

Immune Gene Networks from Lung Cancer Patients Treated with Immune Checkpoint Inhibitors

Abstract
1. Introduction
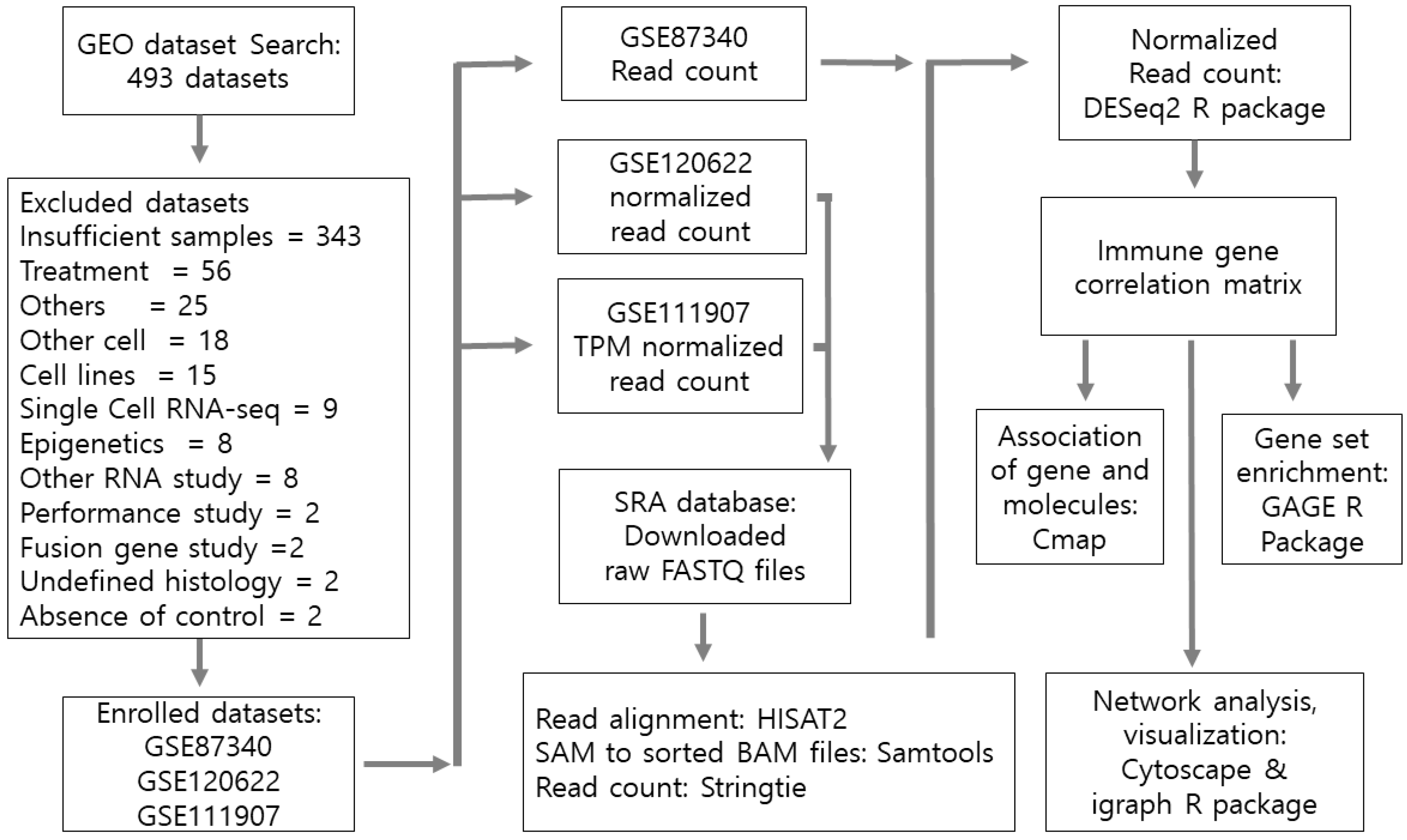
