viscosum (CVL) was from Toyo Jozo Enzymes (Tokyo, Japan). Bovine serum albumin (BSA) standard solutions were from Thermo Scientific (Rockford, IL, USA).
2.2. Methods
2.2.1. Solid-Phase Synthesis of Ligand 3′/11
Preparation of Dichlorotriazinyl Agarose
Sepharose CL-6B was activated with epichlorohydrin (0.1 mL/g gel, in 1 M NaOH, overnight in a rotary shaker, at 30 °C) and aminated by a protocol described in [23]. Epoxy-activated gel was suspended in 1.5 mL of ammonia per gram of moist gel. The slurry was incubated overnight at 30 °C with gentle agitation in a rotary shaker. The aminated support was washed utterly in a sintered funnel with distilled water to remove any traces of ammonia. Washing was carried out until the pH of the washing solution was lowered to the pH of the distilled water and no odor could be detected. Aminated supports were either used immediately and activated with cyanuric chloride or stored in 20% (v/v) ethanol between 0 and 4 °C.
Preparation of dichlorotriazinyl agarose was performed as described in [23]. Aminated Sepharose CL-6B was suspended in a solution of acetone/water 50% (v/v)—1 mL per gram of gel. The slurry was maintained at 0 °C in an ice bath on a shaker. An amount corresponding to 5 molar equivalents of cyanuric chloride, relative to the extent of amination (≈20 μmol amine groups/g gel), was dissolved in 8.6 mL of acetone per gram of cyanuric chloride and divided into four aliquots. Each aliquot was added to the aminated gel at intervals of about 30 min, maintaining the mixture at 0 °C with constant shaking. The pH was monitored with the aid of pH-indicator strips and maintained neutral by addition of a NaOH 1 M solution. The gel was then washed with 2 × 10 gel volumes of each acetone/distilled water mixture (v/v)—1:1, 1:3, 0:1, 1:1, 3:1, 1:0—and then with abundant water to remove unreacted cyanuric chloride. The cyanuric chloride activated gel (dichlorotriazinyl agarose) was immediately used for the substitution of the two remaining chlorine atoms, designated as R1 and R2.
Nucleophilic Substitution of Chlorine Atoms with Selected Amines
The R1 and R2 reactive chlorines on the triazine scaffold and on dichlorotriazinyl agarose were sequentially substituted with selected amines mimicking the side chains of some amino acids. Cyanuric chloride activated gel was divided into aliquots. Each aliquot was used for the substitution of R1 chlorine in the triazine ring with two different aminated compounds: ammonia-amine 0- or 4-aminobenzoic acid-amine 3′ from a previous combinatorial library [18]. An amount corresponding to 2 molar equivalents of each amine, relative to the determined density of amine groups in the support (≈20 μmol/g gel), was dissolved in distilled water [19]. For amine 3′, one equivalent of sodium bicarbonate was also added. The volume of solvent used was 1 mL per gram of gel. Each aliquot of dichlorotriazinyl agarose with the respective amine solution was incubated at 30 °C for 24 h in a rotary shaker. After this, each gel was thoroughly washed with distilled water on a sintered funnel. The R1 monosubstituted ligands were used immediately for the substitution with amino compounds in R2.
The following R2 substitution was performed using ammonia-amine 0- or 2-methylbutylamine-amine 11 from a previous combinatorial library [18]. For this step, a 5-fold molar equivalent of aminated compounds, relative to the determined density of amine groups in the support (≈20 μmol/g gel), were used, and the volume of distilled water was 3 mL per gram of gel. The substitution was performed at 83 °C for 72 h in a hybridization oven/shaker from Amersham Pharmacia Biotech. The gels were thoroughly washed with distilled water and stored in 20% (v/v) ethanol at 0–4 °C.
Solid-phase affinity ligands 3/3′ and 5/3′ were available from a previously synthesized combinatorial library following a similar methodology and contained ≈20 μmol ligand/g gel [18].
2.2.2. Enzyme Adsorption to Solid-Phase Synthesized Ligands
Enzyme adsorption to solid-phase synthesized ligands was performed by affinity chromatographic assays at room temperature as described in [18,19]. Sepharose CL-6B with bound ligand (0.5–0.75 mL) was packed into 4 mL columns and washed with 3 × 2 mL regeneration solution, 0.1 M NaOH in 30% (v/v) isopropanol, then with water to bring the pH to neutral and finally with equilibration buffer, 20 mM Tris-HCl, pH 8.0 or 20 mM acetate, pH 5.3 in the case of invertase. For each resin, 1–5 mL of enzyme solution with protein concentration between 0.5–1.5 mg/mL, depending on the purity of the sample containing each enzyme—in 20 mM Tris-HCl, pH 8.0—was loaded onto the column. Washing with equilibration buffer was performed, and 1 mL fractions were collected until the absorbance at 280 nm became lower than 0.005. Finally, the columns were washed with regeneration solution, followed by distilled water, and stored between 0 and 4 °C in 20% (v/v) ethanol. Adsorption yields (ηprot and ηact) were calculated based on the percentage of total protein adsorbed (measured by absorbance at 280 nm) and on the percentage of activity adsorbed according to the following equations (Equations (1) and (2)):
where mprot,n is the mass of protein of the fraction n and mprot,0 is the total mass of the initial enzyme solution.
where Actn is the total activity of fraction n and Act0 is the total activity of the initial enzyme solution.
2.2.3. Thermostability Assays with Free and Adsorbed Enzymes
Thermostability assays were performed by measuring the irreversible loss of activity upon incubation at 60 °C of both free enzymes and enzymes adsorbed to selected solid-phase synthesized ligands. The concentrations of free and immobilized enzymes were adjusted in each assay in order to measure activity in a reproducible mode with minimal error.
For thermostability assessment with adsorbed enzymes, the resins were removed from the column after the adsorption/washing steps, suspended in 1 mL of 20 mM Tris-HCl, pH 8.0, or 20 mM acetate, pH 5.3 in the case of invertase, and then centrifuged at 11,500 rpm for 5 min, allowing the complete settling of the gel.
The solutions/suspensions containing both free and bound enzymes were incubated in a dry bath, AccublockTM Digital Dry Bath from Labnet International, Lda (Edison, NJ, USA), at 60 °C. Samples (15–100 μL, depending on the assays) were taken at several times of incubation and added directly to an Eppendorf (Hamburg, Germany) already containing a solution of 20 mM Tris-HCl buffer, pH 7.0–8.0 or 20 mM acetate buffer, pH 5.3 in the case of invertase, for activity determination. Samples were immediately diluted in a large volume (1.0–1.5 mL) of buffer at room temperature. After a period of 15 min, to assure refolding of all molecules reversibly unfolded, enzyme activity was measured [18].
Deactivations observed were not always first-order processes. Therefore, activity decays were fitted either to a simple first-order kinetic model (exponential decay) or to four-parameter biexponential models [27,28] (see Supplementary Materials) using the method of least squares and the Solver tool of Microsoft® Excel® 2016. Fitting to these models was made to provide a phenomenological description of the data as well as to guide the eye on the graphic representation of the inactivation curves. However, mechanistic elucidation was not possible with the available data, and considerably different deactivation mechanisms can have the same formalism (Supplementary Materials). Nevertheless, simplifications of this type are of interest when little is known about the inactivation mechanisms, such as in the present work, to compare enzyme deactivation under similar conditions [29].
2.2.4. Enzyme Activity Assays
Lipase Activity Assays
The lipase activity assays followed the release of p-nitrophenol by reading the absorbance at 400 nm (ε = 15,400 M−1 cm−1) [30]. For all activity measurements, an electronic stirrer Model 300 from Rank Brothers Ltd. was adapted on the cuvette holder, and a Thermomix® MM thermocouple from B. Braun (Melsungen, Germany) was used to set and measure the temperature during the assay. Spectrophotometric readings were performed in 1 cm pathlength Hellma Cuvettes (Basel, Switzerland), in a Hitachi U-2000 spectrophotometer (Tokyo, Japan). The optimal synthetic substrate for each lipase was determined based on information gathered from the literature. In all assays, non-enzymatic hydrolysis of the substrate was measured and subtracted from the measured activity. One enzyme activity unit (U) was defined as the activity corresponding to formation of 1 μmol of p-nitrophenol per minute in standard assay conditions.
Activity towards p-NPA: For AOL, cutinase and CVL, the activity assays were performed in 20 mM Tris-HCl buffer at pH 7.0 or 8.0 at either 30 or 37 °C in a stirred cuvette, with a total reaction volume of 1.5 mL. AOL sample was added to reach a final concentration of 14 μg/mL of total protein; cutinase and CVL samples were added to obtain a final concentration of 16 nM of enzyme. In the case of RML, a volume was added to have a final concentration of 22 μg/mL of total protein. For all activity assays, the concentration was the same in free and immobilized enzyme assays. The reaction was initiated by the addition of 15 μL of stock substrate solution. The release of p-nitrophenol was monitored every 6 s for one minute.
Activity towards p-NPB: For RNL, activity assays were performed in 20 mM Tris-HCl buffer at pH 7.0 or 8.0 at either 30 or 37 °C in a stirred cuvette, with a total reaction volume of 1.5 mL. RNL sample was added to a final concentration of 6.4 μM for the free enzyme and 4 μM for the adsorbed enzyme. The release of p-nitrophenol was monitored for two minutes in the case of free enzymes or for five minutes in the case of the adsorbed enzymes.
Activity towards p-NPP: Activity of CRL towards p-nitrophenyl palmitate (p-NPP) was determined by a spectrophotometric assay adapted from [31].
This activity assay required two solutions: solution 1, or stock substrate, 8 mM of p-NPP in propane-1-ol, and solution 2, 4.4% (v/v) Triton X-100 (Dramstadt, Germany), 1.1% (w/v) gum arabic in 20 mM Tris-HCl buffer at pH 7.0 or 8.0. The assay was performed at 30 or 37 °C in a stirred cuvette, with a total reaction volume of 2 mL. A mixture containing 1780 μL of solution 2 and 180 μL of solution 1 was added to the cuvette, and the reaction was started by addition of 40 μL of enzyme solution, either in the case of free or adsorbed enzymes. The release of p-nitrophenol was monitored every 6 s for one minute in the case of free enzymes or every second during three minutes in the case of the adsorbed enzyme.
Invertase Activity Assays
Invertase activity was measured by end-point assays using the 3,5-dinitrosalicylic acid (DNS) method for the quantification of reducing sugars in samples taken at different incubation times at 60 °C. In a 96-deep well plate (U96 MicroWell plate, NUNC™ (Thermo Fisher, Waltham, MA, USA), each well was filled with 980 μL of sucrose 5% (w/v) in 20 mM acetate buffer pH 5.3 and 20 μL of an invertase sample, either from free enzyme or adsorbed enzyme thermostability assay. The plate was covered and incubated for 30 min at 50 °C. After, 20 μL of each well was added to 80 μL of MilliQ water in another 96-deep well plate, and 100 μL of DNS reagent was added to each well. The plate was covered and incubated at 100 °C for 5 min. After cooling to room temperature (in a tap water bath), 500 μL of MilliQ water (Merck, Rahway, NJ, USA) was added to each well and the plate was shaken; 200 μL of the content of each well was transferred to a reading plate, and the absorbance was read at 540 nm on a SpectroMax384 Plus microplate reader from Molecular Devices (San Jose, CA, USA). A calibration curve, with glucose standards in a suitable concentration range, was used for determination of the content in reducing sugars and calculation of the activity. One enzyme activity unit (U) was defined as the activity corresponding to hydrolysis of 1 μmol of sucrose to inverted sugar per minute under assay conditions [32].
2.2.5. Protein Determination by the BCA Assay
A microplate assay was used to determine protein concentrations by the BCA method [33] according to the instructions in the BCATM Protein Assay kit (suitable for concentrations ranging from 20 to 2000 μg/mL) or the Micro BCATM Protein Assay kit (for concentrations between 2 and 40 μg/mL) from Thermo Scientific (Waltham, MA, USA).
3. Results and Discussion
Biomimetic ligands were assessed as probes to enhance protein stability by immobilization based upon affinity-like interactions. Different lipases were adsorbed to selected dipeptide-mimic triazine-scaffolded affinity ligands and tested for improved thermal stability. In previous studies with the same rationale, cutinase from the phytopathogen fungus Fusarium solani pisi, a 197-residue protein with a molecular weight of 22 kDa [34] and an isoelectric point of 7.8 [35], was chosen as a model protein.
