Artificially Expanded Genetic Information Systems for New Aptamer Technologies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Non-Natural Nucleotides in Functional Nucleic Acids (DNA, RNA, Collectively xNA)
3. The Artificially Expanded Genetic Information System (AEGIS)
- Is designed, using the best chemical theory, to include functional groups that intrinsically deliver the desired reactivity. Given the limitations of current chemical theory, this design can be only “coarse” [60,61]; chemical theory is not adequate to “finely tune” those properties. However, and inaccessible to natural biopolymers, the functional groups need not be constrained to those that have been delivered to us by prebiotic chemistry and natural history; should chemical theory direct, they may include groups that manage the intrinsic difficulties of binding difficult targets or catalyzing difficult reactions, such as cleaving peptide bonds.
- Supports laboratory Darwinism, in order to allow the power of Darwinism to “finely tune” molecular systems to convert poor binders or catalysts into good ones. Darwinism cannot act prospectively. Therefore, it cannot anticipate what functional groups will be needed to solve future problems in binding and catalysis. Thus, the limits of Darwinism (it is a bad innovator, but an excellent refiner) complement the limits of design (a good innovator, but a bad refiner).
- Has added building blocks to ensure unique folds, but where:
- Only some of the building blocks carry functional groups to avoid “over decoration”.
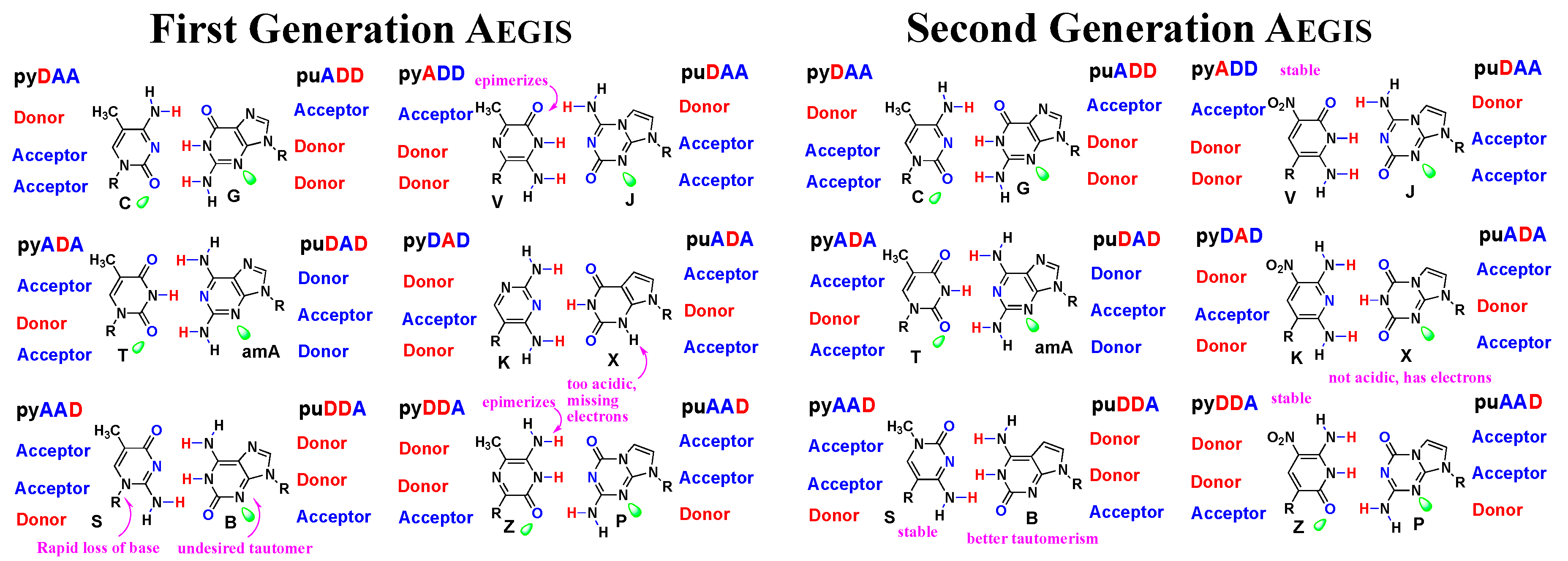
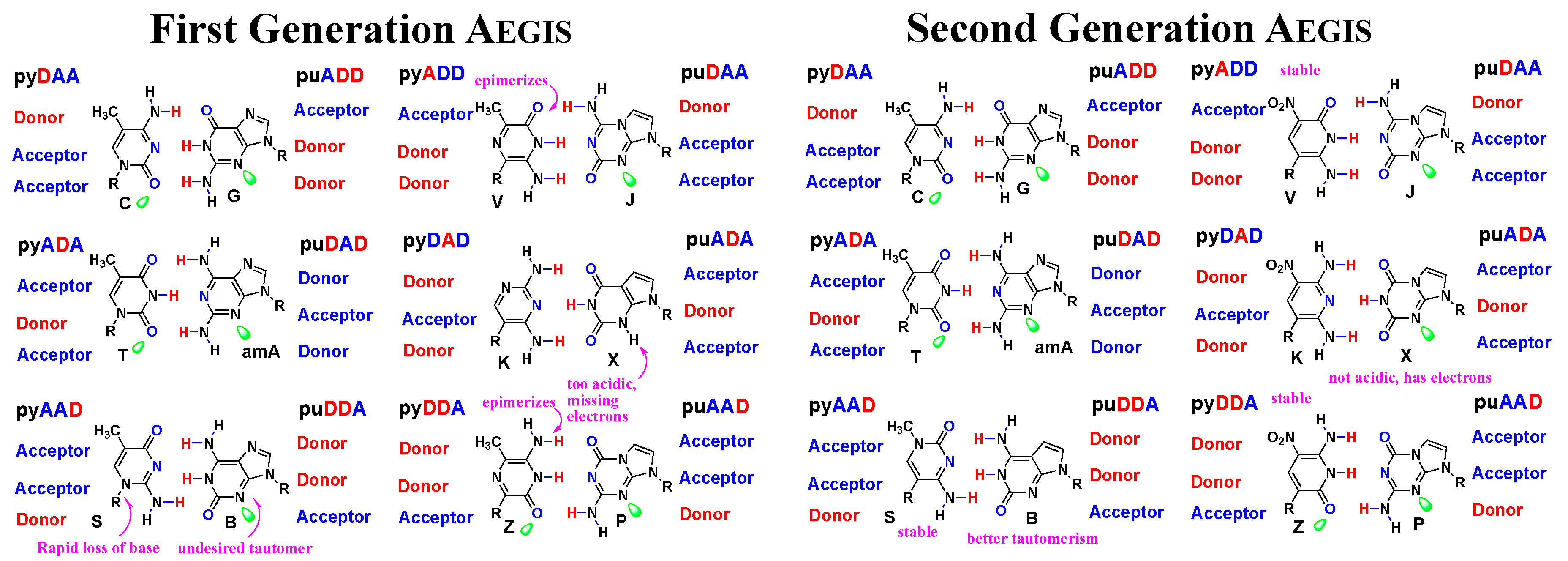
3.1. First and Second Generation AEGIS
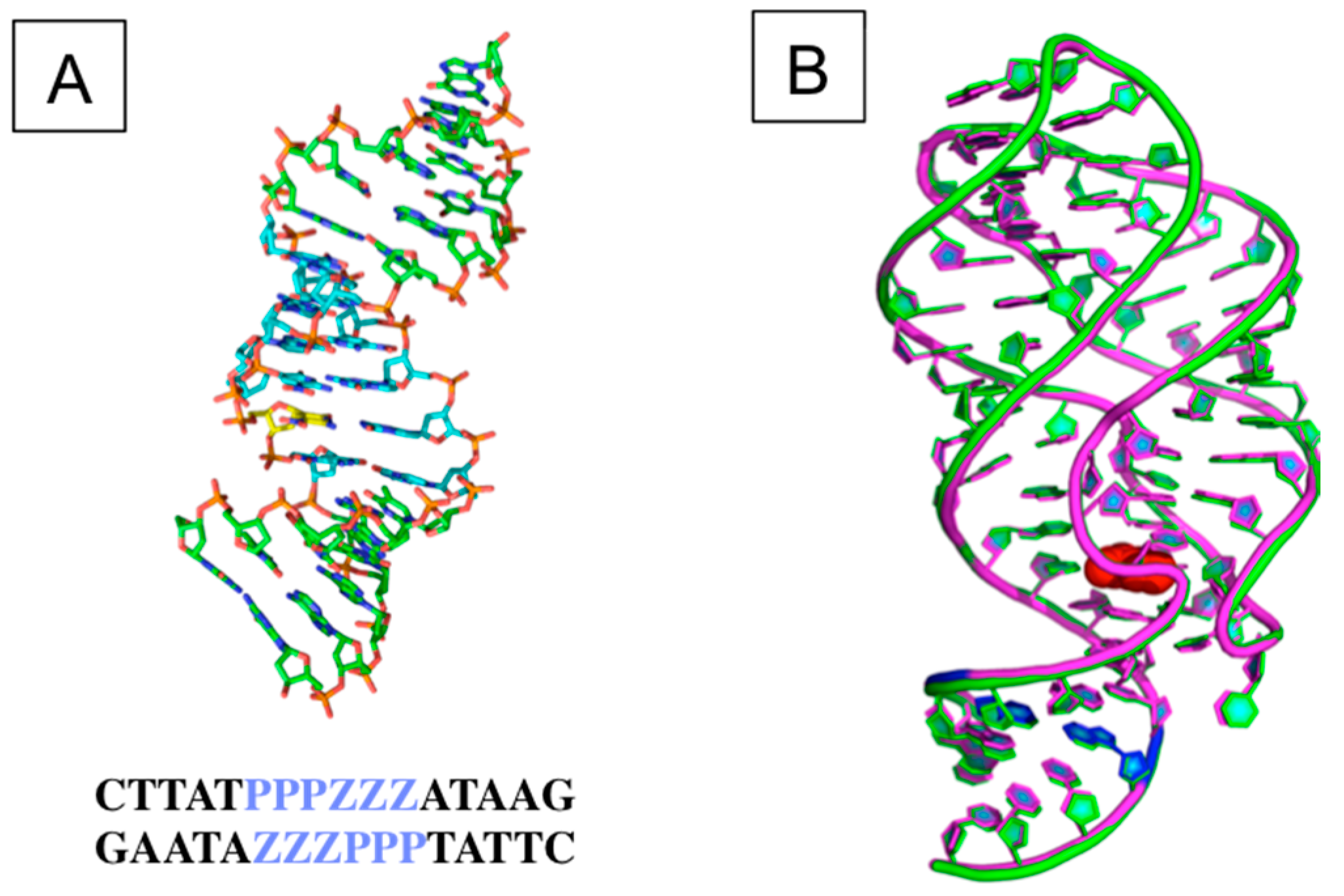
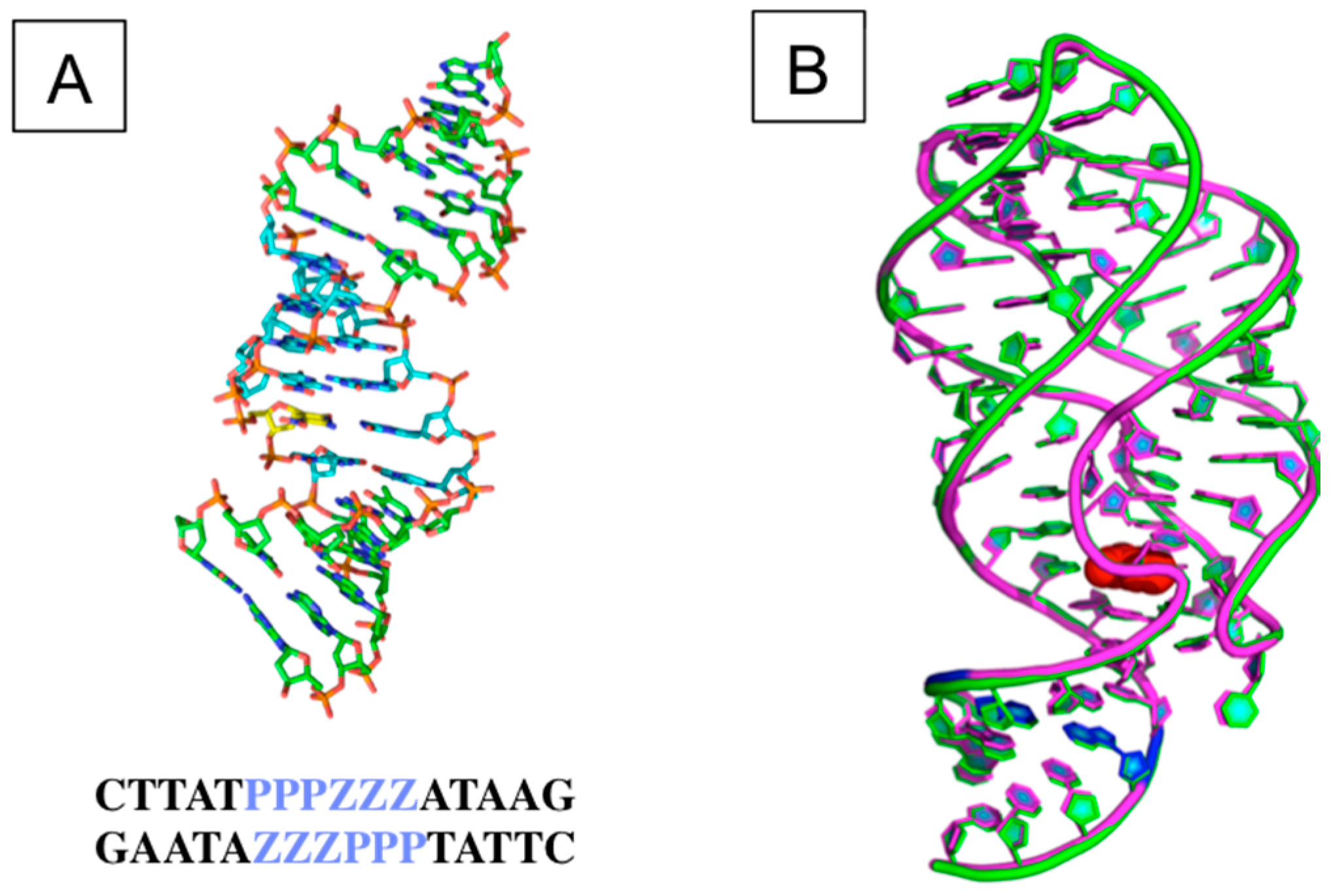
3.2. The Structural and Molecular Biology of AEGIS
4. AEGIS Laboratory In Vitro Evolution (AEGIS-LIVE)
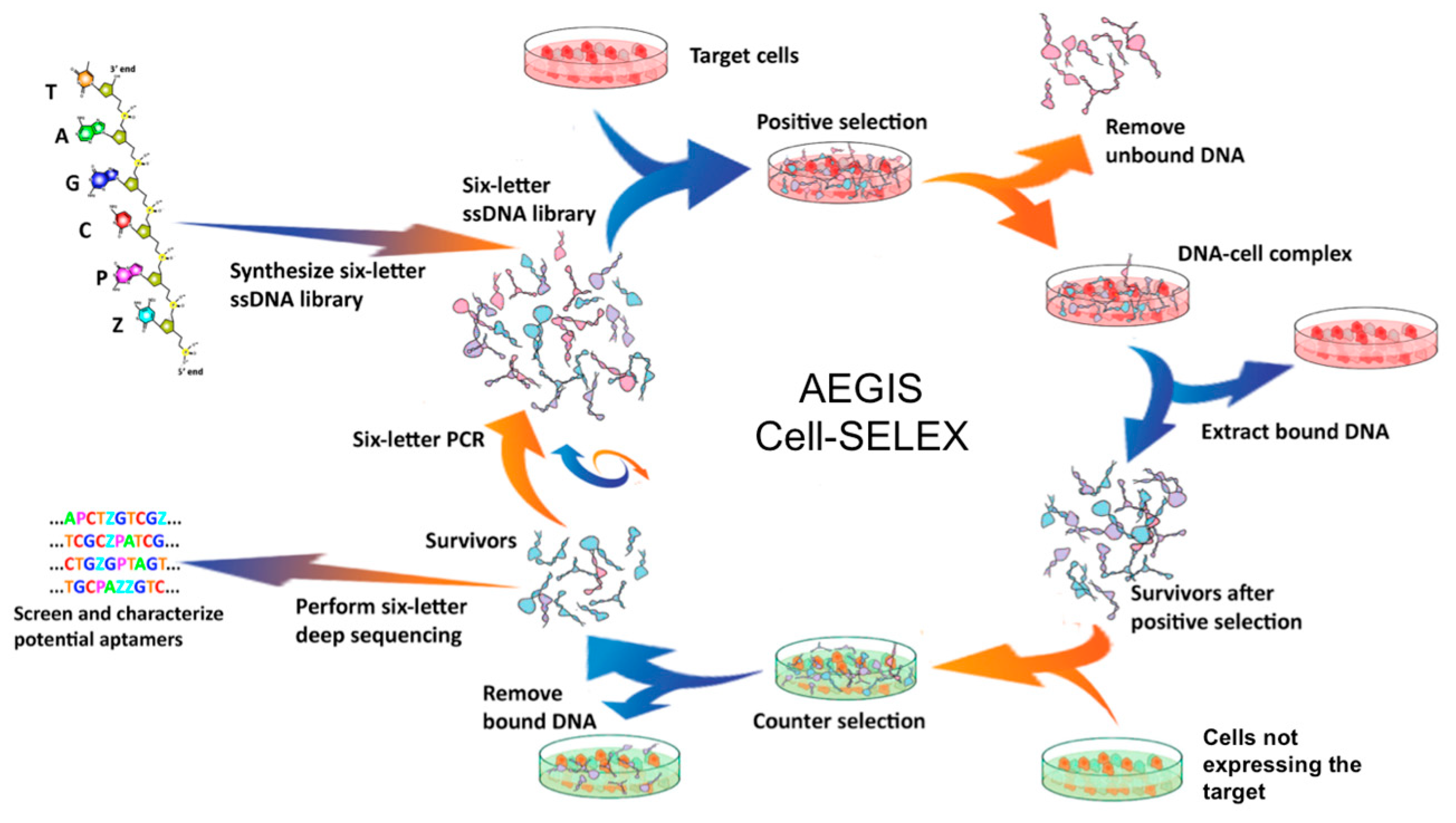
4.1. Cell-SELEX (Sistematic Evolution of Ligands by Exponential Enrichment)
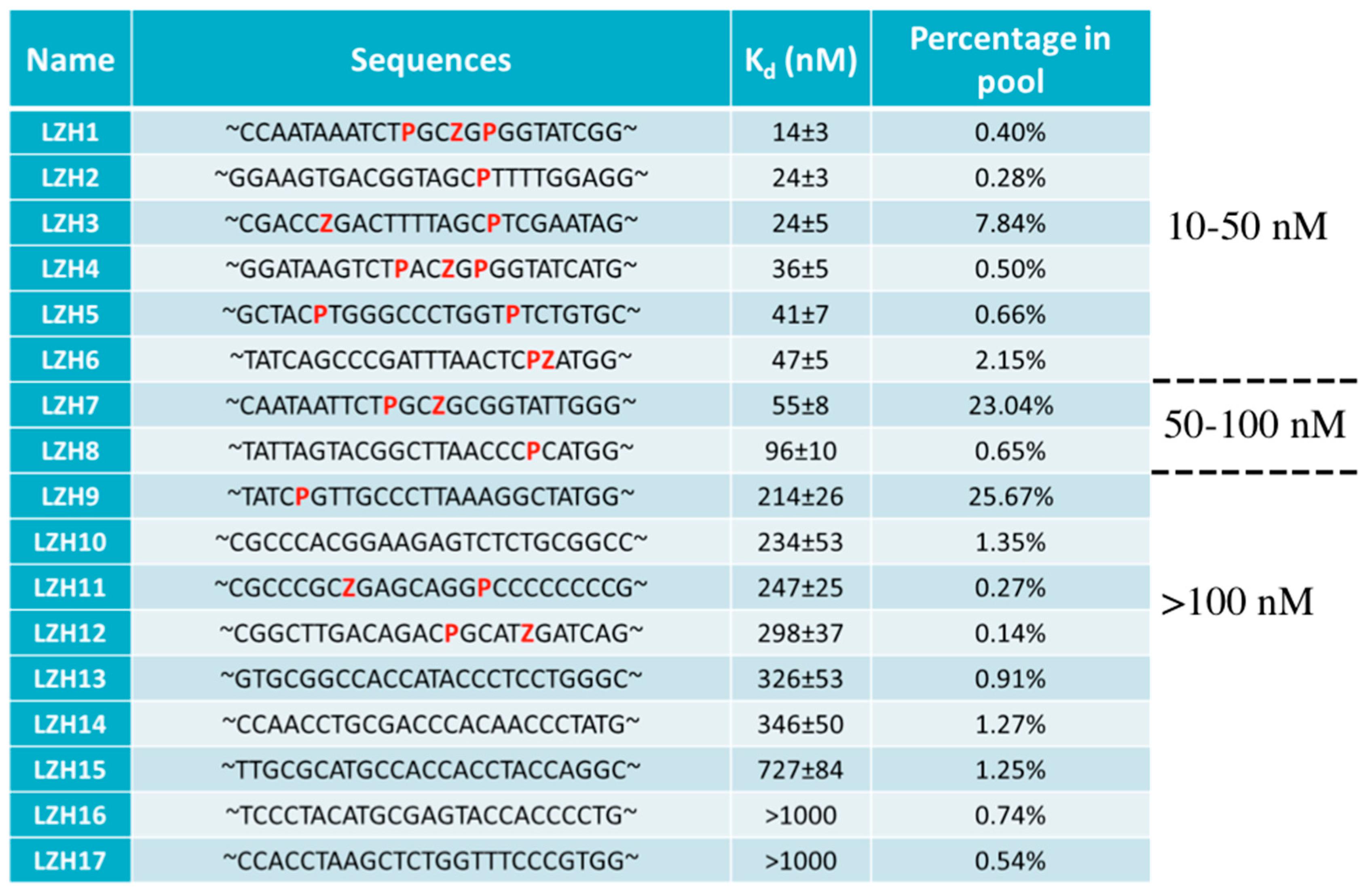
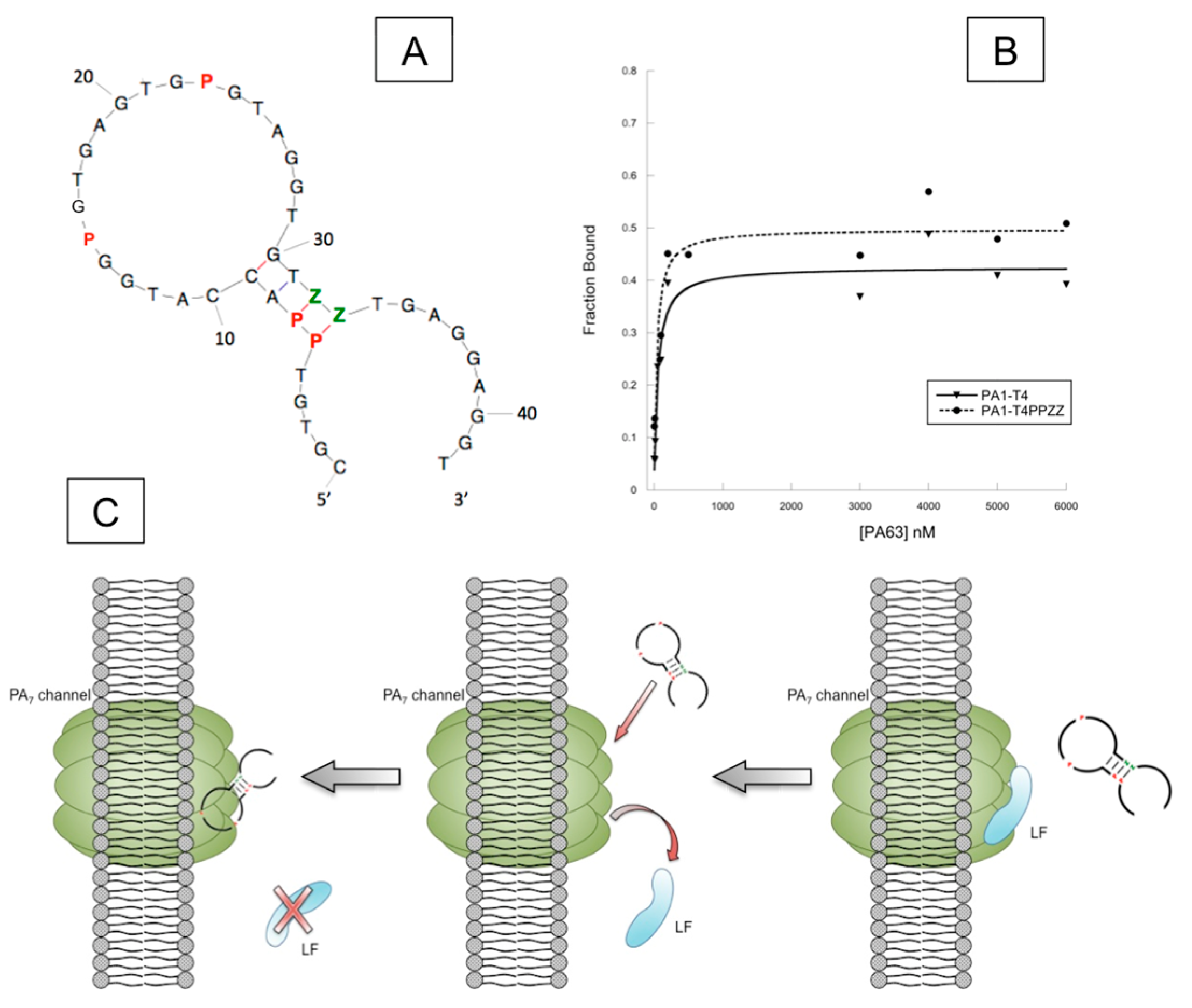
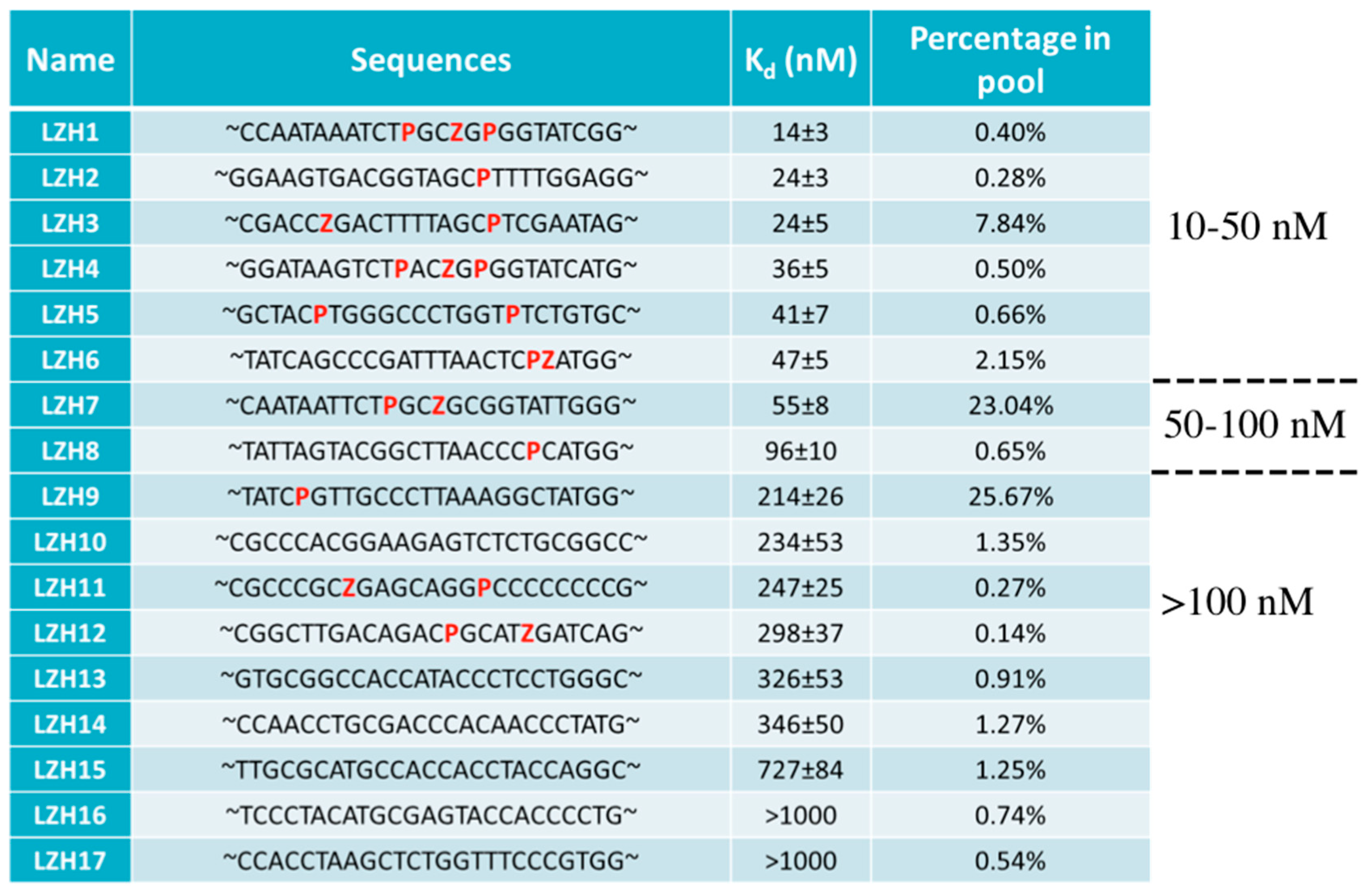
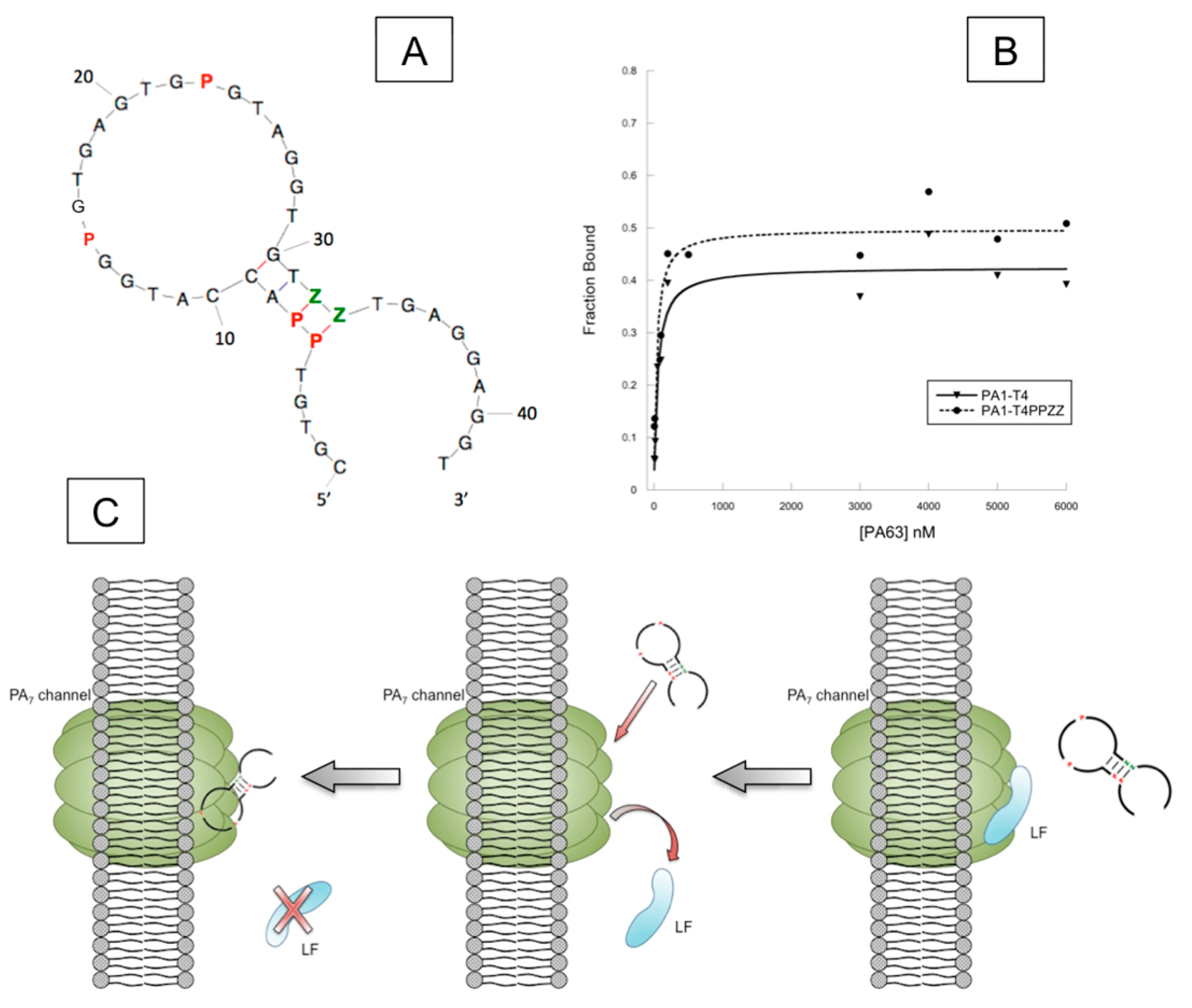
4.2. AEGIS-LIVE on Anthrax Protective Antigen
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Lorsch, J.R.; Szostak, J.W. Chance and necessity in the selection of nucleic acid catalysts. Acc. Chem. Res. 1996, 29, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Rich, A. On the problems of evolution and biochemical information transfer. In Horizons in Biochemistry; Kasha, M.P.B., Ed.; Academic Press: New York, NY, USA, 1962. [Google Scholar]
- Benner, S.A.; Ellington, A.D.; Tauer, A. Modern metabolism as a palimpsest of the RNA world. Proc. Natl. Acad. Sci. USA 1989, 86, 7054–7058. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R. Structural biology. The ribosome is a ribozyme. Science 2000, 289, 878–879. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Du, L.; Li, M. Aptamer-based carbohydrate recognition. Curr. Pharm. Des. 2010, 16, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- Famulok, M. Oligonucleotide aptamers that recognize small molecules. Curr. Opin. Struct. Biol. 1999, 9, 324–329. [Google Scholar] [CrossRef]
