
Aberrant AHRR, ADAMTS2 and FAM184 DNA Methylation: Candidate Biomarkers in the Oral Rinse of Heavy Smokers
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
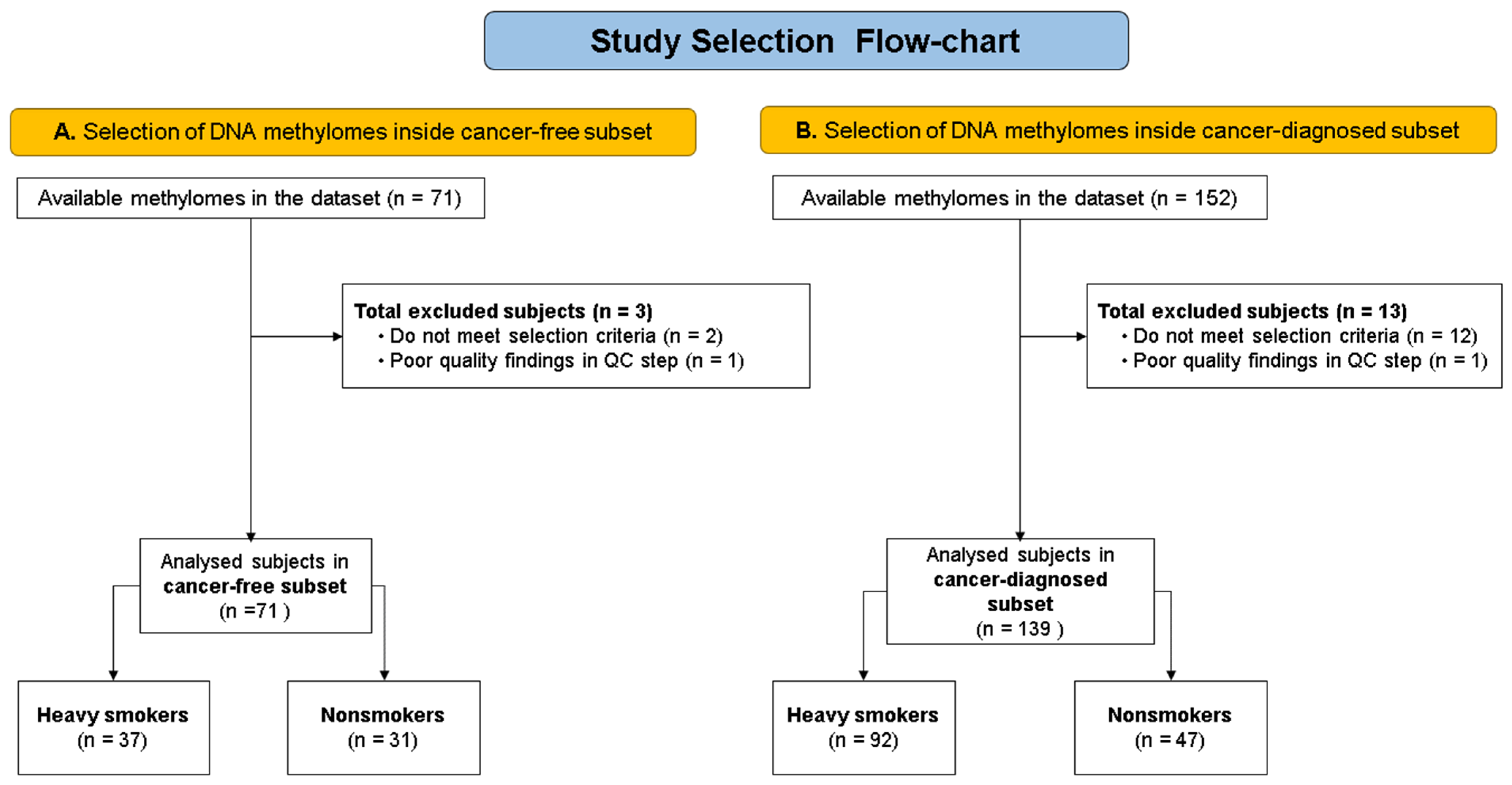
2.1. Available Data (Population)
2.2. Data Importation, Cleansing, and Quality Control
2.3. Statistical and Bioinformatic Analyses
2.4. Genome-Wide Differential Methylation Positions Analysis for Heavy Smokers
2.5. Genome-Wide Differentially Methylated Regions Analysis for Heavy Smokers
2.6. Gene Prioritization for Analysis Focused on Specific Genes
2.7. Endeavour Parameters Were Set As Follows:
- (a)
- Input 1 (list of training): genes identified in the nondirected analysis (AHRR, ADAMTS2, FAM184B) and with an OMIM assignation for “Orolaryngeal cancer” (CDKN2A).