Cutinase is an enzyme that catalyzes the hydrolysis of insoluble biopolyester cutin, a structural component of plants, being the smallest member of serine hydrolases family with dual properties of lipase and esterase. In addition to cutin, cutinase hydrolyses a variety of soluble and insoluble synthetic polymer esters, extending from p-nitrophenyl esters to insoluble long-chain triglycerides (lipolytic activity) [36,37,38,39].
3.1. Characterization of Enzyme Preparations by SDS-PAGE
Cutinase from Fusarium solani pisi and lipases from Aspergillus oryzae (AOL), Candida rugosa (CRL), Chromobacterium viscosum (CVL), Rhizomucor miehei (RML) and Rhizopus niveus (RNL) have been fully characterized in previous studies. The enzyme preparations were analyzed by SDS-PAGE (see Figure S1), and the molecular weight of the protein bands observed were compared with those reported in the literature. Although some of the commercial preparations were less pure than others, this variability was not a limitation to conduct the planned experiments, as it remained possible to distinguish the bands corresponding to each lipase in all cases.
Cutinase from F.s.pisi was produced in our laboratory and purified by a well-established protocol as described in [26]. The purified enzyme presented a molecular weight of ≈23 kDa, a value identical to that previously reported [40,41,42]. The commercial lipase designated as Chromobacterium viscosum lipase (which is identical to the lipase from Pseudomonas glumae [43]) is an enzyme with 319 amino acid residues, a molecular mass of 33 kDa and an isoelectric point of 7.1. The C. viscosum lipase presented a major protein band at 33 kDa, as expected. Some bands of low molecular weight could also be detected and are due to proteolytic degradation during purification and/or storage [43]. Lipase from A. oryzae was characterized as a monomeric enzyme with a molecular mass of 41 kDa [44]. The solution containing Aspergillus oryzae lipase presented two thick bands at approximately 50 kDa, and one band at approximately 41.5 kDa, which was attributed to the lipase. For the Rhizomucor miehei preparation, the protein band located at 31 kDa was identified as the lipase, according to the molecular weight described in the literature for this enzyme [45]. Two types of lipases were previously reported for R. niveus: lipase I and lipase II. Lipase I consists of two polypeptide chains: a small peptide of 52 amino acids with a sugar moiety (A-chain) and a large peptide of 297 amino acids. Lipase II is described as a single polypeptide chain with a molecular weight of 30 kDa [46]. The information provided by the manufacturer for the commercial preparation of R. niveus utilized in our study indicates an expected molecular weight of 83 kDa for the lipase. The strong band observed at around 81 kDa was therefore considered representative of the lipolytic enzyme. Finally, in the Candida rugosa preparation the band appearing at 63 kDa corresponds to the lipase, which has been characterized as a single polypeptide chain with 543 amino acids and a molecular weight of 60 kDa [47].
3.2. Enzymatic Activity Screening
The enzymatic activity of the different enzyme preparations was measured at two different temperatures, 30 and 37 °C, and two distinct pH values, 7.0 and 8.0 (Table 1), in order to select between these two conditions the best combination of pH and temperature for comparing free and adsorbed enzymes. The choice of such conditions was based on the knowledge that most lipases are typically stable and have their highest activity at pH values between 6.0 and 7.5 [48] and at temperatures between 30 and 50 °C. However, above 40 °C, the conformational stability of many lipases is compromised, leading often to a decrease in enzymatic activity [48]. Based on this preliminary assessment, a pair of conditions was then established for measuring the activity of each lipolytic enzyme under study. In the case of cutinase, a temperature of 30 °C was chosen for the activity assays to enable comparison with previous results.
3.3. Assessment of Binding of Ligand 3′/11 to Different Lipases
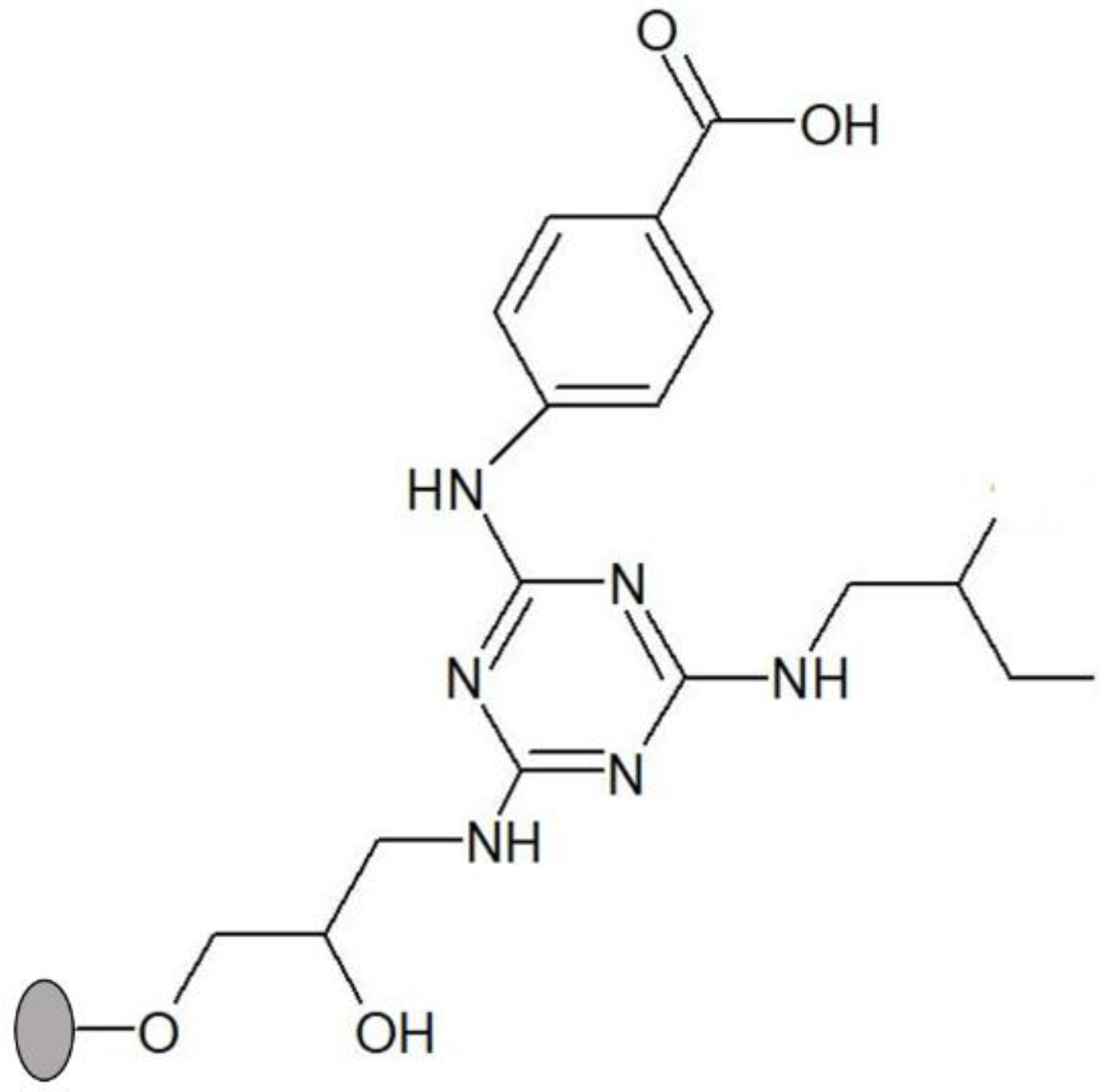
Screening of a biased second-generation 36-member solid-phase combinatorial library of triazine-scaffolded affinity ligands yielded several compounds as potential stabilizers for cutinase from F.s.pisi [18]. These synthetic affinity ligands, directly synthesized in agarose, contain cyanuric chloride as a scaffold to display two substituent groups, which consist of different aliphatic and aromatic amines mimicking the side chain of certain amino acids [17,23]. As such, these mimics are expected to interact with residues from the surface of the enzyme by intermolecular interactions, similar to those responsible for the maintenance of a protein’s tertiary structure. The enzyme’s stability is likely to be affected and potentially improved by these interactions. Amongst the ligands tested, solid-phase ligand 3′/11 (Figure 1) was chosen as a lead for subsequent thermal stability studies. This lead was proven to bind cutinase with high affinity while stabilizing bound cutinase at temperatures between 60 and 80 °C [18,19]. Ligand 3′/11 bears 4-aminobenzoic acid at the R1 position and 2-methylbutylamine at the R2 position and mimics the interaction of aspartic acid–isoleucine or glutamic acid–isoleucine dipeptides with the protein surface. Solid-phase synthesized supports with ligand 3′/11 enabled adsorbed cutinase to retain most of its activity (≈90%) after an incubation of 90 min at 60 °C when compared with the free enzyme, which retained only 16% of activity after incubation during the same period of time [18].
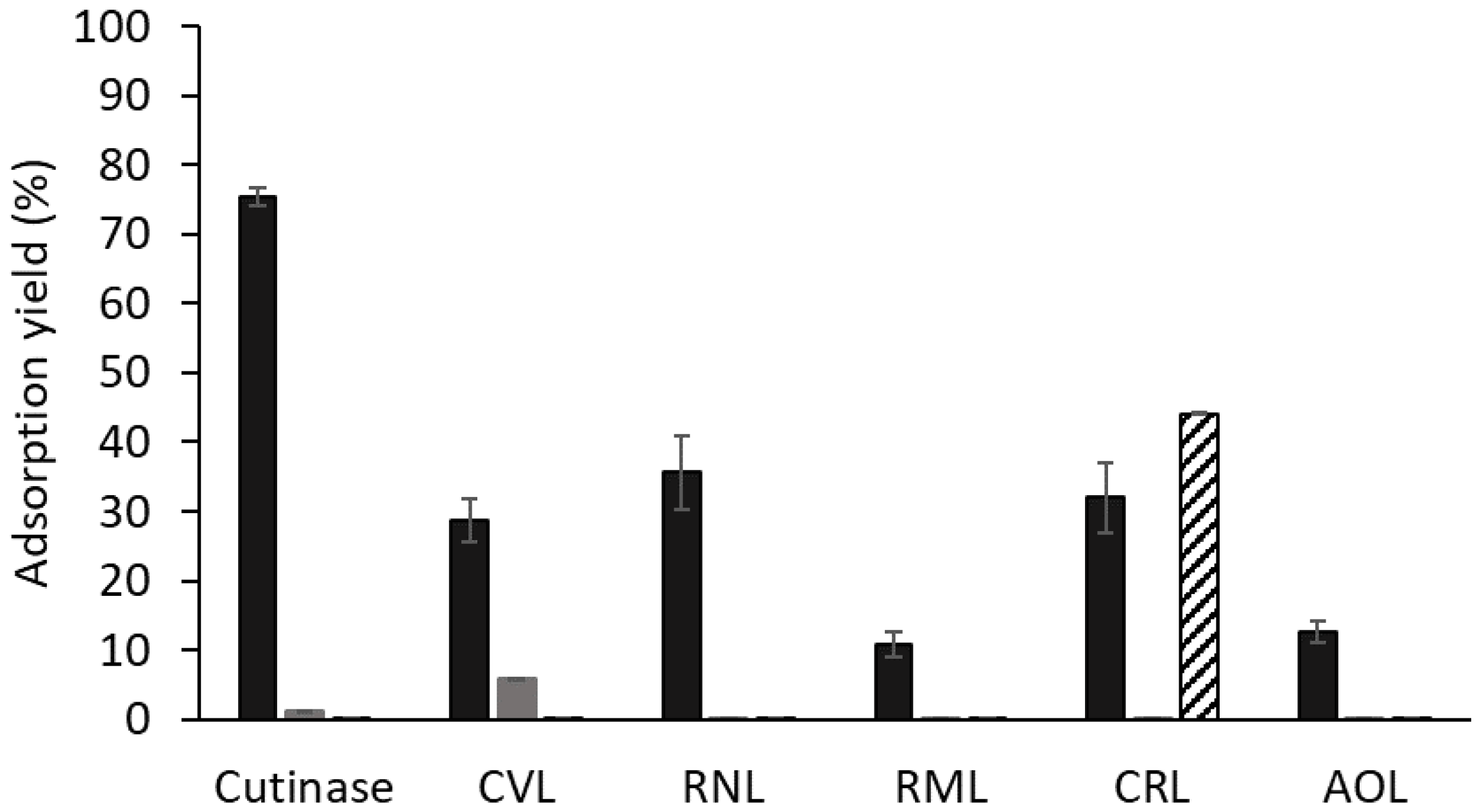
The adsorption of different preparations containing microbial lipases to solid-phase ligand 3′/11 (≈20 μmol ligand/g gel) was assessed, and those that consistently gave good results in terms of binding to this support were selected to perform thermal stability assays with free and bound enzymes. Figure 2 summarizes the protein adsorption yields (ηprot) to ligand 3′/11–Sepharose CL-6B and to both Sepharose CL-6B and ligand 0/0 (support in which the two substituents in the triazine ring (R1 and R2) are NH2 groups) as controls. Cutinase presented the highest adsorption yield of all tested enzymes, as expected.