- Gopinath, S.C. Methods developed for SELEX. Anal. Bioanal. Chem. 2007, 387, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Annemans, L.; White, R.; Gallagher, M.; Thomas, S. Cost effectiveness of treatments for wet age-related macular degeneration. Pharmacoeconomics 2011, 29, 107–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batool, S.; Bhandari, S.; George, S.; Okeoma, P.; Van, N.; Zumrut, H.E.; Mallikaratchy, P. Engineered aptamers to probe molecular interactions on the cell surface. Biomedicines 2017, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Tawiah, K.D.; Porciani, D.; Burke, D.H. Toward the selection of cell targeting aptamers with extended biological functionalities to facilitate endosomal escape of cargoes. Biomedicines 2017, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Catuogno, S.; Esposito, C.L. Aptamer cell-based selection: Overview and advances. Biomedicines 2017, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Kruspe, S.; Giangrande, P.H. Aptamer-siRNA chimeras: Discovery, progress, and future prospects. Biomedicines 2017, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Volk, D.E.; Lokesh, G.L.R. Development of phosphorothioate DNA and DNA thioaptamers. Biomedicines 2017, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Nik Kamarudin, N.A.A.; Mohammed, N.A.; Mustaffa, K.M.F. Aptamer technology: Adjunct therapy for malaria. Biomedicines 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, S.D. Aptamers: An emerging class of molecules that rival antibodies in diagnostics. Clin. Chem. 1999, 45, 1628–1650. [Google Scholar] [PubMed]
- Proske, D.; Blank, M.; Buhmann, R.; Resch, A. Aptamers-basic research, drug development, and clinical applications. Appl. Microbiol. Biotechnol. 2005, 69, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, J.W.; Hamula, C.L.; Zhang, H.; Le, X.C. Assays for cytokines using aptamers. Methods 2006, 38, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ebright, J.N.; Stovall, G.M.; Chen, X.; Nguyen, H.H.; Singh, A.; Syrett, A.; Ellington, A.D. Technical and biological issues relevant to cell typing with aptamers. J. Proteome Res. 2009, 8, 2438–2448. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.; Worland, S.; Gold, L. Isolation of high-affinity RNA ligands to HIV-1 integrase from a random pool. Virology 1995, 209, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.; Collins, B.; Brown, D.; Hostomsky, Z.; Gold, L. A specific RNA structural motif mediates high affinity binding by the HIV-1 nucleocapsid protein (NCp7). Virology 1996, 225, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Burke, D.H.; Scates, L.; Andrews, K.; Gold, L. Bent pseudoknots and novel RNA inhibitors of type 1 human immunodeficiency virus (HIV-1) reverse transcriptase. J. Mol. Biol. 1996, 264, 650–666. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.J.; Feigon, J.; Hostomsky, Z.; Gold, L. High-affinity ssDNA inhibitors of the reverse transcriptase of type 1 human immunodeficiency virus. Biochemistry 1995, 34, 9599–9610. [Google Scholar] [CrossRef] [PubMed]