- (b)
- Input 2 (list of candidates): genes evaluated/annotated by Infinium Illumina DNA Methylation 450 K.
- (c)
- For the gene prioritization analysis, the statistical threshold for identification was p < 0.01; otherwise, the settings in Endeavour were set to default [32].
2.8. Analyses of Differentially Methylated Positions (DMPs) and Regions (DMRs) of Specific Loci
2.9. Effect of Cell Composition Sensitivity Analysis
3. Results
Gene Prioritization Results and Corresponding DNA Methylation Analysis
4. Discussion
4.1. AHRR
4.2. ADAMTS2
4.3. FAM184B
4.4. MAPK14 and TFAP2A Genes Found by Gene Prioritization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ALHarthi, S.S.Y.; Natto, Z.S.; Midle, J.B.; Gyurko, R.; O’Neill, R.; Steffensen, B. Association between time since quitting smoking and periodontitis in former smokers in the National Health and Nutrition Examination Surveys (NHANES) 2009 to 2012. J. Periodontol. 2019, 90, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.O.; Gumus, Z.H.; Kacker, A.; Choksi, V.L.; Bocker, J.M.; Zhou, X.K.; Yantiss, R.K.; Hughes, D.B.; Du, B.; Judson, B.L.; et al. Effects of cigarette smoke on the human oral mucosal transcriptome. Cancer Prev. Res. 2010, 3, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Canova, C.; Richiardi, L.; Merletti, F.; Pentenero, M.; Gervasio, C.; Tanturri, G.; Garzino-Demo, P.; Pecorari, G.; Talamini, R.; Barzan, L.; et al. Alcohol, tobacco and genetic susceptibility in relation to cancers of the upper aerodigestive tract in northern Italy. Tumori 2010, 96, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.K.; Gour, N.; Pandey, A.; Wallia, D. Epidemiological study of risk factors for oral, laryngeal and esophageal cancers at a tertiary care hospital in India. Asian Pac. J. Cancer Prev. 2011, 12, 1215–1218. [Google Scholar] [PubMed]
- Cox, L.S.; Nollen, N.L.; Mayo, M.S.; Faseru, B.; Greiner, A.; Ellerbeck, E.F.; Krebill, R.; Tyndale, R.F.; Benowitz, N.L.; Ahluwalia, J.S. Effect of Varenicline Added to Counseling on Smoking Cessation Among African American Daily Smokers: The Kick It at Swope IV Randomized Clinical Trial. JAMA 2022, 327, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Galan, I.; Ortiz, C.; Perez-Rios, M.; Ayuso-Alvarez, A.; Rodriguez-Blazquez, C.; Damian, J.; Fernandez-Escobar, C.; Garcia-Esquinas, E.; Lopez-Cuadrado, T. Light cigarette smoking and all-cause mortality in Spain. A national population-based cohort study. Ann. Epidemiol. 2023, 78, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Toporcov, T.N.; Znaor, A.; Zhang, Z.F.; Yu, G.P.; Winn, D.M.; Wei, Q.; Vilensky, M.; Vaughan, T.; Thomson, P.; Talamini, R.; et al. Risk factors for head and neck cancer in young adults: A pooled analysis in the INHANCE consortium. Int. J. Epidemiol. 2015, 44, 169–185. [Google Scholar] [CrossRef] [Green Version]