Lipases from A. oryzae and R. miehei lipases exhibited low adsorption yields to ligand 3′/11, (13 ± 2%) and (11 ± 2%), respectively. SDS-PAGE analysis of the fractions collected in the breakthrough and washing steps of the column-binding assay of the R. miehei lipolytic preparation confirmed that the enzyme adsorption to ligand 3′/11 was low either at pH 7.0 (Figure S2A) or pH 8.0 (Figure S2B). Additionally, free A. oryzae lipase displayed a high thermal stability at 60 °C, preserving its biological activity over 24 h of incubation at pH 8.0. Therefore, further studies were not conducted with these two lipases.
Amongst the other enzymes screened, lipases from R. niveus and C. rugosa exhibited higher adsorption yields to ligand 3′/11 compared to lipase from C. viscosum (Figure 2). These lipases, along with lipase from C. viscosum, were chosen as potential candidates for further investigation aiming at enhancing their thermal stability through binding to solid-phase ligand 3′/11. The adsorption to controls was negligible for all enzymes except for the lipase from C. rugosa. The high adsorption of CRL to ligand 0/0 (control) indicates that other interactions in addition to those between the dipeptide mimetic ligand and functional groups in the protein surface may be involved in the adsorption process. At pH 8.0, CRL lipase (pI ≈ 4.5) is negatively charged, while diaminetriazinyl groups in ligand 0/0 support are neutrally charged (pKa around 5.0) [49]. Therefore, we can hypothesize that adsorption to ligand 0/0 is probably not due to ionic interactions with amine groups in R1 and R2 positions but likely to other type of non-specific interactions (e.g., between the triazine ring (scaffold) and hydrophobic surface groups).
The adsorption of each enzyme to ligand 3′/11 was also quantified by the enzymatic activity adsorption yield. The protein (ηprot) and activity (ηact) adsorption yields are summarized in Table 2.
The high ηprot (100%) and ηact (75%) obtained for cutinase are in accordance with the purity of the enzyme preparation used and the results reported by Sousa et al. [17,18]. The ηact (14.7%) for the lipase from C. viscosum was almost half of the protein adsorption yield (29%) obtained with this lipase (Table 2). This fact may be explained by two reasons: (i) proteins/peptides of low molecular weight present in the CVL preparation (see Figure S1), resulting from unspecific proteolysis during the purification and/or storage of CVL [43] are adsorbed but are likely inactive, thus increasing the value of protein adsorption yield (obtained by measurement of absorption at 280 nm); (ii) and/or the process of loading and washing affects lipase activity, thus reducing the value of ηact. Oppositely, in the case of lipase from C. rugosa, the ηact (70.9%) was more than two times higher (Table 2) than the protein adsorption yield (36%). According to the SDS-PAGE densitometry analysis of the breakthrough fractions collected during the CRL adsorption assay, it was found that approximately 30% of CRL did not bind to the ligand. This finding is consistent with the approximate ηact value of 70% obtained for this specific lipase.
The ηprot for RNL was low (32%), but activity could be clearly measured in the agarose support after enzyme adsorption. However, the yield in activity could not be accurately quantified since the measuring of activity was only possible for protein concentrations above 5.0 μM (or 0.42 mg/mL), and not all fractions collected from the column-binding assay contained protein concentration in this range. Therefore, the fractions collected in the RNL assay were analyzed by SDS-PAGE (Figure S3). The intensity of the bands representing the lipase of R. niveus in the breakthrough, first and second washings were significantly weaker when compared to the load with similar total protein mass loading. In light of this analysis and based on the fact that activity could be clearly measured with the bound enzyme, the ηact (activity recovery) for this lipase was estimated to be approximately 100% (Table 2).
3.4. Thermal Stability of Lipases Free and Adsorbed to Ligand 3′/11
The thermal stability of different enzymes free and adsorbed to solid-phase ligand 3′/11 (≈20 μmol ligand/g moist gel) was studied at 60 °C and pH 8.0. The selection of these temperature and pH conditions aimed to enable a straightforward comparison with cutinase, which served as the model protein in previous studies. Under these conditions, it has been shown that the bound enzyme exhibited a significantly slower rate of irreversible activity loss in comparison to the free enzyme [18,19].
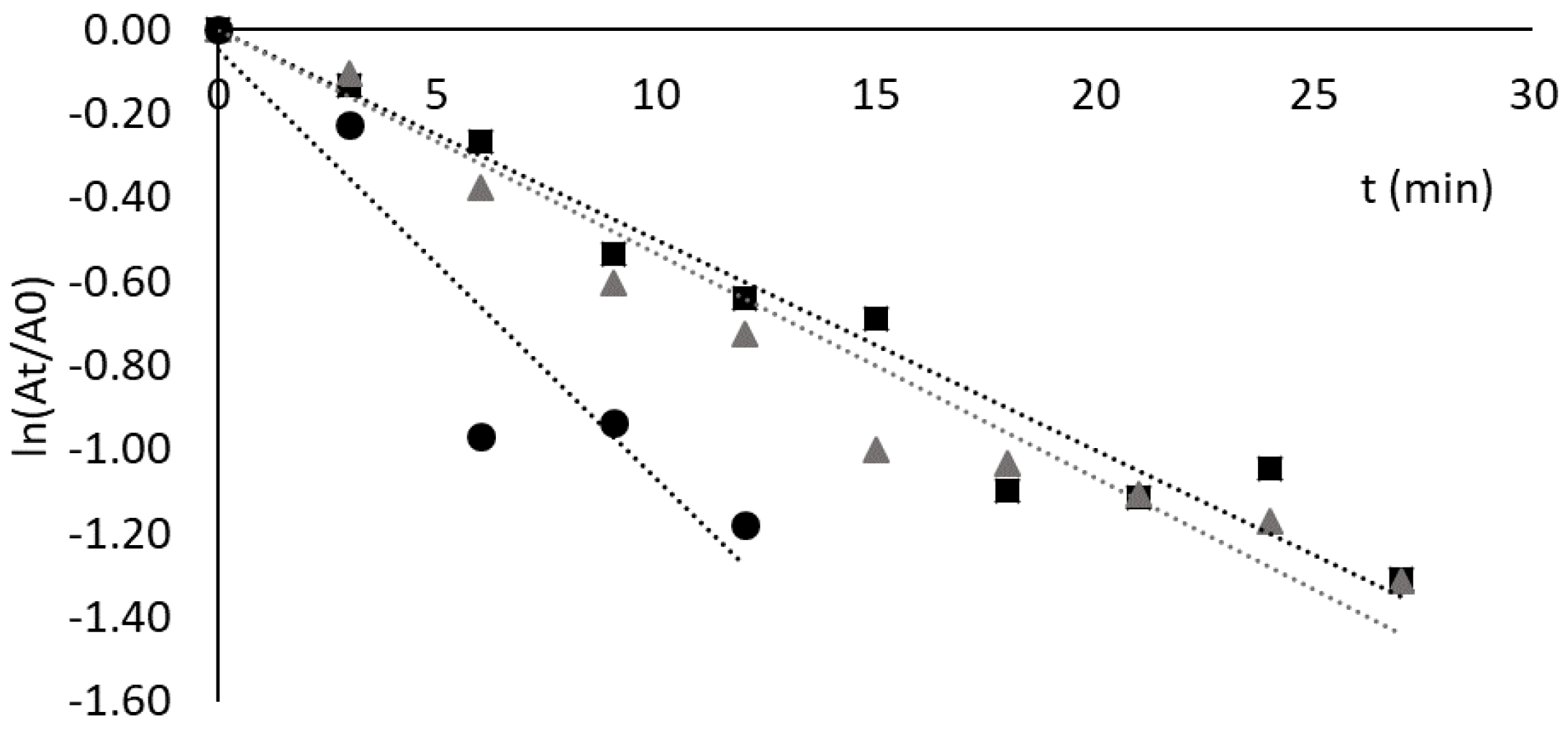
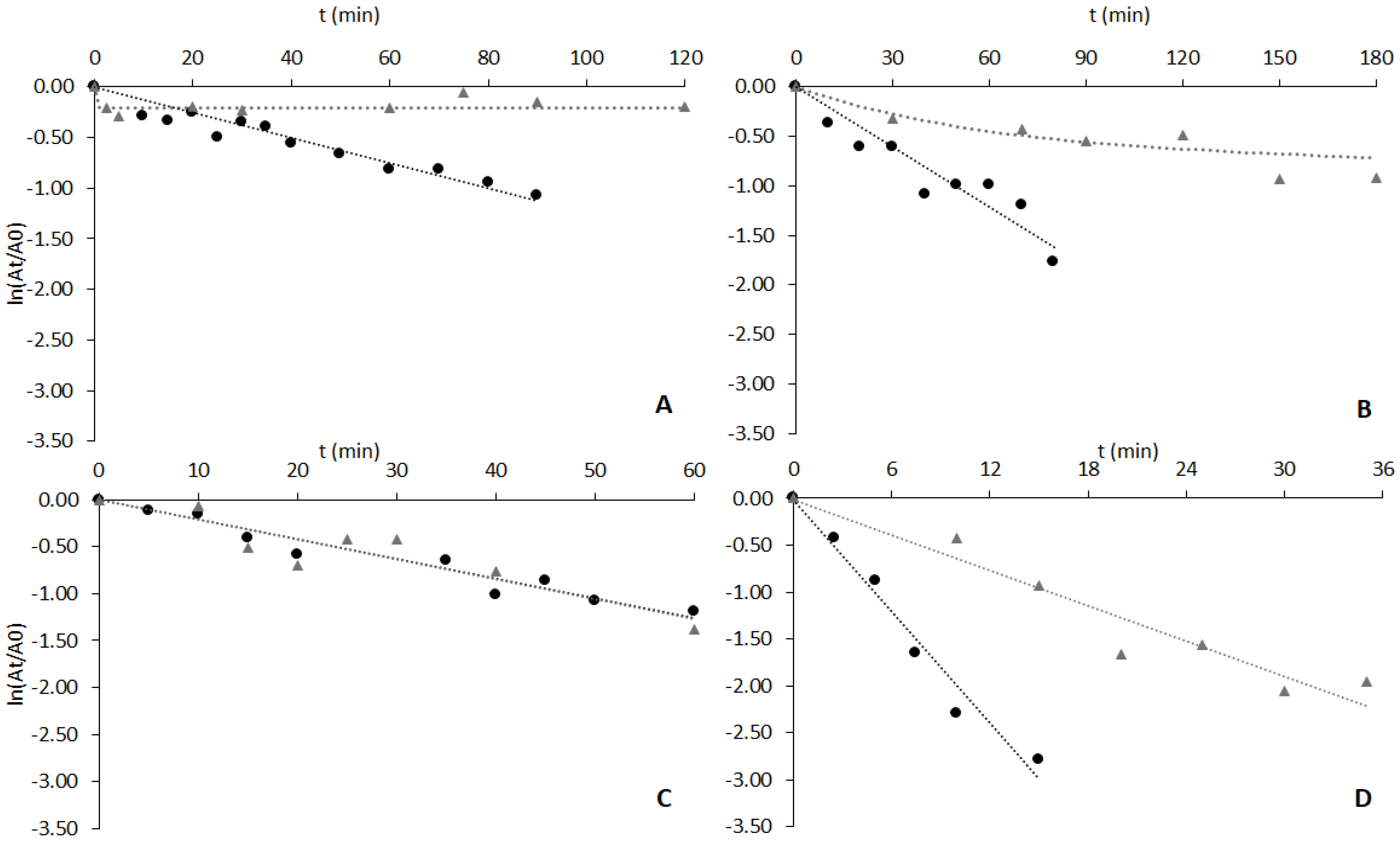
The data on Figure 3A show that deactivation of free cutinase follows a first-order kinetics characteristic of free enzymes. The half-life time (t1/2) obtained was around 50 min, a value slightly higher than the one reported in [19]—35 min—at the same pH and temperature. In the case of cutinase adsorbed to solid-phase ligand 3′/11, the deactivation profile was fitted to a four-parameter biexponential model of Henley and Sadana [27]. The values obtained for the coefficients and constants of the model (Supplementary Materials) are in accordance with the biphasic deactivation that was observed. The deactivation profile obtained shows an initial significant decrease in the relative enzyme activity, which was not observed by Sousa et al. under identical conditions [19]. The specific activity of adsorbed cutinase in this work (105 U/mg) was different from that reported in [19] (50 U/mg), which may account for slight differences in the activity decay. Nevertheless, in both studies, the residual relative activity of adsorbed cutinase was very similar after 90 min of incubation at 60 °C and pH 8.0 [18], with values of 87% and 94%, respectively.