2. Materials and Methods
2.1. Gene Set Enrichment for Pathway Analysis
2.2. Network Construction
2.3. Network Topological Analysis
2.4. Connectivity Map Analysis
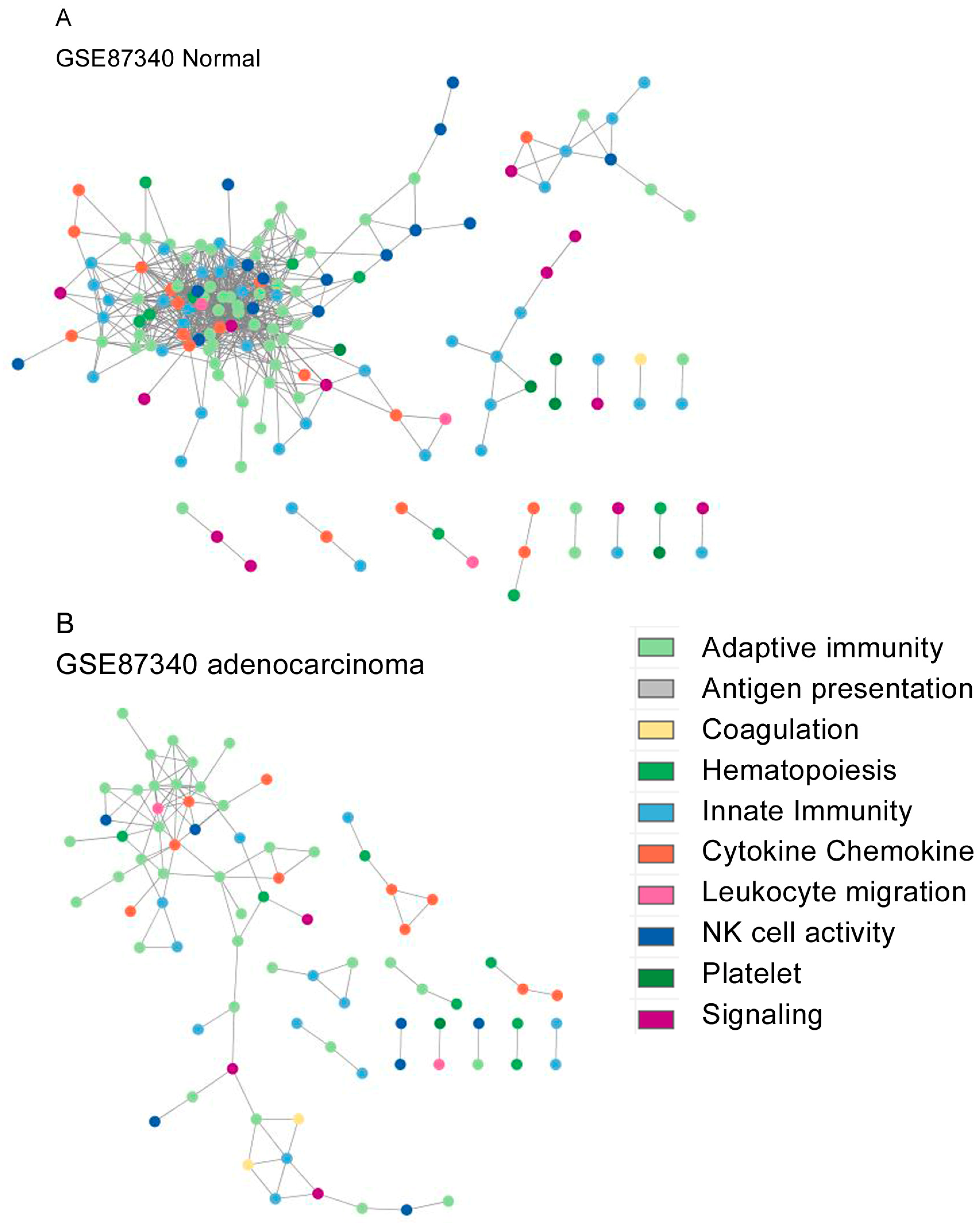
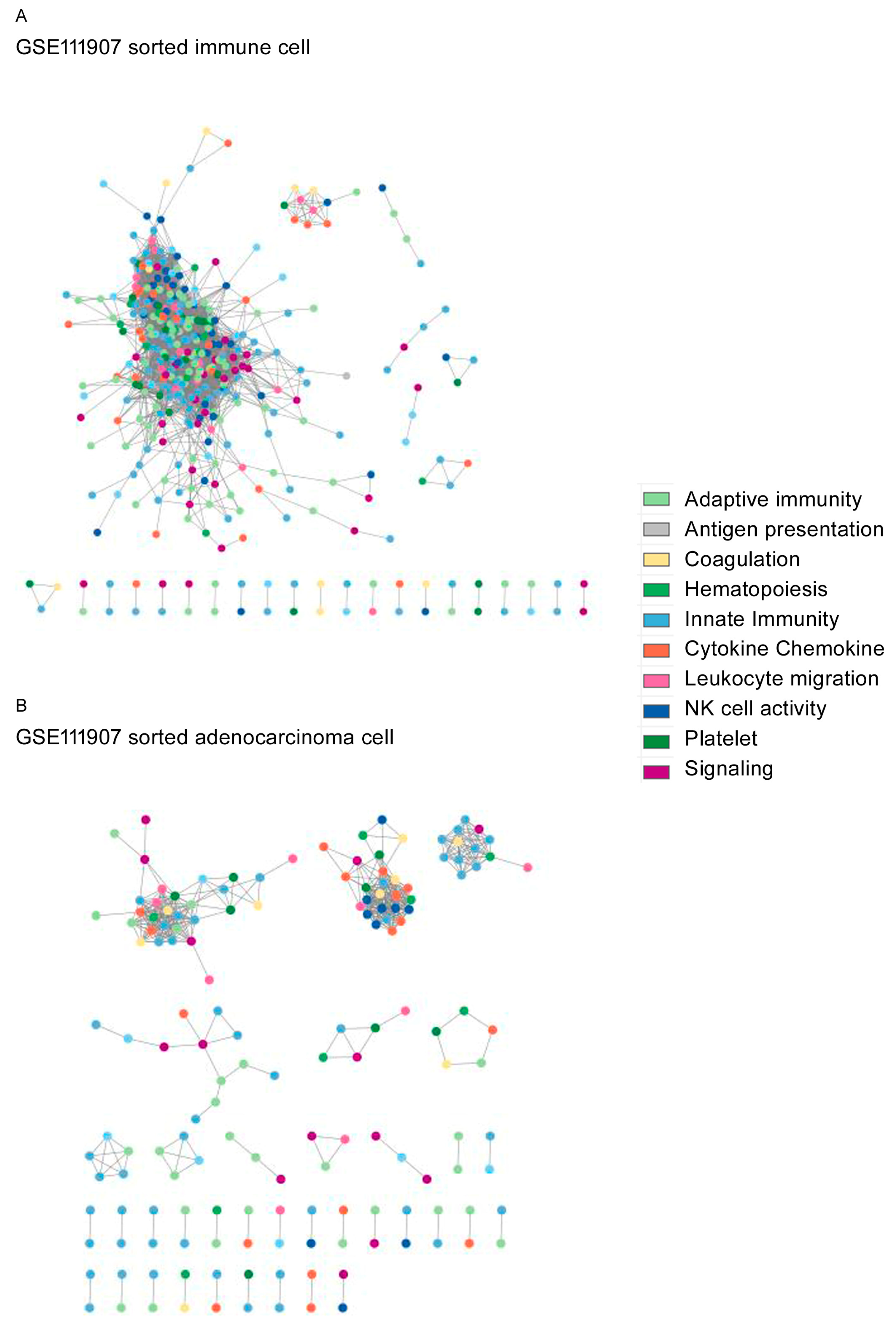
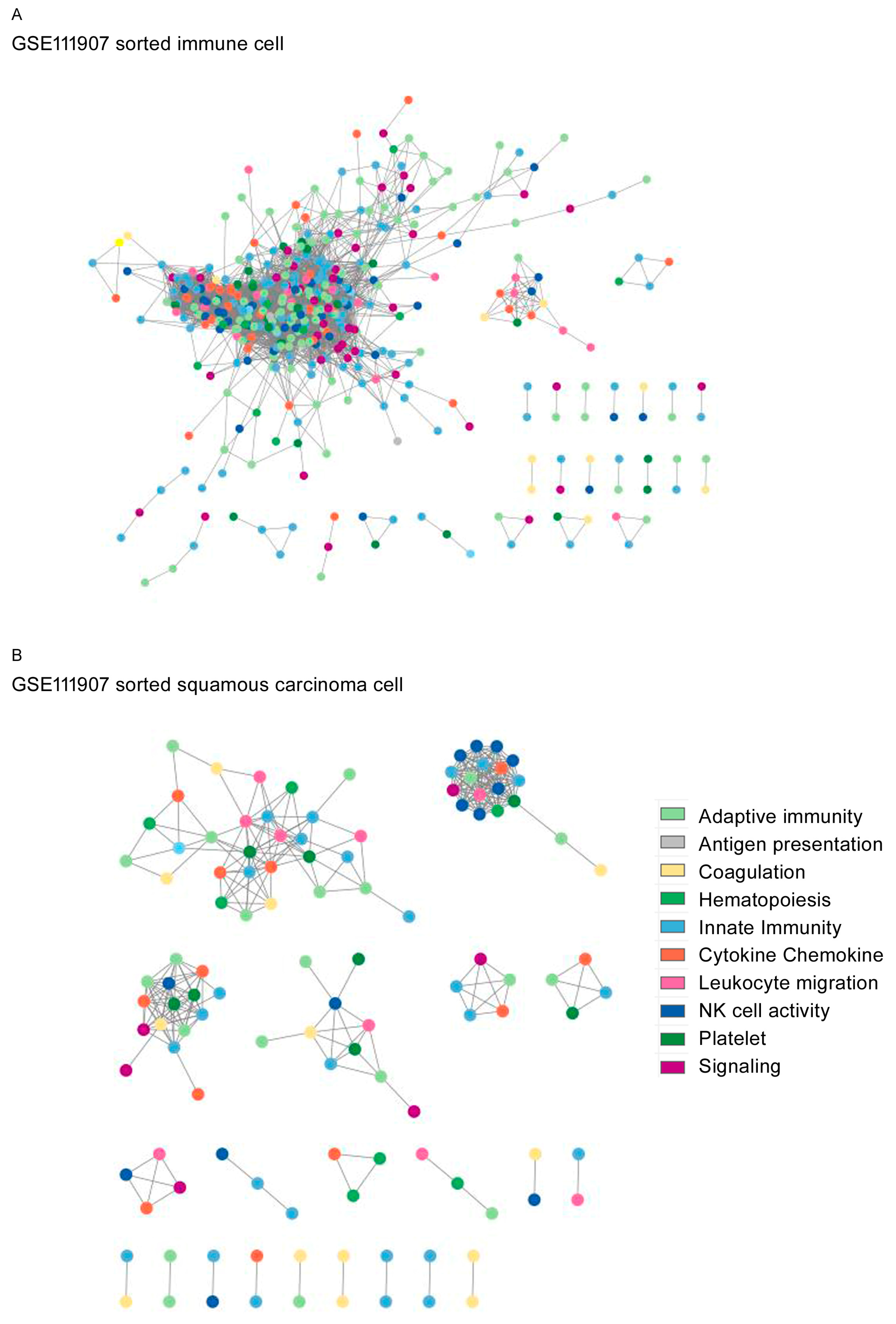
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Globocan 2020. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/15-Lung-fact-sheet.pdf (accessed on 21 January 2023).