- Kawakita, D.; Hosono, S.; Ito, H.; Oze, I.; Watanabe, M.; Hanai, N.; Hasegawa, Y.; Tajima, K.; Murakami, S.; Tanaka, H.; et al. Impact of smoking status on clinical outcome in oral cavity cancer patients. Oral. Oncol. 2012, 48, 186–191. [Google Scholar] [CrossRef]
- Galvin, S.; Moran, G.P.; Healy, C.M. Influence of site and smoking on malignant transformation in the oral cavity: Is the microbiome the missing link? Front. Oral Health 2023, 4, 1166037. [Google Scholar] [CrossRef]
- Zieba, S.; Maciejczyk, M.; Zalewska, A. Ethanol- and Cigarette Smoke-Related Alternations in Oral Redox Homeostasis. Front. Physiol. 2021, 12, 793028. [Google Scholar] [CrossRef]
- Sreekumar, V.N. Global Scenario of Research in Oral Cancer. J. Maxillofac. Oral Surg. 2019, 18, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.I.; Harris, C.C. Integration of multiple “OMIC” biomarkers: A precision medicine strategy for lung cancer. Lung. Cancer 2017, 107, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedzwiecki, M.M.; Walker, D.I.; Vermeulen, R.; Chadeau-Hyam, M.; Jones, D.P.; Miller, G.W. The Exposome: Molecules to Populations. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Vacante, M.; Borzi, A.M.; Basile, F.; Biondi, A. Biomarkers in colorectal cancer: Current clinical utility and future perspectives. World J. Clin. Cases 2018, 6, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Um, S.W.; Kim, Y.; Lee, B.B.; Kim, D.; Lee, K.J.; Kim, H.K.; Han, J.; Kim, H.; Shim, Y.M.; Kim, D.H. Genome-wide analysis of DNA methylation in bronchial washings. Clin. Epigenetics 2018, 10, 65. [Google Scholar] [CrossRef] [Green Version]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Chen, D.; Liu, P.; Xu, S.; Lin, X.; Zeng, R. Secondary analysis of existing microarray data reveals potential gene drivers of cutaneous squamous cell carcinoma. J. Cell Physiol. 2019, 234, 15270–15278. [Google Scholar] [CrossRef] [Green Version]
- Beltran-Garcia, J.; Osca-Verdegal, R.; Mena-Molla, S.; Garcia-Gimenez, J.L. Epigenetic IVD Tests for Personalized Precision Medicine in Cancer. Front. Genet. 2019, 10, 621. [Google Scholar] [CrossRef]
- Joehanes, R.; Just, A.C.; Marioni, R.E.; Pilling, L.C.; Reynolds, L.M.; Mandaviya, P.R.; Guan, W.; Xu, T.; Elks, C.E.; Aslibekyan, S.; et al. Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc. Genet. 