The ability of cutinase, when bound to ligand 3′/11 through affinity-like interactions, to remain stable at temperatures above 60 °C has been attributed to a balanced distribution of polar, charged and non-polar residues on its surface. These properties likely create suitable binding sites for a ligand such as 3′/11, bearing a combination of moieties (charged and hydrophobic), facilitating complementary interactions [50]. Ligand density in the solid matrix was found to be a determinant parameter for stabilization, which was not achieved for resins with ligand densities lower than approximately 14 μmol/g moist gel [19]. Palanisamy et al. described a similar effect when studying the interaction of glucose oxidase with a triazine-based synthetic ligand at various ligand densities. At ligand densities lower than 11 μmol/g moist gel, ligand molecules were spaced too far apart to create an effective ligand–macromolecule complex [51]. Scoble et al. also found, when studying the adsorption of human IgG to divinylsulfone (DVS)-based gels, that a minimum ligand density was required for efficient IgG binding to occur [52]. The fact that a minimum ligand density has been identified as a requisite for stabilization of bound cutinase suggests that binding to ligand molecules close enough to each other is necessary to allow a favorable and stabilizing attachment of the enzyme molecule. Small dipeptide-mimic ligand molecules with a triazine scaffold are likely able to find more than one site on the protein surface where binding with more or less affinity is possible, leading to effective protein–ligand macromolecular configurations that can restrict structural motions for unfolding and enhance stability [17,18].
Figure 3B shows the results of the thermal stability assays with CVL free and adsorbed to solid-phase ligand 3′/11. The deactivation of free lipase followed a first-order kinetics, with a half-life time (t1/2) of 39 min. The deactivation profile obtained for CVL adsorbed to ligand 3′/11 (Figure 3B) was fitted to a four-parameter biexponential model of Aymard and Belarbi [28]. The immobilized enzyme retained approximately 60% of initial activity after 3 h of incubation at 60 °C and was, therefore, clearly stabilized by the ligand. However, the specific activity of adsorbed lipase was only 8% of that of free enzyme (an activity retention much lower than in the case of cutinase). It is conceivable that the reduction in specific activity may be surpassed by optimizing both ligand composition and density on the support [19]. It is worth noting that the ligand 3′/11 may not be the most specific or optimal for CVL stabilization compared to cutinase.
The deactivation profiles of lipase from C. rugosa free and adsorbed to Sepharose CL-6B derivatized with ligand 3′/11 are depicted in Figure 3C. The half-life time obtained for the free enzyme (by a first-order kinetics) was 35 min, and after 60 min the lipase presented 30% of residual activity in contrast to the value of 10% reported by Soares et al. [53]. The difference between the two results can be explained by the slightly different conditions used in the enzymatic assays, namely 20 mM Tris-HCl, pH 8.0 in the present work and 100 mM phosphate, pH 7.5 in the study of Soares et al. [54].
The deactivation of CRL adsorbed to ligand 3′/11 could also be fitted to a first-order kinetic law with a half-life time of 35 min (Figure 3C).
As a control, protein was quantified in the supernatants of the resin suspensions after the thermal stability assays to verify if deactivation of bound enzymes could partially be explained by leaking from the adsorbent. No significant amount of protein was detected in the supernatants. This means that in the experimental conditions used in the assay, CRL is tightly bound and does not leak from the solid support. It is interesting to notice that the specific activity of adsorbed CRL was higher than that of the free lipase (0.8 ± 0.2 U/mg of free lipase vs. 2.8 ± 0.8 U/mg of adsorbed enzyme). This result might indicate that CRL is aggregated in solution, which corroborates the very low activity measured for this enzyme (Table 1). Aggregation may also have a stabilizing effect on proteins, and this possibly explains the similarity in the activity decay of both free and immobilized enzymes.
The results obtained with lipase from R. niveus free and adsorbed to ligand 3′/11 are presented in Figure 3D. A half-life time of 3.5 min was obtained for the free enzyme (exponential decay). This value is not comparable, however, with other values reported in the literature [46,54] where lipases from the same organism were used. The adsorbed lipase activity decay (Figure 3D) could also be fitted by a first-order kinetic law with a half-life time of 11 min, three times higher than that of the free enzyme. However, after 35 min, the enzymatic activity of bound enzyme was only 14% of the initial value (a stabilizing effect much less effective than in the cases of cutinase and CVL). The measurement of the quantity of protein in solution during the thermal stability assay confirmed that the lipase was tightly adsorbed to the support (less than 10% of the bound protein could be detected in the supernatant after 30 min of incubation at 60 °C).
3.5. Adsorption and Thermostability Assays with CRL Adsorbed to Other Dipeptide-Mimic Ligands
The deactivation profiles of lipase from Candida rugosa free and adsorbed to ligand 3′/11 were similar (Figure 3C). Therefore, other available ligands from the previous 36-member combinatorial library [18] were tested for adsorption and potential stabilization of C. rugosa lipase.
The criteria for choosing ligands alternative to 3′/11 were as follows: (i) mixed ligands containing both a hydrophobic and an acidic group that were reported to display a stabilizing effect on cutinase and (ii) availability in an amount sufficient to be used in binding and thermal stability assays. Following these criteria, two additional ligands were selected, designated as 3/3′ and 5/3′, respectively (Figure 4). Ligand 3/3′ comprises tyramine as substituent in the R1 position (mimic of tyrosine) and 4-aminobenzoic acid in the R2 position (mimic of aspartic/glutamic acids); ligand 5/3′ comprises isoamylamine as substituent in the R1 position (mimic of Leucine) and 4-aminobenzoic acid in the R2 position [17,18].
Adsorption yields of CRL to solid-phase ligands 3/3′ and 5/3′ were (43 ± 2)% and (31 ± 1)%, respectively. The highest adsorption yield of CRL was achieved with ligand 3/3′ (containing as substituents two amines with an aromatic ring), while the value obtained with ligand 5/3′ was similar to that obtained with ligand 3′/11 (both with only one amine containing an aromatic ring). However, the adsorption yield of CRL to ligand 0/0 (control) was also high (44%) and comparable to that obtained with ligand 3/3′. This result seems to indicate that in the case of CRL the triazine ring (and not only R1 and R2 substituents) may contribute to the adsorption process.
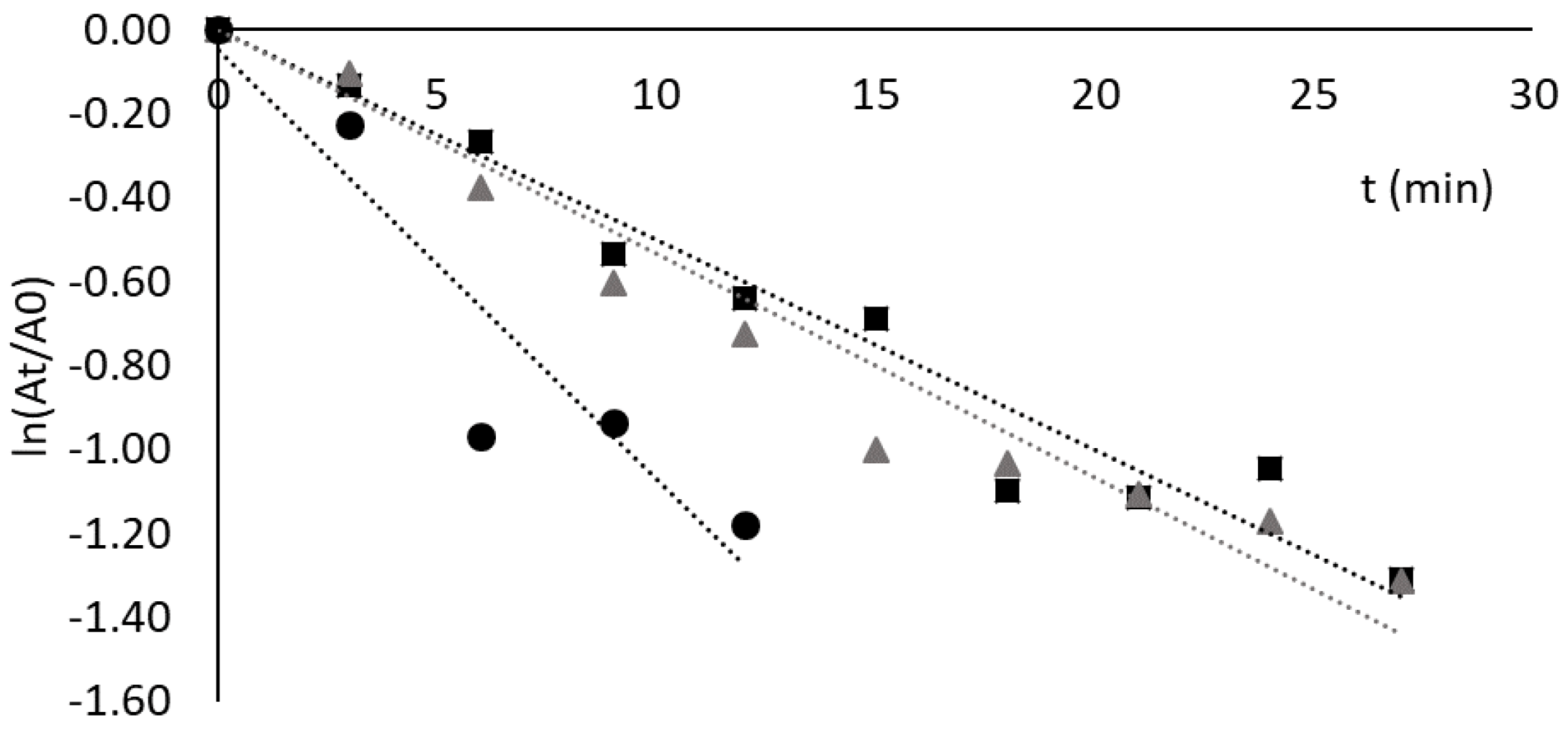
The deactivation profile of lipase from C. rugosa free and adsorbed to ligand 3/3′ at pH 8.0 and 60 °C was studied, and the results are shown in Figure 5. As can be observed, adsorption to solid-phase ligand 3′/3 resulted in significant stabilization of this lipase. The deactivation kinetics could be fitted by the model of Henley and Sadana [27], with two phases of deactivation. The difference observed in estimated rate constants k1 and k2 (see Supplementary Materials) indicates that an intermediate conformation, E1, is produced 10 times slower than the final enzyme conformation, E2. However, the initial deactivation and relative decline in enzyme activity are likely due to the exclusive formation of the intermediate conformation, E1, in the first 20–30 min of incubation. After 40 min of incubation, the enzyme conformation E2 increases its concentration relative to E1 and reaches a plateau (yielding approximately 57% of the initial activity).