- Geiger, A.; Burgstaller, P.; von der Eltz, H.; Roeder, A.; Famulok, M. RNA aptamers that bind l-Arginine with sub-micromolar dissociation constants and high enantioselectivity. Nucleic Acids Res. 1996, 24, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Huizenga, D.E.; Szostak, J.W. A DNA aptamer that binds adenosine and ATP. Biochemistry 1995, 34, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Kiga, D.; Futamura, Y.; Sakamoto, K.; Yokoyama, S. An RNA aptamer to the xanthine/guanine base with a distinctive mode of purine recognition. Nucleic Acids Res. 1998, 26, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Ellis, L.M. Drug development: Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998, 284, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Stumpp, M.T.; Amstutz, P. Darpins: A true alternative to antibodies. Curr. Opin. Drug Disc. 2007, 10, 153–159. [Google Scholar]
- Bloom, L.; Calabro, V. FN3: A new protein scaffold reaches the clinic. Drug Discov. Today 2009, 14, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T.; et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 2010, 5, e15004. [Google Scholar] [CrossRef] [PubMed]
- Tarasow, T.M.; Eaton, B.E. Dressed for success: Realizing the catalytic potential of RNA. Biopolymers 1998, 48, 29–37. [Google Scholar] [CrossRef]
- Tolle, F.; Mayer, G. Dressed for success—Applying chemistry to modulate aptamer functionality. Chem. Sci. 2013, 4, 60–67. [Google Scholar] [CrossRef]
- Battersby, T.R.; Ang, D.N.; Burgstaller, P.; Jurczyk, S.C.; Bowser, M.T.; Buchanan, D.D.; Kennedy, R.T.; Benner, S.A. Quantitative analysis of receptors for adenosine nucleotides obtained via in vitro selection from a library incorporating a cationic nucleotide analog. J. Am. Chem. Soc. 1999, 121, 9781–9789. [Google Scholar] [CrossRef] [PubMed]
- Rothlisberger, P.; Gasse, C.; Hollenstein, M. Nucleic acid aptamers: Emerging applications in medical imaging, nanotechnology, neurosciences, and drug delivery. Int. J. Mol. Sci. 2017, 18, 2430. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.R.; Jimenez, R.M.; Chaput, J.C. Analysis of aptamer discovery and technology. Nat. Rev. Chem. 2017, 1. [Google Scholar] [CrossRef]
- Zhou, J.H.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M.; Hipolito, C.J.; Lam, C.H.; Perrin, D.M. A DNAzyme with three protein-like functional groups: Enhancing catalytic efficiency of M2+-independent RNA cleavage. Chembiochem 2009, 10, 1988–1992. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M.; Hipolito, C.J.; Lam, C.H.; Perrin, D.M. A self-cleaving DNA enzyme modified with amines, guanidines and imidazoles operates independently of divalent metal cations (M2+). Nucleic Acids Res. 2009, 37, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Silverman, S.K. Pursuing DNA catalysts for protein modification. Accounts Chem. Res. 2015, 48, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Avins, J.L.; Klauser, P.C.; Brandsen, B.M.; Lee, Y.; Silverman, S.K. DNA-catalyzed amide hydrolysis. J. Am. Chem. Soc. 2016, 138, 2106–2109. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.C.; Demesmaeker, A.; Joyce, G.F. Cleavage of an amide bond by a ribozyme. Science 1995, 267, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Joyce, G.F.; Dai, X.C.; DeMesmaeker, A. Cleavage of an amide bond by a ribozyme. Science 1996, 272, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Ameta, S.; Jaschke, A. An RNA catalyst that reacts with a mechanistic inhibitor of serine proteases. Chem. Sci. 2013, 4, 957–964. [Google Scholar] [CrossRef]
- Chandra, M.; Sachdeva, A.; Silverman, S.K. DNA-catalyzed sequence-specific hydrolysis of DNA. Nat. Chem. Biol. 2009, 5, 718–720. [Google Scholar] [CrossRef] [PubMed]
- Mohan, U.; Burai, R.; McNaughton, B.R. In vitro evolution of a friedel-crafts deoxyribozyme. Org. Biomol. Chem. 2013, 11, 2241–2244. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R.; Joyce, G.F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223–229. [Google Scholar] [CrossRef]
- Carrigan, M.A.; Ricardo, A.; Ang, D.N.; Benner, S.A. Quantitative analysis of a RNA-cleaving DNA catalyst obtained via in vitro selection. Biochemistry 2004, 43, 11446–11459. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.A.; Francklyn, C.S.; Robey-Bond, S.M. Transfer RNA and human disease. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.C.; Mecozzi, S. Identification of a 14mer RNA that recognizes and binds flavin mononucleotide with high affinity. Nucleic Acids Res. 2005, 33, 6992–6999. [Google Scholar] [CrossRef] [PubMed]
- Uhlenbeck, O.C. Keeping RNA happy. RNA 1995, 1, 4–6. [Google Scholar] [PubMed]
- Bhaskaran, H.; Taniguchi, T.; Suzuki, T.; Suzuki, T.; Perona, J.J. Structural dynamics of a mitochondrial tRNA possessing weak thermodynamic stability. Biochemistry 2014, 53, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, A.; Illangkoon, H.; Hendrickson, C.L.; Benner, S.A. 2′-deoxycytidines carrying amino and thiol functionality: Synthesis and incorporation by vent (exo(−)) polymerase. Org. Lett. 2004, 6, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Brakmann, S.; Nieckchen, P. The large fragment of escherichia coli DNA polymerase I can synthesize DNA exclusively from fluorescently labeled nucleotides. Chembiochem 2001, 2, 773–777. [Google Scholar] [CrossRef]
- Brakmann, S.; Lobermann, S. A further step towards single-molecule sequencing: Escherichia coli exonuclease III degrades DNA that is fluorescently labeled at each base pair. Angew. Chem. Int. Ed. 2002, 41, 3215–3217. [Google Scholar] [CrossRef]