- Siegel, R.L.; Miller, K.D.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Qin, C.; Hu, H.; Liu, T.; He, Y.; Guo, H.; Yan, H.; Zhang, J.; Tang, S.; Zhou, H. Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer: Progress, Challenges, and Prospects. Cells 2022, 11, 11030320. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, R.; Imbimbo, M.; Malouf, R.; Paget-Bailly, S.; Calais, F.; Marchal, C.; Westeel, V. Single or combined immune checkpoint inhibitors compared to first-line platinum-based chemotherapy with or without bevacizumab for people with advanced non-small cell lung cancer. Cochrane Database Syst. Rev. 2020, 12, Cd013257. [Google Scholar] [PubMed]
- Sullivan, M.R.; Ugolini, G.S.; Sarkar, S.; Kang, W.; Smith, E.C.; Mckenney, S.; Konry, T. Quantifying the efficacy of checkpoint inhibitors on CD8+ cytotoxic T cells for immunotherapeutic applications via single-cell interaction. Cell Death Dis. 2020, 11, 979. [Google Scholar] [CrossRef]
- Watson, R.A.; Tong, O.; Cooper, R.; Taylor, C.A.; Sharma, P.K.; de Los Aires, A.V.; Mahé, E.A.; Ruffieux, H.; Nassiri, I.; Middleton, M.R.; et al. Immune checkpoint blockade sensitivity and progression-free survival associates with baseline CD8(+) T cell clone size and cytotoxicity. Sci. Immunol. 2021, 6, eabj8825. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Forde, P.M.; Spicer, J.; Lu, S.; Provencio, M.; Mitsudomi, T.; Awad, M.M.; Felip, E.; Broderick, S.R.; Brahmer, J.R.; Swanson, S.J.; et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N. Engl. J. Med. 2022, 386, 1973–1985. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Borghaei, H.; Ramalingam, S.S.; Horn, L.; Carpeño, J.D.C.; Pluzanski, A.; Burgio, M.A.; Garassino, M.; Chow, L.Q.; Gettinger, S.; et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: A pooled analysis. Lancet Oncol. 2019, 20, 1395–1408. [Google Scholar] [CrossRef]
- Xiong, W.; Zhao, Y.; Du, H.; Guo, X. Current Status of Immune Checkpoint Inhibitor Immunotherapy for LungCancer. Front. Oncol. 2021, 11, 704336. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhai, S.; Lv, Z.; Yuan, L.; Wang, S.; Jin, D.; Yi, H.; Fu, L.; Mao, Y. Effect of histology on the efficacy of immune checkpoint inhibitors in advanced non-small cell lung cancer: A systematic review and meta-analysis. Front. Oncol. 2022, 12, 968517. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A. Network Science; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Newman, M.E.J. Networks; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Koutrouli, M.; Karatzas, E.; Paez-Espino, D.; Pavlopoulos, G.A. A Guide to Conquer the Biological Network Era Using Graph Theory. Front. Bioeng. Biotechnol. 2020, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Jekarl, D.W.; Yoo, J.; Lee, S.; Kim, M.; Kim, Y. Immune gene expression networks in sepsis: A network biology approach. PLoS ONE 2021, 16, e0247669. [Google Scholar] [CrossRef] [PubMed]
- Jekarl, D.W.; Kim, K.S.; Lee, S.; Kim, M.; Kim, Y. Cytokine and molecular networks in sepsis cases: A network biology approach. Eur. Cytokine Netw. 2018, 29, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Jekarl, D.W.; Lee, S.; Kwon, J.H.; Nam, S.W.; Kim, M.; Kim, Y.; Jang, J.W. Complex interaction networks of cytokines after transarterial chemotherapy in patients with hepatocellular carcinoma. PLoS ONE 2019, 14, e0224318. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, L.; Eckloff, B.W.; Deng, B.; Wang, Y.; Wampfler, J.A.; Jang, J.; Wieben, E.D.; Jen, J.; You, M.; et al. Conserved recurrent gene mutations correlate with pathway deregulation and clinical outcomes of lung adenocarcinoma in never-smokers. BMC Med. Genom. 2014, 7, 32. [Google Scholar] [CrossRef]
- Lu, H.H.; Lin, S.Y.; Weng, R.R.; Juan, Y.H.; Chen, Y.W.; Hou, H.H.; Hung, Z.C.; Oswita, G.A.; Huang, Y.J.; Guu, S.Y.; et al. Fucosyltransferase 4 shapes oncogenic glycoproteome to drive metastasis of lung adenocarcinoma. EBioMedicine 2020, 57, 102846. [Google Scholar] [CrossRef]
- Gentles, A.J.; Hui, A.B.Y.; Feng, W.; Azizi, A.; Nair, R.V.; Bouchard, G.; Knowles, D.A.; Yu, A.; Jeong, Y.; Bejnood, A.; et al. A human lung tumor microenvironment interactome identifies clinically relevant cell-type cross-talk. Genome Biol. 2020, 21, 107. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New pers- pectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Consortium, T.G.O.; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef]
- Harrell, F.E. Package ‘Hmisc’. Available online: https://cran.r-project.org/web/packages/Hmisc/Hmisc.pdf (accessed on 25 March 2023).