When comparing the stabilizing effect of ligands on different lipases, while ligand 3′/11 has been shown to have a stabilizing effect on adsorbed cutinase, CVL and RNL, the lipase of C. rugosa (CRL) was not stabilized when bound to ligand 3′/11. However, a significant stabilization was observed when CRL was adsorbed to solid-phase ligand 3′/3. The main difference between the two dipeptide-mimic ligands is that ligand 3′/3 comprises two mimic substituents with aromatic rings (tyramine and 4-aminobenzoic acid), while in ligand 3′/11 one of the substituents (2-methylbutylamine) does not contain an aromatic ring. The structure of lipase from C. rugosa was determined and refined at 2.1 Å resolution. Two main conformational states were reported for the lipase in aqueous solution. The first, designated as ‘open’, represents the enzyme’s active conformation, and the second, designated as ‘closed’, where a loop with an elongated shape lies flat on the protein surface above the active site, corresponds to the inactivated form of the lipase. The open conformation is a result of the movement of this loop or flap, turning the active site accessible to the solvent and presumably to the substrate [55]. This loop has an amphipathic character, with hydrophobic residues in the inner part over the active site and with an upper side (exposed to the solvent) distinctly hydrophilic. The stabilization achieved when C. rugosa lipase was adsorbed to a ligand with phenyl groups in both arms may suggest that phenylalanine residues located on the hydrophobic side of the flap might interact with such groups and play a major role in the attained stabilization of the lipase with ligand 3/3′. However, hypothetical explanations about the mechanism of stabilization of enzymes by biomimetic/dipeptide-mimic affinity ligands must be further confirmed by (ongoing) in silico molecular modeling studies that shall provide valuable information to explain the results obtained and unveil likely scenarios for protein–ligand interaction at the molecular level.
It is, however, relevant to emphasize that the adsorption yield and the observed stabilizing effect were diverse for the same ligand (3′/11) and different lipases, as well as the fact that slightly different biomimetic ligands had a significantly different stabilizing effect on C. rugosa lipase.
3.6. Adsorption and Thermostability of Invertase Free and Adsorbed to Different Biomimetic Ligands
Ligand 3′/11, 3/3′ and 5/3′ were also screened as affinity-like binders to invertase from S. cerevisiae, a protein functionally and structurally more distant from cutinase than the other tested enzymes. Invertase has relevant applications in the food industry and catalyzes the hydrolysis of sucrose into an equimolar mixture of glucose and fructose, known as inverted sugars [56]. The main structure of the external invertase from S. cerevisiae is a homodimer with a molecular weight of 270 kDa and an isoelectric point between 3.4 and 4.4.
Adsorption yields were high for the three ligands tested, with values of (93.5 ± 0.2)%, (90 ± 2)% and (82 ± 5)% for ligands 3′/11, 3/3′ and 5/3′, respectively. Ligands with an amine having a phenyl group in position R1 (3′/11 and 3/3′) presented slightly higher adsorption yields for invertase than the ligands with an aliphatic amine in the same position (5/3′). Adsorption to Sepharose CL-6B (used as control) was also significant for invertase (68 ± 6)%, indicating that that adsorption to ligand-derivatized supports might not be entirely specific but also mediated by non-specific interactions with the matrix.
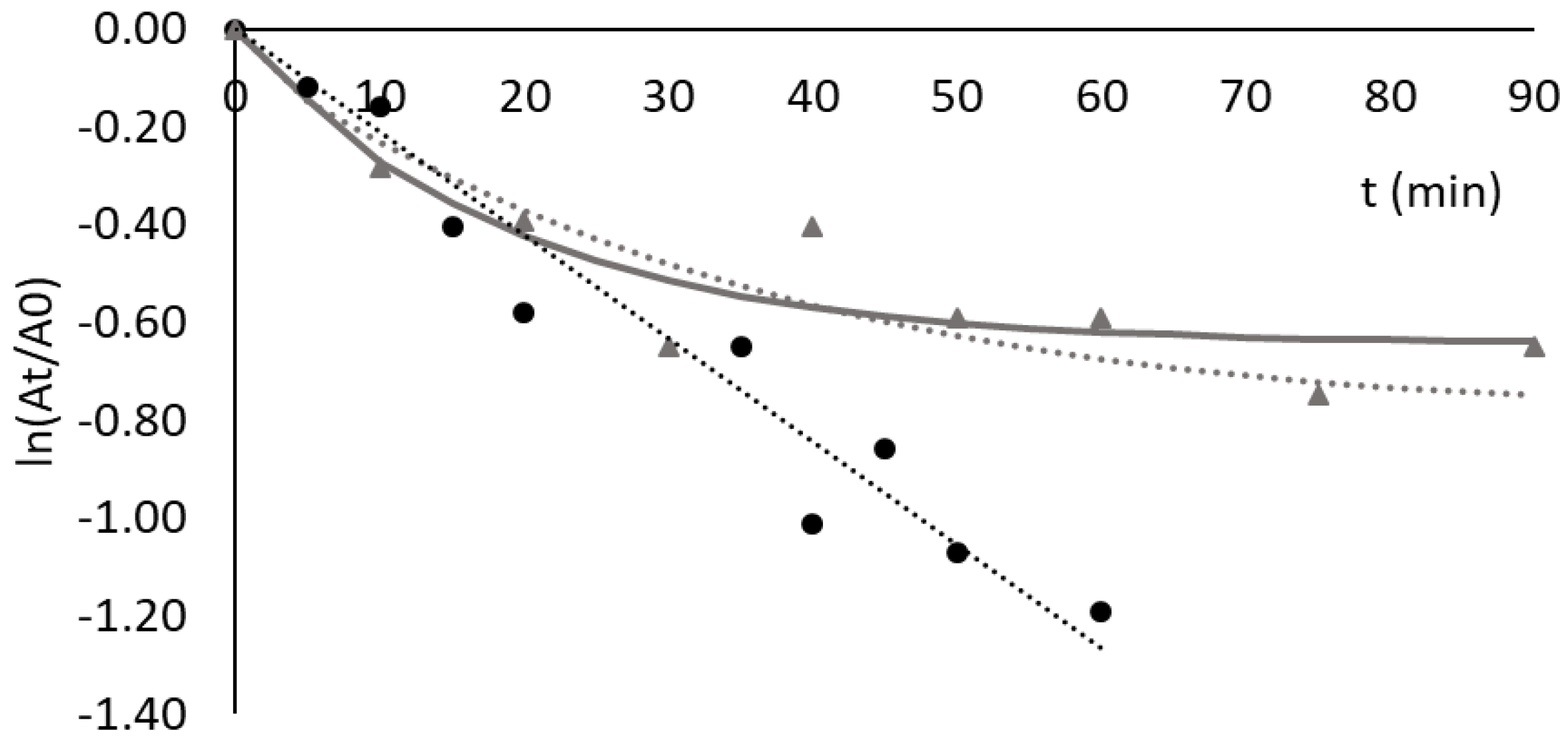
The deactivation of the free enzyme was studied at 60 °C and pH 5.3. This pH value was chosen to favor enzyme activity while preventing any desorption from the support. The activity decay followed a first-order exponential law, with a t1/2 of 14 min (Figure 6). This value is comparable to the one described by Andjelković et al. (12 min at 60 °C in deionized water) [57]. In the present work, invertase incubated in 20 mM acetate buffer, pH 5.3 at 60 °C, after a period of 25 min presented 35% of initial enzyme activity, while Andjelković et al. reported a value of 20%, in deionized water at 60 °C, under similar conditions [57].
Given the significant adsorption (68 ± 6%) of invertase onto Sepharose CL-6B gel (control), an additional experiment was conducted to ascertain whether this adsorption also resulted in the stabilization of the enzyme. The data in Figure 6 show that binding to Sepharose CL-6B had a destabilizing effect on bound invertase (t1/2 of 6.8 min). Furthermore, it was observed that ligands 3′/11, 5/3′ and 3/3′ showed no stabilizing effect on adsorbed invertase. The deactivation profile of invertase bound to ligand 3′/11 was very similar to the one observed for the free enzyme, with a t1/2 of 13 min (Figure 6). In the case of the other two ligands, after 30 min the residual activity of invertase immobilized in solid-phase 5/3′ and 3/3′ ligands was 14 and 16%, respectively, both below the corresponding value of free enzymes (27%).
These results indicate that a high adsorption yield does not always result in a stabilizing effect on bound enzymes, thus evidencing that the composition of the ligand and specificity of binding on the protein’s surface are definitely key factors for achieving stabilization.
4. Conclusions
Adsorption to solid-phase dipeptide-mimic synthetic affinity ligands was formerly proposed and found to be effective in the thermal stabilization of cutinase from F. solani pisi at 60 °C. Such biomimetic ligands, bearing structural motifs mimicking the side chains of different amino acids, can interact with specific residues on the surface of the enzyme by intermolecular interactions similar to those responsible for the maintenance of a protein’s structure. If this interaction takes place at unfolding-prone regions, structural stabilization is expected to occur. In the present study, triazine-scaffolded solid-phase lead ligands with a thermal stabilizing effect on cutinase were screened as potential binders/thermostabilizers of some microbial lipases, enzymes functionally related to cutinase. Adsorption of C. viscosum and R. niveus lipases to ligand 3′/11 showed that although the binding yield was much lower when compared to cutinase, stabilization of bound enzyme at 60 °C was still observed. Oppositely, lipase from C. rugosa was bound with high yield but was not stabilized by ligand 3′/11. Stabilization of this lipase was, however, achieved with an alternative ligand (3′/3), with a different mimetic composition compared to ligand 3′/11. An entirely distinct enzyme, invertase from Saccharomyces cerevisiae, was also assessed for binding to the same set of ligands and was not stabilized by any of the tested compounds.
The results obtained provide additional proof of concept that triazine-scaffolded affinity ligands may be used as a new tool to modulate enzymes’ stability upon binding to target regions on the protein surface without affecting the enzymes’ functionality. It became clear that a high adsorption yield does not always result in a stabilizing effect on the bound enzyme, thus evidencing that the composition of the ligand and the binding to specific amino acid residues on the protein’s surface are definitely key factors for achieving stabilization. Nevertheless, additional investigations involving in silico modeling studies and a broader array of ligands, specifically designed/selected for different proteins, are being conducted to complement the preliminary findings herein presented. These studies shall provide comprehensive insight into the binding modes of triazine-based ligands to diverse enzymes and their correlation with observed stabilizing effects.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pr12020371/s1. Figure S1. SDS-PAGE analysis of different enzyme preparations. Lane 1: Precision Plus ProteinTM Dual Color Standards (kDa); lane 2: cutinase (10 μg of total protein); lane 3: CVL (10 μg of total protein); lane 4: AOL (30 μg of total protein); lane 5: RML (30 μg of total protein); lane 6: RNL (30 μg of total protein); lane 7: CRL (35 μg of total protein). The arrows indicate the band with the molecular weight corresponding to the lipase protein in each sample. Figure S2. SDS-PAGE analysis of the load and fractions collected in the adsorption assay of RML. Lane 1: Molecular Weight Markers. Assay at pH 7 (A): lane 2: load (8 μg of total protein); lane 3: breakthrough (4 μg of total protein); lane 4: first wash (3 μg of total protein). Assay at pH 8 (B): lane 5: load (12 μg of total protein); lane 6: breakthrough (3 μg of total protein); lane 7 (8 μg of total protein): wash 1. The box surrounds the molecular weight zone of RML. Table S1. Values obtained by fitting the models of Aymard and Belarbi and of Henley and Sadana to the activity decay of the different enzymes adsorbed to triazine-based synthetic affinity ligands.
Author Contributions
M.Â.T. conceptualized/designed the study, provided resources and supervised the work; D.F.-F. executed the experiments within the framework of a M.Sc. thesis, processed the data and prepared figures and tables; D.F.-F. and M.Â.T. analyzed the data and wrote the manuscript; M.Â.T. edited and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Portuguese Foundation for Science and Technology (FCT). The authors acknowledge FCT funds in the scope of projects UIDB/04565/2020 and UIDP/04565/2020 of iBB and project LA/P/0140/2020 of i4HB.