2016, 9, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.K.; Hong, Y.; Kim, S.Y.; London, S.J.; Kim, W.J. DNA methylation and smoking in Korean adults: Epigenome-wide association study. Clin. Epigenetics 2016, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Langevin, S.M.; Eliot, M.; Butler, R.A.; Cheong, A.; Zhang, X.; McClean, M.D.; Koestler, D.C.; Kelsey, K.T. CpG island methylation profile in non-invasive oral rinse samples is predictive of oral and pharyngeal carcinoma. Clin. Epigenetics 2015, 7, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, S.; Kang, M.-I.; Ohtake, F. Hyperbolic Discounting, the Sign Effect, and the Body Mass Index. In Behavioral Economics of Preferences, Choices, and Happiness; Ikeda, S., Kato, H.K., Ohtake, F., Tsutsui, Y., Eds.; Springer: Japan, Tokyo, 2016; pp. 277–313. [Google Scholar]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- Fortin, J.P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivorra, C.; Fraga, M.F.; Bayon, G.F.; Fernandez, A.F.; Garcia-Vicent, C.; Chaves, F.J.; Redon, J.; Lurbe, E. DNA methylation patterns in newborns exposed to tobacco in utero. J. Transl. Med. 2015, 13, 25. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, H.G.; Sandoval-Hernandez, A.G.; Garrido-Gil, P.; Labandeira-Garcia, J.L.; Zelaya, M.V.; Bayon, G.F.; Fernandez, A.F.; Fraga, M.F.; Arboleda, G.; Arboleda, H. Alzheimer’s disease DNA methylome of pyramidal layers in frontal cortex: Laser-assisted microdissection study. Epigenomics 2018, 10, 1365–1382. [Google Scholar] [CrossRef]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, H.G.; Hernandez-Castaneda, A.A.; Pieschacon, M.P.; Arboleda, H. ZNF718, HOXA4, and ZFP57 are differentially methylated in periodontitis in comparison with periodontal health: Epigenome-wide DNA methylation pilot study. J. Periodontal. Res. 2021, 56, 710–725. [Google Scholar] [CrossRef]
- Carnielli, C.M.; Macedo, C.C.S.; De Rossi, T.; Granato, D.C.; Rivera, C.; Domingues, R.R.; Pauletti, B.A.; Yokoo, S.; Heberle, H.; Busso-Lopes, A.F.; et al. Combining discovery and targeted proteomics reveals a prognostic signature in oral cancer. Nat. Commun. 2018, 9, 3598. [Google Scholar] [CrossRef] [Green Version]
- Tranchevent, L.C.; Ardeshirdavani, A.; ElShal, S.; Alcaide, D.; Aerts, J.; Auboeuf, D.; Moreau, Y. Candidate gene prioritization with Endeavour. Nucleic Acids Res. 2016, 44, W117–W121. [Google Scholar] [CrossRef]
- Middleton, L.Y.M.; Dou, J.; Fisher, J.; Heiss, J.A.; Nguyen, V.K.; Just, A.C.; Faul, J.; Ware, E.B.; Mitchell, C.; Colacino, J.A.; et al. Saliva cell type DNA methylation reference panel for epidemiological studies in children. Epigenetics 2022, 17, 161–177. [Google Scholar] [CrossRef]
- Fisher, J.; Middleton, L.; Bakulski, K. BeadSorted.Saliva.EPIC: Illumina DNA Methylation Data on Sorted Saliva Cell Populations. R Package. Available online: https://bioconductor.org/packages/devel/data/experiment/vignettes/BeadSorted.Saliva.EPIC/inst/doc/BeadSorted.Saliva.EPIC.html (accessed on 1 March 2023).