- Grolemund, G.; Wickham, H. R for Data Science: Import, Tidy, Transform, Visualize, and Model Data; O’Reilly: Sebastopol, CA, USA, 2017. [Google Scholar]
- Franz, M.; Lopes, C.T.; Fong, D.; Kucera, M.; Cheung, M.; Siper, M.C.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape.js 2023 update: A graph theory library for visualization and analysis. Bioinformatics 2023, 39, btad031. [Google Scholar] [CrossRef]
- NetworkAnalyzer. Available online: https://med.bioinf.mpi-inf.mpg.de/netanalyzer/index.php (accessed on 25 March 2023).
- Gillespie, C. Package “powRlaw”. Available online: https://cran.r-project.org/web/packages/poweRlaw/poweRlaw.pdf (accessed on 25 March 2023).
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- R Developoment Core Team. R: A language and environment for statistical computing. Available online: http://cran.r-project.org/ (accessed on 22 August 2023).
- Carlin, D.E.; Demchak, B.; Pratt, D.; Sage, E.; Ideker, T. Network propagation in the cytoscape cyberinfrastructure. PLoS Comput. Biol. 2017, 13, e1005598. [Google Scholar] [CrossRef]
- Pavlopoulos, G.A.; Secrier, M.; Moschopoulos, C.N.; Soldatos, T.G.; Kossida, S.; Aerts, J.; Schneider, R.; Bagos, P.G. Using graph theory to analyze biological networks. BioData Min. 2011, 4, 10. [Google Scholar] [CrossRef]
- Kalna, G.; Higham, D.J. A clustering Coefficient for weightd networks, with application to gene expression data. AI commun. 2007, 20, 263–271. [Google Scholar]
- Zimmermann, S.; Peters, S.; Owinokoko, T.; Gadgeel, S.M. Immune Checkpoint Inhibitors in the Management of Lung Cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Musa, A.; Ghoraie, L.S.; Zhang, S.D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Brief Bioinform. 2018, 19, 506–523. [Google Scholar] [PubMed]
- Wang, Y.; Han, H.; Zhang, F.; Lv, T.; Zhan, P.; Ye, M.; Song, Y.; Liu, H. Immune checkpoint inhibitors alone vs. immune checkpoint inhibitors—Combined chemotherapy for NSCLC patients with high PD-L1 expression: A network meta-analysis. Br. J. Cancer 2022, 127, 948–956. [Google Scholar] [CrossRef]
- Kim, K.S.; Moon, S.W.; Moon, M.H.; Hyun, K.Y.; Kim, S.J.; Kim, Y.K.; Kim, K.Y.; Jekarl, D.W.; Oh, E.J.; Kim, Y. Metabolic profiles of lung adenocarcinoma via peripheral blood and diagnostic model construction. Sci. Rep. 2023, 13, 7304. [Google Scholar] [CrossRef]
- De Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef]
- Chen, J.C.; Perez-Lorenzo, R.; Saenger, Y.M.; Drake, C.G.; Christiano, A.M. IKZF1 Enhances Immune Infiltrate Recruitment in Solid Tumors and Susceptibility to Immunotherapy. Cell. Syst. 2018, 7, 92–103. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSE87340 | GSE120622 | GSE111907 | ||
|---|---|---|---|---|
| LUAD | LUAD | LUSC | LUAD/LUSC | |
| Cases (n) | 27 | 43 | 37 | 24/12 |
| sex (M/F), n | 4/23 | 24/19 | 35/2 | 7/29 |
| Age, yr (sd) | 66.1 (12.6) | 81.5 (12.1) | 89.1 (8.1) | 70 (32–87) |
| Stage (n) a | IA (10)/IB (17) | IA (2), IB (16), IIA (4) | IA (3), IB (16), IIA (4) | I (14), II (12) |