- Ramsay, N.; Jemth, A.S.; Brown, A.; Crampton, N.; Dear, P.; Holliger, P. Cydna: Synthesis and replication of highly cy-dye substituted DNA by an evolved polymerase. J. Am. Chem. Soc. 2010, 132, 5096–5104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.K.; Lamb, B.M.; Feldman, A.W.; Zhou, A.X.; Lavergne, T.; Li, L.J.; Romesberg, F.E. A semisynthetic organism engineered for the stable expansion of the genetic alphabet. Proc. Natl. Acad. Sci. USA 2017, 114, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, M.; Yamashige, R.; Matsunaga, K.; Yokoyama, S.; Hirao, I. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat. Biotechnol. 2013, 31, 453. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Kimoto, M.; Hirao, I. High-affinity DNA aptamer generation targeting von willebrand factor a1-domain by genetic alphabet expansion for systematic evolution of ligands by exponential enrichment using two types of libraries composed of five different bases. J. Am. Chem. Soc. 2017, 139, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Zepik, H.H.; Benner, S.A. Catalysts, anticatalysts, and receptors for unactivated phosphate diesters in water. J. Org. Chem. 1999, 64, 8080–8083. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, W.W. Synthetic life. Sci. Am. 2004, 290, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A.; Karalkar, N.B.; Hoshika, S.; Laos, R.; Shaw, R.W.; Matsuura, M.; Fajardo, D.; Moussatche, P. Alternative watson-crick synthetic genetic systems. Cold Spring Harbor Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A. Understanding nucleic acids using synthetic chemistry. Accounts Chem. Res. 2004, 37, 784–797. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Leal, N.A.; Hoshika, S.; Benner, S.A. Ribonucleosides for an artificially expanded genetic information system. J. Org. Chem. 2014, 79, 3194–3199. [Google Scholar] [CrossRef] [PubMed]
- Zubay, G. A case for an additional RNA base pair in early evolution. In The Roots of Modern Biochemistry; Walter de Gruiter and Co.: Berlin, Germany, 1988; pp. 911–916. [Google Scholar]
- Voegel, J.J.; Altorfer, M.M.; Benner, S.A. The donor-acceptor-acceptor purine analog—Transformation of 5-aza-7-deaza-1H-isoguanine (= 4-aminoimidazo-[1,2-a]-1,3,5-triazin-2(1H)-one) to 2′-deoxy-5-aza-7-deaza-isoguanosine using purine nucleoside phosphorylase. Helv. Chim. Acta 1993, 76, 2061–2069. [Google Scholar] [CrossRef]
- Voegel, J.J.; Vonkrosigk, U.; Benner, S.A. Synthesis and tautomeric equilibrium of 6-amino-5-benzyl-3-methylpyrazin-2-one—An acceptor-donor-donor nucleoside base analog. J. Org. Chem. 1993, 58, 7542–7547. [Google Scholar] [CrossRef]
- Voegel, J.J.; Benner, S.A. Nonstandard hydrogen-bonding in duplex oligonucleotides—The base-pair between an acceptor-donor-donor pyrimidine analog and a donor-acceptor-acceptor purine analog. J. Am. Chem. Soc. 1994, 116, 6929–6930. [Google Scholar] [CrossRef]
- Voegel, J.J.; Benner, S.A. Synthesis, molecular recognition, and enzymology of oligonucleotides containing the non-standard base pair between 5-aza-7-deazaisoguanine and 6-amino-3-methylpyrazin-2(1H)-one, a donor-acceptor-acceptor purine analog and an acceptor-donor-donor pyrimidine analog. Helv. Chim. Acta 1996, 79, 1881–1898. [Google Scholar]