Data Availability Statement
Data are included within this article and its Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Petersen, S.B.; Harald Jonson, P.; Fojan, P.; Petersen, E.I.; Neves Petersen, M.T.; Hansen, S.; Ishak, R.J.; Hough, E. Protein engineering the surface of enzymes. J. Biotechnol. 1998, 66, 11–26. [Google Scholar] [CrossRef]
- Jaenicke, R. Stability and stabilization of globular proteins in solution. J. Biotechnol. 2000, 79, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Melo, E.P.; Faria, T.Q.; Martins, L.O.; Gonçalves, A.M.; Cabral, J.M. Cutinase unfolding and stabilization by trehalose and mannosylglycerate. Proteins 2001, 42, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Pyne, P.; Samanta, N.; Ebbinghaus, S.; Mitra, R.K. Thermal stability modulation of the native and chemically-unfolded state of bovine serum albumin by amino acids. Phys. Chem. Chem. Phys. 2019, 22, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Guzik, U.; Hupert-Kocurek, K.; Wojcieszyńska, D. Immobilization as a strategy for improving enzyme properties-application to oxidoreductases. Molecules 2014, 19, 8995–9018. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lopez, L.; Pedrero, S.G.; Lopez-Carrobles, N.; Gorines, B.C.; Virgen-Ortíz, J.J.; Fernandez-Lafuente, R. Effect of protein load on stability of immobilized enzymes. Enzym. Microb. Technol. 2017, 98, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Boudrant, J.; Woodley, J.M.; Fernandez-Lafuente, R. Parameters necessary to define an immobilized enzyme preparation. Process Biochem. 2020, 90, 66–80. [Google Scholar] [CrossRef]
- Bilal, M.; Zhao, Y.; Rasheed, T.; Iqbal, H.M.N. Magnetic nanoparticles as versatile carriers for enzymes immobilization: A review. Int. J. Biol. Macromol. 2018, 120, 2530–2544. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.; Ripoll, M.; Jackson, E.; Grazú, V.; Betancor, L. Stabilization of b-Glucuronidase by Immobilization in Magnetic-Silica Hybrid Supports. Catalysts 2020, 10, 669. [Google Scholar] [CrossRef]
- Mansfeld, J.; Vriend, G.; Van den Burg, B.; Eijsink, V.G.H.; Ulbrich-Hofmann, R. Probing the unfolding region in a thermolysin-like protease by site-specific immobilization. Biochemistry 1999, 38, 8240–8245. [Google Scholar] [CrossRef]
- Mansfeld, J.; Ulbrich-Hofmann, R. Site-specific and random immobilization of thermolysin-like proteases reflected in the thermal inactivation kinetics. Biotechnol. Appl. Biochem. 2000, 32, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, J.M.; Nidetzky, B. Positively charged mini-protein zbasic2 as a highly efficient silica binding module: Opportunities for enzyme immobilization on unmodified silica supports. Langmuir 2012, 28, 10040–10049. [Google Scholar] [CrossRef] [PubMed]
- García-García, P.; Guisan, J.M.; Fernandez-Lorente, G. A mild intensity of the enzyme-support multi-point attachment promotes the optimal stabilization of mesophilic multimeric enzymes: Amine oxidase from Pisum sativum. J. Biotechnol. 2020, 318, 39–44. [Google Scholar] [CrossRef]
- Matos, M.J.B.; Pina, A.S.; Roque, A.C.A. Rational design of affinity ligands for bioseparation. J. Chromatogr. A 2020, 1619, 460871. [Google Scholar] [CrossRef]
- Ye, L.; Xu, A.; Cheng, C.; Zhang, L.; Huo, C.; Huang, F.; Xu, H.; Li, R. Design and synthesis of affinity ligands and relation of their structure with adsorption of proteins. J. Sep. Sci. 2011, 34, 3145–3150. [Google Scholar] [CrossRef]
- Kim, J.Y.H.; Lee, J.W.; Lee, W.S.; Ha, H.-H.; Vendrell, M.; Bork, J.T.; Lee, Y.; Chang, Y.-T. Combinatorial Solid-Phase Synthesis of 4,6-Diaryl and 4-Aryl, 6-Alkyl-1,3,5-triazines and Their Application to Efficient Biofuel Production. ACS Comb. Sci. 2012, 14, 395–398. [Google Scholar] [CrossRef]
- Ruiu, L.; Roque, A.C.A.; Taipa, M.A.; Lowe, C.R. De novo design, synthesis and screening of a combinatorial library of complementary ligands directed towards the surface of cutinase from Fusarium solani pisi. J. Mol. Recognit. 2006, 19, 372–378. [Google Scholar] [CrossRef]
- Sousa, I.T.; Ruiu, L.; Lowe, C.R.; Taipa, M.A. Synthetic affinity ligands as a novel tool to improve protein stability. J. Mol. Recognit. 2009, 22, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sousa, I.T.; Lourenço, N.M.T.; Afonso, C.A.M.; Taipa, M.A. Protein stabilization with a dipeptide-mimic triazine-scaffolded synthetic affinity ligand. J. Mol. Recognit. 2013, 26, 104–112. [Google Scholar] [CrossRef]
- Chen, S.; Su, L.; Chen, J.; Wu, J. Cutinase: Characteristics, preparation, and application. Biotechnol. Adv. 2013, 31, 1754–1767. [Google Scholar] [CrossRef]
- Roussel, A.; Amara, S.; Nyyssölä, A.; Mateos-Diaz, E.; Blangy, S.; Kontkanen, H.; Westerholm-Parvinen, A.; Carrière, F.; Cambillau, C. A cutinase from Trichoderma reesei with a lid-covered active site and kinetic properties of true lipases. J. Mol. Biol. 2014, 426, 3757–3772. [Google Scholar] [CrossRef]
- Creveld, L.D.; Amadei, A.; van Schaik, R.C.; Pepermans, H.A.M.; de Vlieg, J.; Berendsen, H.J.C. Identification of functional and unfolding motions of cutinase as obtained from molecular dynamics computer simulations. Proteins Struct. Funct. Bioinform. 1998, 33, 253–264. [Google Scholar] [CrossRef]
- Sousa, I.T.; Taipa, M.A. Biomimetic Affinity Ligands for Protein Purification. In Protein Downstream Processing: Design, Development and Application of High and Low-Resolution Methods; Labrou, N., Ed.; Methods in Molecular Biology Series; Springer Science: Berlin/Heidelberg, Germany, 2021; Volume 1129, Chapter 14; pp. 231–262. [Google Scholar] [CrossRef]
- Zucca, P.; Fernandez-Lafuente, R.; Sanjust, E. Agarose and Its Derivatives as Supports for Enzyme Immobilization. Molecules 2016, 21, 1577. [Google Scholar] [CrossRef] [PubMed]
- Bernal, C.; Rodríguez, K.; Martínez, R. Integrating enzyme immobilization and protein engineering: An alternative path for the development of novel and improved industrial biocatalysts. Biotechnol. Adv. 2018, 36, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Sebastião, M.J.; Cabral, J.M.S.; Aires-Barros, M.R. Improved purification protocol of a Fusarium solani pisi recombinant cutinase by phase partitioning in aqueous two-phase systems of polyethylene glycol and phosphate. Enzym. Microb. Technol. 1996, 18, 251–260. [Google Scholar] [CrossRef]
- Henley, J.P.; Sadana, A. Categorization of enzyme deactivations using a series-type mechanism. Enzym. Microb. Technol. 1985, 7, 50–60. [Google Scholar] [CrossRef]
- Aymard, C.; Belarbi, A. Kinetics of thermal deactivation of enzymes: A simple three parameters phenomenological model can describe the decay of enzyme activity, irrespectively of the mechanism. Enzym. Microb. Technol. 2000, 27, 612–618. [Google Scholar] [CrossRef]
- Correa, S.; Puertas, S.; Gutiérrez, L.; Asín, L.; de la Fuente, J.M.; Grazú, V.; Betancor, L. Design of stable magnetic hybrid nanoparticles of Si-entrapped HRP. PLoS ONE 2019, 14, e0214004. [Google Scholar] [CrossRef]