| IIB (4), IIIa (12), IIIb (4) | IIB (7), IIIA (4), IIIB (0) | III (8) | ||
| IV(1) | IV(3) | IV (1) | ||
| Differentiation (n) | Well (20) | NA | NA | Well (7), Well/Mod (1) |
| Moderate (7) | NA | NA | Mod (15), Mod/poor (5) | |
| NA | NA | poor (9) | ||
| Sequential selection | 26 | 19 | 19 | 11/10 |
| of control cases (n) | ||||
| Sequential selection | 26 | 19 | 19 | 11/10 |
| of cancer cases (n) | ||||
| Platform | GPL111154 | GPL20301 | GPL17553 | |
| Illumina | Illumina | Illumina | ||
| HiSeq 2000 | HiSeq 4 000 | HiSeq 2000 | ||
| Reference | [21] | [22] | [23] | |
| GSE87340 | GSE120622 | GSE111907-1 | GSE111907-2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | LUAD | N | LUAD | LUSC | CD45+ | CD10+ | CD31+ | LUAD | CD45+ | CD10+ | CD31+ | LUSC | |
| Nodes (N) | 155 | 79 | 385 | 228 | 156 | 385 | 150 | 118 | 158 | 400 | 159 | 151 | 116 |
| Edge (N) | 805 | 109 | 2704 | 1069 | 319 | 4839 | 670 | 1579 | 472 | 4820 | 1149 | 517 | 327 |
| Average neighbor | 14.07 | 3.49 | 19.81 | 11.544 | 10 | 30.369 | 12 | 39.128 | 10.8 | 29.11 | 22.813 | 18.857 | 6.069 |
| Network diameter | 9 | 14 | 22 | 10 | 5 | 11 | 6 | 9 | 5 | 12 | 8 | 6 | 5 |
| Network radius | 5 | 7 | 11 | 5 | 3 | 6 | 3 | 5 | 3 | 6 | 4 | 3 | 3 |
| Path length | 2.852 | 4.994 | 6.463 | 3.765 | 2.2 | 3.167 | 2.177 | 2.11 | 2.032 | 4.148 | 2.393 | 1.782 | 2.365 |
| Clustering coefficient | 0.552 | 0.358 | 0.544 | 0.545 | 0.762 | 0.554 | 0.627 | 0.801 | 0.614 | 0.523 | 0.761 | 0.89 | 0.513 |
| Network density | 0.13 | 0.07 | 0.078 | 0.064 | 0.325 | 0.097 | 0.444 | 0.508 | 0.372 | 0.090 | 0.362 | 0.555 | 0.217 |
| Hetero- geneity | 0.834 | 0.682 | 1.097 | 0.766 | 0.507 | 0.788 | 0.612 | 0.564 | 0.604 | 0.803 | 0.678 | 0.518 | 0.59 |
| Network centrality | 0.273 | 0.136 | 0.216 | 0.121 | 0.273 | 0.182 | 0.279 | 0.225 | 0.34 | 0.174 | 0.282 | 0.192 | 0.304 |
| Connected components | 15 | 11 | 32 | 22 | 30 | 28 | 25 | 12 | 36 | 25 | 21 | 39 | 21 |
| Assortativity | 0.351 | 0.512 | 0.605 | 0.375 | 0.858 | 0.422 | 0.894 | 0.873 | 0.791 | 0.529 | 0.781 | 0.931 | 0.833 |
| Modularity | 0.293 | 0.677 | 0.333 | 0.571 | 0.765 | 0.383 | 0.778 | 0.107 | 0.7246 | 0.502 | 0.559 | 0.549 | 0.751 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.S.; Kang, T.; Jekarl, D.W. Immune Gene Networks from Lung Cancer Patients Treated with Immune Checkpoint Inhibitors. Biomedicines 2024, 12, 628. https://doi.org/10.3390/biomedicines12030628
Kim KS, Kang T, Jekarl DW. Immune Gene Networks from Lung Cancer Patients Treated with Immune Checkpoint Inhibitors. Biomedicines. 2024; 12(3):628. https://doi.org/10.3390/biomedicines12030628
Chicago/Turabian StyleKim, Kyung Soo, Taewon Kang, and Dong Wook Jekarl. 2024. "Immune Gene Networks from Lung Cancer Patients Treated with Immune Checkpoint Inhibitors" Biomedicines 12, no. 3: 628. https://doi.org/10.3390/biomedicines12030628
APA StyleKim, K. S., Kang, T., & Jekarl, D. W. (2024). Immune Gene Networks from Lung Cancer Patients Treated with Immune Checkpoint Inhibitors. Biomedicines, 12(3), 628. https://doi.org/10.3390/biomedicines12030628