- Voegel, J.J.; Benner, S.A. Synthesis and characterization of non-standard nucleosides and nucleotides bearing the acceptor-donor-donor pyrimidine analog 6-amino-3-methylpyrazin-2(1H)-one. Helv. Chim. Acta 1996, 79, 1863–1880. [Google Scholar] [CrossRef]
- von Krosigk, U.B.; Benner, S.A. Expanding the genetic alphabet. Pyrazine nucleosides that support a donor-donor-acceptor hydrogen bonding pattern. Helv. Chim. Acta 2004, 87, 1299–1324. [Google Scholar] [CrossRef]
- Sepiol, J.; Kazimierczuk, Z.; Shugar, D. Tautomerism of isoguanosine and solvent-induced keto-enol equilibrium. Z. Naturforsch. C 1976, 31, 361–370. [Google Scholar] [PubMed]
- Sismour, A.M.; Benner, S.A. The use of thymidine analogs to improve the replication of an extra DNA base pair: A synthetic biological system. Nucleic Acids Res. 2005, 33, 5640–5646. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Leal, N.A.; Benner, S.A. 2′-deoxy-1-methylpseudocytidine, a stable analog of 2′-deoxy-5-methylisocytidine. Bioorg. Med. Chem. 2009, 17, 3728–3732. [Google Scholar] [CrossRef] [PubMed]
- Martinot, T.A.; Benner, S.A. Artificial genetic systems: Exploiting the “aromaticity” formalism to improve the tautomeric ratio for isoguanosine derivatives. J. Org. Chem. 2004, 69, 3972–3975. [Google Scholar] [CrossRef] [PubMed]
- Karalkar, N.B.; Khare, K.; Molt, R.; Benner, S.A. Tautomeric equilibria of isoguanine and related purine analogs. Nucleosides Nucleotides Nucleic Acids 2017, 36, 256–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hoshika, S.; Peterson, R.J.; Kim, M.J.; Benner, S.A.; Kahn, J.D. Biophysics of artificially expanded genetic information systems. Thermodynamics of DNA duplexes containing matches and mismatches involving 2-amino-3-nitropyridin-6-one (Z) and imidazo[1,2-a]-1,3,5-triazin-4(8H)one (P). Acs. Synth. Biol. 2017, 6, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Winiger, C.B.; Kim, M.J.; Hoshika, S.; Shaw, R.W.; Moses, J.D.; Matsuura, M.F.; Gerloff, D.L.; Benner, S.A. Polymerase interactions with wobble mismatches in synthetic genetic systems and their evolutionary implications. Biochemistry 2016, 55, 3847–3850. [Google Scholar] [CrossRef] [PubMed]
- Winiger, C.B.; Shaw, R.W.; Kim, M.J.; Moses, J.D.; Matsuura, M.F.; Benner, S.K. Expanded genetic alphabets: Managing nucleotides that lack tautomeric, protonated, or deprotonated versions complementary to natural nucleotides. ACS Synth. Biol. 2017, 6, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Laos, R.; Thomson, J.M.; Benner, S.A. DNA polymerases engineered by directed evolution to incorporate non-standard nucleotides. Front. Microbiol. 2014, 5, 565. [Google Scholar] [CrossRef] [PubMed]