- Gonçalves, A.M.; Serro, A.P.; Aires-Barros, M.R.; Cabral, J.M.S. Effects of ionic surfactants used in reversed micelles on cutinase activity and stability. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 2000, 1480, 92–106. [Google Scholar] [CrossRef]
- Vorderwülbecke, T.; Kieslich, K.; Erdmann, H. Comparison of lipases by different assays. Enzym. Microb. Technol. 1992, 14, 631–639. [Google Scholar] [CrossRef]
- Nunes, M.A.P.; Vila-Real, H.; Fernandes, P.C.B.; Ribeiro, M.H.L. Immobilization of Naringinase in PVA–Alginate Matrix Using an Innovative Technique. Appl. Biochem. Biotechnol. 2010, 160, 2129–2147. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Longhi, S.; Czjzek, M.; Lamzin, V.; Nicolas, A.; Cambillau, C. Atomic resolution (1.0 A) crystal structure of Fusarium solani cutinase: Stereochemical analysis. J. Mol. Biol. 1997, 268, 779–799. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.B.; Fojan, P.; Petersen, E.I.; Petersen, M.T.N. The thermal stability of the Fusarium solani pisi cutinase as a function of pH. BioMed Res. Int. 2001, 1, 62–69. [Google Scholar] [CrossRef]
- Dutta, K.; Sen, S.; Veeranki, V.D. Production, characterization and applications of microbial cutinases. Process Biochem. 2009, 44, 127–134. [Google Scholar] [CrossRef]
- Martínez, A.; Maicas, S. Cutinases: Characteristics and Insights in Industrial Production. Catalysts 2021, 11, 1194. [Google Scholar] [CrossRef]
- Nyyssölä, A. Which properties of cutinases are important for applications? Appl. Microbiol. Biotechnol. 2015, 99, 4931–4942. [Google Scholar] [CrossRef] [PubMed]
- Nikolaivits, E.; Kanelli, M.; Dimarogona, M.; Topakas, E. A Middle-Aged Enzyme Still in Its Prime: Recent Advances in the Field of Cutinases. Catalysts 2018, 8, 612. [Google Scholar] [CrossRef]
- Martinez, C.; De Geus, P.; Lauwereys, M.; Matthyssens, G.; Cambillau, C. Fusarium solani cutinase is a lipolytic enzyme with a catalytic serine accessible to solvent. Nature 1992, 356, 615–618. [Google Scholar] [CrossRef]
- Egmond, M.R.; de Vlieg, J. Fusarium solani pisi cutinase. Biochimie 2000, 82, 1015–1021. [Google Scholar] [CrossRef]
- Longhi, S.; Cambillau, C. Structure-activity of cutinase, a small lipolytic enzyme. Biochim. Biophys. Acta 1999, 1441, 185–196. [Google Scholar] [CrossRef]
- Lang, D.; Hofmann, B.; Haalck, L.; Hecht, H.-J.; Spener, F.; Schmid, R.D.; Schomburg, D. Crystal Structure of a Bacterial Lipase from Chromobacterium viscosum ATCC 6918 Refined at 1.6 Å Resolution. J. Mol. Biol. 1996, 259, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Toida, J.; Kondoh, K.; Fukuzawa, M.; Ohnishi, K.; Sekiguchi, J. Purification and Characterization of a Lipase from Aspergillus oryzae. Biosci. Biotechnol. Biochem. 1995, 59, 1199–1203. [Google Scholar] [CrossRef]
- Wu, X.Y.; JÄÄskelÄinen, S.; Linko, W.Y. Purification and partial characterization of Rhizomucor miehei lipase for ester synthesis. Appl. Biochem. Biotechnol. 1996, 59, 145–158. [Google Scholar] [CrossRef]
- Kohno, M.; Kugimiya, W.; Hashimoto, Y.; Morita, Y. Purification, characterization, and crystallization of two types of lipase from Rhizopus niveus. Biosci. Biotechnol. Biochem. 1994, 58, 1007–1012. [Google Scholar] [CrossRef]
- Benjamin, S.; Pandey, A. Candida rugosa lipases: Molecular biology and versatility in biotechnology. Yeast 1998, 14, 1069–1087. [Google Scholar] [CrossRef]
- Ghosh, P.K.; Saxena, R.K.; Gupta, R.; Yadav, R.P.; Davidson, S. Microbial lipases: Production and applications. Sci. Prog. 1996, 79 Pt 2, 119–157. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.H.; Hwang, S.; Chang, S.B.; Ku, J.; Chung, D.S. Acid dissociation constants of melamine derivatives from density functional theory calculations. J. Phys. Chem. A 2009, 113, 13036–13040. [Google Scholar] [CrossRef]
- Petersen, M.T.; Martel, P.; Petersen, E.I.; Drabløs, F.; Petersen, S.B. Surface and electrostatics of cutinases. Methods Enzym. 1997, 284, 130–154. [Google Scholar] [CrossRef]
- Palanisamy, U.D.; Winzor, D.J.; Lowe, C.R. Synthesis and evaluation of affinity adsorbents for glycoproteins: An artificial lectin. J. Chromatogr. B Biomed. Sci. Appl. 2000, 746, 265–281. [Google Scholar] [CrossRef]
- Scoble, J.A.; Scopes, R.K. Ligand structure of the divinylsulfone-based T-gel. J. Chromatogr. A 1997, 787, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Soares, C.M.F.; De Castro, H.F.; De Moraes, F.F.; Zanin, G.M. Characterization and utilization of Candida rugosa lipase immobilized on controlled pore silica. Appl. Biochem. Biotechnol. 1999, 79, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Enatsu, M.; Takee, R.; Kugimiya, W. Thermal stability of Rhizopus niveus lipase expressed in a kex2 mutant yeast. J. Biotechnol. 2000, 81, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Cygler, M. Two conformational states of Candida rugosa lipase. Protein Sci. 1994, 3, 82–91. [Google Scholar] [CrossRef]
- Danisman, T.; Tan, S.; Kacar, Y.; Ergene, A. Covalent immobilization of invertase on microporous pHEMA–GMA membrane. Food Chem. 2004, 85, 461–466. [Google Scholar] [CrossRef]
- Andjelković, U.; Pićurić, S.; Vujčić, Z. Purification and characterisation of Saccharomyces cerevisiae external invertase isoforms. Food Chem. 2010, 120, 799–804. [Google Scholar] [CrossRef]
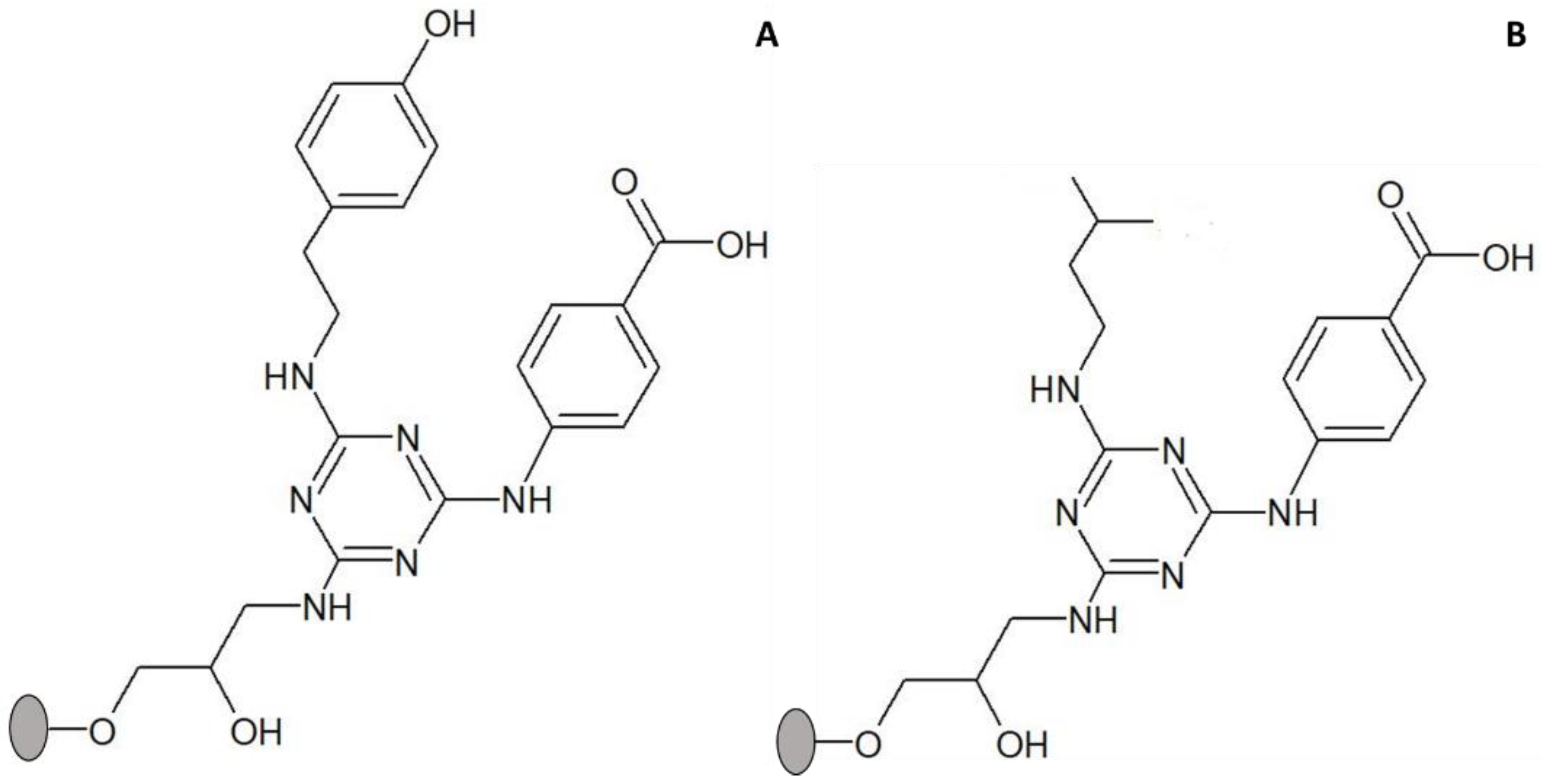
Figure 1.
Structure of solid-phase ligand 3′/11, comprising 4-aminobenzoic acid as substituent in R1 position (mimic of aspartic and glutamic acids) and 2-methylbutylamine in R2 position (mimic of isoleucine). The ligand was synthesized directly on agarose, which is represented by a grey circle.
Figure 1.
Structure of solid-phase ligand 3′/11, comprising 4-aminobenzoic acid as substituent in R1 position (mimic of aspartic and glutamic acids) and 2-methylbutylamine in R2 position (mimic of isoleucine). The ligand was synthesized directly on agarose, which is represented by a grey circle.
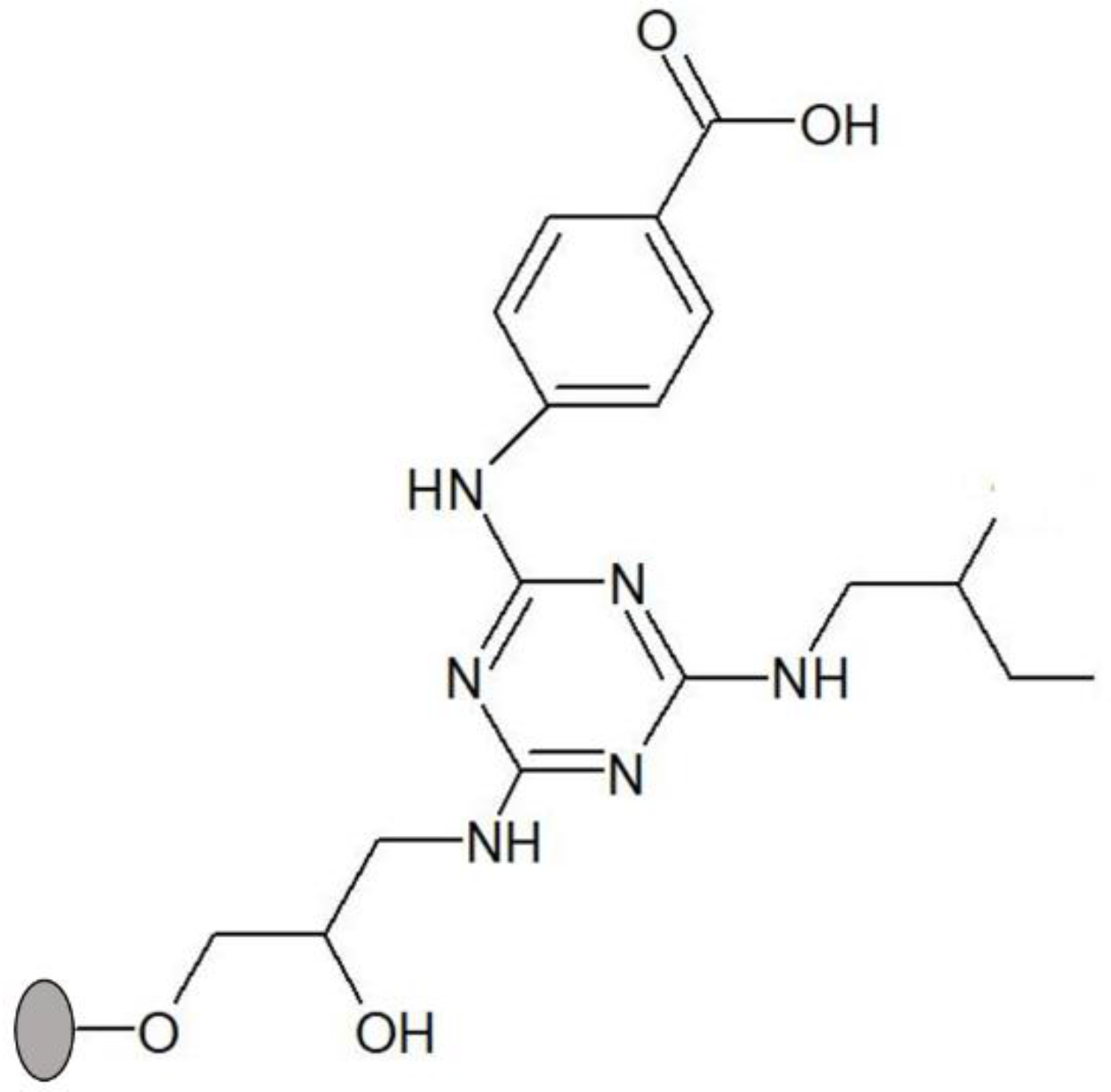
Figure 2.
Protein adsorption yields of different lipases to biomimetic solid-phase ligand 3′/11 (black), Sepharose CL-6B (dark grey) and ligand 0/0 (diagonal stripes). All the assays were performed in triplicate in 20 mM Tris-HCl pH 8.0. Abbreviations represent lipases from Chromobacterium viscosum (CVL), Rhizopus niveus (RNL), Rhizomucor miehei (RML), Candida rugosa (CRL), and Aspergillus oryzae (AOL).
Figure 2.
Protein adsorption yields of different lipases to biomimetic solid-phase ligand 3′/11 (black), Sepharose CL-6B (dark grey) and ligand 0/0 (diagonal stripes). All the assays were performed in triplicate in 20 mM Tris-HCl pH 8.0. Abbreviations represent lipases from Chromobacterium viscosum (CVL), Rhizopus niveus (RNL), Rhizomucor miehei (RML), Candida rugosa (CRL), and Aspergillus oryzae (AOL).
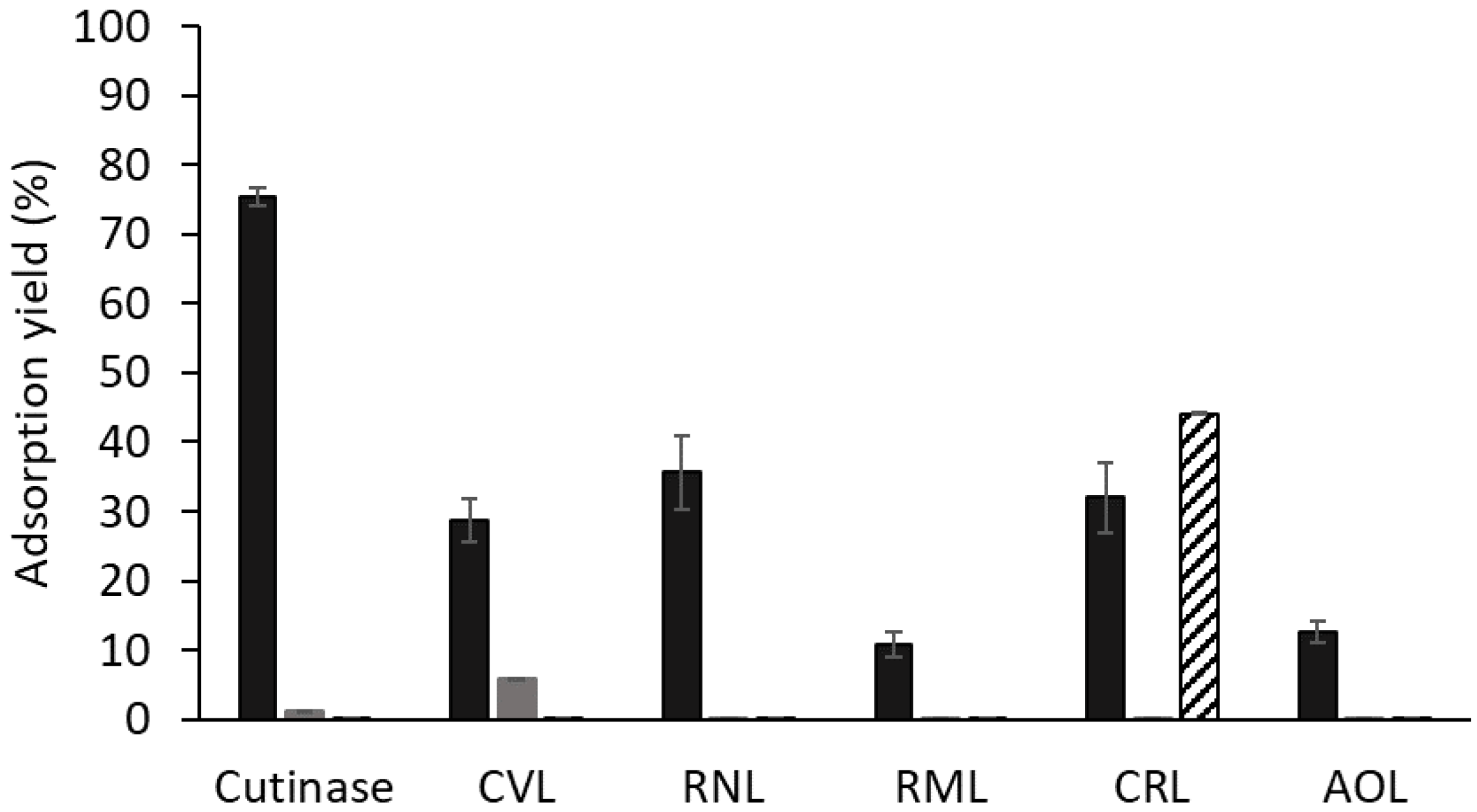
Figure 3.