- Laos, R.; Shaw, R.; Leal, N.A.; Gaucher, E.; Benner, S. Directed evolution of polymerases to accept nucleotides with nonstandard hydrogen bond patterns. Biochemistry 2013, 52, 5288–5294. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, M.M.; Singh, I.; Kellett, W.F.; Hoshika, S.; Benner, S.A.; Richards, N.G.J. Structural basis for a six nucleotide genetic alphabet. J. Am. Chem. Soc. 2015, 137, 6947–6955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiadis, M.; Kellett, W.; Singh, I.; Hoshika, S.; Benner, S.; Richards, N. Structural characterization of non-natural ZP base pairs in duplex DNA. In Abstracts of Papers of the American Chemical Society; American Chemical Society: Washington, DC, USA, 2015. [Google Scholar]
- Singh, I.; Kim, M.J.; Molt, R.W.; Hoshika, S.; Benner, S.A.; Georgiadis, M.M. Structure and biophysics for a six letter DNA alphabet that includes imidazo[1,2-a]-1,3,5-triazine-2(8H)-4(3H)-dione (x) and 2,4-diaminopyrimidine (k). ACS Synth. Biol. 2017, 6, 2118–2129. [Google Scholar] [CrossRef] [PubMed]
- Gonnet, G.H.; Cohen, M.A.; Benner, S.A. Exhaustive matching of the entire protein-sequence database. Science 1992, 256, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Jermann, T.M.; Opitz, J.G.; Stackhouse, J.; Benner, S.A. Reconstructing the evolutionary history of the artiodactyl ribonuclease superfamily. Nature 1995, 374, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, E.A.; Miyamoto, M.M.; Benner, S.A. Function-structure analysis of proteins using covarion-based evolutionary approaches: Elongation factors. Proc. Natl. Acad. Sci. USA 2001, 98, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, M.A.; Uryasev, O.; Frye, C.B.; Eckman, B.L.; Myers, C.R.; Hurley, T.D.; Benner, S.A. Hominids adapted to metabolize ethanol long before human-directed fermentation. Proc. Natl. Acad. Sci. USA 2015, 112, 458–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrigan, M.A.; Uryasev, O.; Davis, R.P.; Zhai, L.M.; Hurley, T.D.; Benner, S.A. The natural history of class I primate alcohol dehydrogenases includes gene duplication, gene loss, and gene conversion. PLoS ONE 2012, 7, e41175. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A.E.; Ellington, A.D. Evolution and structural theory. The frontier between chemistry and biochemistry. In Bioorganic Chemistry Frontiers; Springer: Berlin/Heidelberg, NY, USA, 1990; pp. 1–70. [Google Scholar]
- Geyer, C.R.; Battersby, T.R.; Benner, S.A. Nucleobase pairing in expanded Watson-Crick-like genetic information systems. Structure 2003, 11, 1485–1498. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.R.; Shao, Y.; Hoshika, S.; Yang, Z.; Shelke, S.A.; Herrou, J.; Kim, H.J.; Kim, M.J.; Piccirilli, J.A.; Benner, S.A. A crystal structure of a functional RNA molecule containing an artificial nucleobase pair. Angew. Chem. Int. Ed. Engl. 2015, 54, 9853–9856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Z.; Sefah, K.; Bradley, K.M.; Hoshika, S.; Kim, M.J.; Kim, H.J.; Zhu, G.; Jimenez, E.; Cansiz, S.; et al. Evolution of functional six-nucleotide DNA. J. Am. Chem. Soc. 2015, 137, 6734–6737. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Sismour, A.M.; Sheng, P.; Puskar, N.L.; Benner, S.A. Enzymatic incorporation of a third nucleobase pair. Nucleic Acids Res. 2007, 35, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Chen, F.; Alvarado, J.B.; Benner, S.A. Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J. Am. Chem. Soc. 2011, 133, 15105–15112. [Google Scholar] [CrossRef] [PubMed]