Deactivation of free enzymes (black circles) and enzymes adsorbed to ligand 3′/11 (grey triangles) at 60 °C and pH 8.0. Enzymes and protein concentration by graph: (A) cutinase, the protein concentration was 35 μg/mL in both cases; (B) CVL, the concentrations used were 52 μg/mL and 160 μg/mL solution for free and adsorbed enzymes, respectively; (C) CRL, the concentrations used were 3.5 mg/mL and 200 μg/mL solution for free and adsorbed enzymes, respectively; (D) RNL, the concentrations used were 8 mg/mL and 5 mg/mL solution for free and adsorbed enzymes, respectively.
Figure 3.
Deactivation of free enzymes (black circles) and enzymes adsorbed to ligand 3′/11 (grey triangles) at 60 °C and pH 8.0. Enzymes and protein concentration by graph: (A) cutinase, the protein concentration was 35 μg/mL in both cases; (B) CVL, the concentrations used were 52 μg/mL and 160 μg/mL solution for free and adsorbed enzymes, respectively; (C) CRL, the concentrations used were 3.5 mg/mL and 200 μg/mL solution for free and adsorbed enzymes, respectively; (D) RNL, the concentrations used were 8 mg/mL and 5 mg/mL solution for free and adsorbed enzymes, respectively.

Figure 4.
Structure of solid-phase ligands alternative to ligand 3′/11, screened for enhancement of thermal stability of adsorbed CRL. (A) ligand 3/3′ comprises tyramine as substituent in R1 position (mimicking tyrosine) and 4-aminobenzoic acid in R2 position (mimicking aspartic and glutamic acids); (B) 5/3′ comprises isoamylamine as substituent in R1 position (mimicking leucine) and 4-aminobenzoic acid in R2 position (mimicking aspartic and glutamic acids). The ligands were synthesized directly on agarose, which is represented by a grey circle.
Figure 4.
Structure of solid-phase ligands alternative to ligand 3′/11, screened for enhancement of thermal stability of adsorbed CRL. (A) ligand 3/3′ comprises tyramine as substituent in R1 position (mimicking tyrosine) and 4-aminobenzoic acid in R2 position (mimicking aspartic and glutamic acids); (B) 5/3′ comprises isoamylamine as substituent in R1 position (mimicking leucine) and 4-aminobenzoic acid in R2 position (mimicking aspartic and glutamic acids). The ligands were synthesized directly on agarose, which is represented by a grey circle.
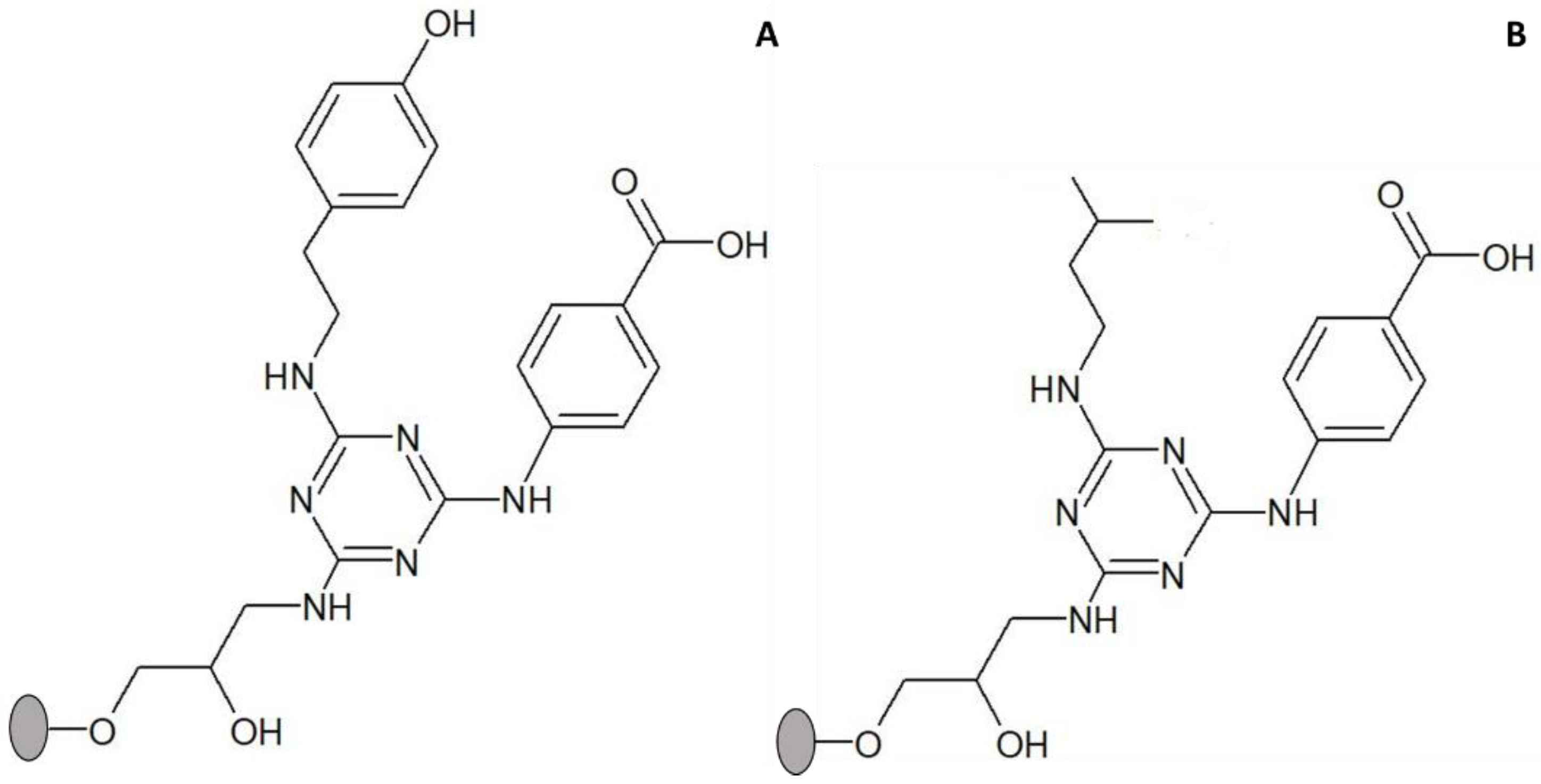
Figure 5.
Deactivation profile of free CRL (black points) and CRL adsorbed to solid-phase ligand 3/3′ (grey triangles) at 60 °C and pH 8.0. The dashed and continuous grey curves are models of Aymard and Belarbi, and Henley and Sadana, respectively. The incubation was performed in 20 mM Tris-HCl, pH 8.0, and the concentrations used were 3.5 mg/mL and 200 μg/mL solution for free and adsorbed CRL, respectively.
Figure 5.
Deactivation profile of free CRL (black points) and CRL adsorbed to solid-phase ligand 3/3′ (grey triangles) at 60 °C and pH 8.0. The dashed and continuous grey curves are models of Aymard and Belarbi, and Henley and Sadana, respectively. The incubation was performed in 20 mM Tris-HCl, pH 8.0, and the concentrations used were 3.5 mg/mL and 200 μg/mL solution for free and adsorbed CRL, respectively.

Figure 6.
Deactivation of invertase free (black squares), adsorbed to Sepharose CL-6B (black circles) and to Sepharose CL-6B bearing ligand 3′/11 (grey triangles) at 60 °C. The incubation was performed in 20 mM acetate buffer, pH 5.3, and the concentration of invertase used was 2.0 mg/mL of solution in all assays. The activity decay followed a first-order exponential law in all cases and is represented by the dotted lines.
Figure 6.
Deactivation of invertase free (black squares), adsorbed to Sepharose CL-6B (black circles) and to Sepharose CL-6B bearing ligand 3′/11 (grey triangles) at 60 °C. The incubation was performed in 20 mM acetate buffer, pH 5.3, and the concentration of invertase used was 2.0 mg/mL of solution in all assays. The activity decay followed a first-order exponential law in all cases and is represented by the dotted lines.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Specific activity of cutinase and other microbial lipases under study, free in solution, at the best pair of selected conditions of pH and temperature.
Table 1.
Specific activity of cutinase and other microbial lipases under study, free in solution, at the best pair of selected conditions of pH and temperature.
| Enzyme | Selected pH | Selected Temperature (°C) | Specific Activity (U/mg) |
|---|---|---|---|
| AOL | 8.0 | 37 | 3.4 ± 0.5 |
| CRL | 0.71 ± 0.07 | ||
| Cutinase * | 160 ± 70 | ||
| CVL | 30 | 98.5 ± 0.9 | |
| RML | 7.0 | 37 | 1.9 ± 0.5 |
| RNL | 8.0 | 0.0352 ± 0.0008 |
* For 30 °C, specific activity = (76 ± 11) U/mg.
Table 2.
Comparison of protein (ηprot) and activity (ηact) adsorption yields of different lipolytic enzymes to solid-phase ligand 3′/11. The load volume in the affinity chromatographic column was adjusted for the several preparations, according to previous characterization and purity analysis by SDS-PAGE, in order to have similar amounts of each enzyme (see Section 2.2.2). Results were obtained in triplicate.
Table 2.
Comparison of protein (ηprot) and activity (ηact) adsorption yields of different lipolytic enzymes to solid-phase ligand 3′/11. The load volume in the affinity chromatographic column was adjusted for the several preparations, according to previous characterization and purity analysis by SDS-PAGE, in order to have similar amounts of each enzyme (see Section 2.2.2). Results were obtained in triplicate.
| Enzyme | ηprot (%) | ηact (%) |
|---|---|---|
| Cutinase | 75 ± 1 | 100 ± 0 |
| CVL | 29 ± 3 | 14.7 ± 0.6 |
| CRL | 36 ± 5 | 70.9 ± 0.6 |
| RNL | 32 ± 5 | ≈100 ± 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ferreira-Faria, D.; Taipa, M.Â. Thermal Stabilization of Lipases Bound to Solid-Phase Triazine-Scaffolded Biomimetic Ligands: A Preliminary Assessment. Processes 2024, 12, 371. https://doi.org/10.3390/pr12020371
AMA Style
Ferreira-Faria D, Taipa MÂ. Thermal Stabilization of Lipases Bound to Solid-Phase Triazine-Scaffolded Biomimetic Ligands: A Preliminary Assessment. Processes. 2024; 12(2):371. https://doi.org/10.3390/pr12020371
Chicago/Turabian StyleFerreira-Faria, Diogo, and M. Ângela Taipa. 2024. "Thermal Stabilization of Lipases Bound to Solid-Phase Triazine-Scaffolded Biomimetic Ligands: A Preliminary Assessment" Processes 12, no. 2: 371. https://doi.org/10.3390/pr12020371
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.