- Leal, N.A.; Kim, H.J.; Hoshika, S.; Kim, M.J.; Carrigan, M.A.; Benner, S.A. Transcription, reverse transcription, and analysis of RNA containing artificial genetic components. ACS Synth. Biol. 2015, 4, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Sismour, A.M.; Lutz, S.; Park, J.H.; Lutz, M.J.; Boyer, P.L.; Hughes, S.H.; Benner, S.A. PCR amplification of DNA containing non-standard base pairs by variants of reverse transcriptase from human immunodeficiency virus-1. Nucleic Acids Res. 2004, 32, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yang, Z.; Yan, M.; Alvarado, J.B.; Wang, G.; Benner, S.A. Recognition of an expanded genetic alphabet by type-II restriction endonucleases and their application to analyze polymerase fidelity. Nucleic Acids Res. 2011, 39, 3949–3961. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Durante, M.; Glushakova, L.G.; Sharma, N.; Leal, N.A.; Bradley, K.M.; Chen, F.; Benner, S.A. Conversion strategy using an expanded genetic alphabet to assay nucleic acids. Anal. Chem. 2013, 85, 4705–4712. [Google Scholar] [CrossRef] [PubMed]
- Sefah, K.; Yang, Z.; Bradley, K.M.; Hoshika, S.; Jimenez, E.; Zhang, L.; Zhu, G.; Shanker, S.; Yu, F.; Turek, D.; et al. In vitro selection with artificial expanded genetic information systems. Proc. Natl. Acad. Sci. USA 2014, 111, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Z.; Le Trinh, T.; Teng, I.T.; Wang, S.; Bradley, K.M.; Hoshika, S.; Wu, Q.; Cansiz, S.; Rowold, D.J.; et al. Aptamers against cells overexpressing glypican 3 from expanded genetic systems combined with cell engineering and laboratory evolution. Angew. Chem. Int. Ed. Engl 2016, 55, 12372–12375. [Google Scholar] [CrossRef] [PubMed]
- Biondi, E.; Lane, J.D.; Das, D.; Dasgupta, S.; Piccirilli, J.A.; Hoshika, S.; Bradley, K.M.; Krantz, B.A.; Benner, S.A. Laboratory evolution of artificially expanded DNA gives redesignable aptamers that target the toxic form of anthrax protective antigen. Nucleic Acids Res. 2016, 44, 9565–9577. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, E. Was ist Leben; Serie Piper 1134; Serie Piper 1134; Cambridge University Press: Cambridge, UK, 1943. [Google Scholar]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biondi, E.; Benner, S.A. Artificially Expanded Genetic Information Systems for New Aptamer Technologies. Biomedicines 2018, 6, 53. https://doi.org/10.3390/biomedicines6020053
Biondi E, Benner SA. Artificially Expanded Genetic Information Systems for New Aptamer Technologies. Biomedicines. 2018; 6(2):53. https://doi.org/10.3390/biomedicines6020053
Chicago/Turabian StyleBiondi, Elisa, and Steven A. Benner. 2018. "Artificially Expanded Genetic Information Systems for New Aptamer Technologies" Biomedicines 6, no. 2: 53. https://doi.org/10.3390/biomedicines6020053