- Yang, S.Y.; Ahmed, S.; Satheesh, S.V.; Matthews, J. Genome-wide mapping and analysis of aryl hydrocarbon receptor (AHR)- and aryl hydrocarbon receptor repressor (AHRR)-binding sites in human breast cancer cells. Arch. Toxicol. 2018, 92, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.A.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor repressor—More than a simple feedback inhibitor of AhR signaling: Clues for its role in inflammation and cancer. Curr. Opin. Toxicol. 2017, 2, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, G.M.; Kruppa, J.; Munz, M.; Wiehe, R.; Hasler, R.; Franke, A.; Martins, O.; Jockel-Schneider, Y.; Bruckmann, C.; Dommisch, H.; et al. A combined epigenome- and transcriptome-wide association study of the oral masticatory mucosa assigns CYP1B1 a central role for epithelial health in smokers. Clin. Epigenetics 2019, 11, 105. [Google Scholar] [CrossRef]
- Su, S.C.; Hsieh, M.J.; Liu, Y.F.; Chou, Y.E.; Lin, C.W.; Yang, S.F. ADAMTS14 Gene Polymorphism and Environmental Risk in the Development of Oral Cancer. PLoS ONE 2016, 11, e0159585. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Rao, N.; Ge, R. Emerging Roles of ADAMTSs in Angiogenesis and Cancer. Cancers 2012, 4, 1252–1299. [Google Scholar] [CrossRef] [Green Version]
- Aguet, F.; Anand, S.; Ardlie, K.G.; Gabriel, S.; Getz, G.A.; Graubert, A.; Hadley, K.; Handsaker, R.E.; Huang, K.H.; Kashin, S.; et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Wiklund, P.; Karhunen, V.; Richmond, R.C.; Parmar, P.; Rodriguez, A.; De Silva, M.; Wielscher, M.; Rezwan, F.I.; Richardson, T.G.; Veijola, J.; et al. DNA methylation links prenatal smoking exposure to later life health outcomes in offspring. Clin. Epigenetics 2019, 11, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khongsti, S.; Lamare, F.A.; Shunyu, N.B.; Ghosh, S.; Maitra, A.; Ghosh, S. Whole genome DNA methylation profiling of oral cancer in ethnic population of Meghalaya, North East India reveals novel genes. Genomics 2018, 110, 112–123. [Google Scholar] [CrossRef]
- Willis-Owen, S.A.G.; Thompson, A.; Kemp, P.R.; Polkey, M.I.; Cookson, W.; Moffatt, M.F.; Natanek, S.A. COPD is accompanied by co-ordinated transcriptional perturbation in the quadriceps affecting the mitochondria and extracellular matrix. Sci. Rep. 2018, 8, 12165. [Google Scholar] [CrossRef] [PubMed]
- NCBI. MAPK14 Mitogen-Activated Protein Kinase 14 [Homo Sapiens (Human)]—Gene. 2022. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=1432 (accessed on 1 October 2022).
- Groeger, S.; Jarzina, F.; Domann, E.; Meyle, J. Porphyromonas gingivalis activates NFkappaB and MAPK pathways in human oral epithelial cells. BMC Immunol. 2017, 18, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Liu, F.Y.; Kirkwood, K.L. The p38/MKP-1 signaling axis in oral cancer: Impact of tumor-associated macrophages. Oral Oncol. 2020, 103, 104591. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Q.; Tang, Y.; Liu, Z.; Sun, G.; Lu, Z.; Chen, Y. Identification and validation of a cigarette smoke-related five-gene signature as a prognostic biomarker in kidney renal clear cell carcinoma. Sci. Rep. 2022, 12, 2189. [Google Scholar] [CrossRef]
- Shi, D.; Xie, F.; Zhang, Y.; Tian, Y.; Chen, W.; Fu, L.; Wang, J.; Guo, W.; Kang, T.; Huang, W.; et al. TFAP2A regulates nasopharyngeal carcinoma growth and survival by targeting HIF-1alpha signaling pathway. Cancer Prev. Res. 2014, 7, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Hallberg, A.R.; Vorrink, S.U.; Hudachek, D.R.; Cramer-Morales, K.; Milhem, M.M.; Cornell, R.A.; Domann, F.E. Aberrant CpG methylation of the TFAP2A gene constitutes a mechanism for loss of TFAP2A expression in human metastatic melanoma. Epigenetics 2014, 9, 1641–1647. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, C.; Castillo-Fernandez, J.E.; Domingo-Relloso, A.; Zhao, W.; El-Sayed Moustafa, J.S.; Tsai, P.C.; Maddock, J.; Haack, K.; Cole, S.A.; Kardia, S.L.R.; et al. Novel DNA methylation signatures of tobacco smoking with trans-ethnic effects. Clin. Epigenetics 2021, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.R.; Da Silva, I.C.; Mariz, B.A.; Pereira, A.M.; De Oliveira, N.F. DNA methylation analysis of cancer-related genes in oral epithelial cells of healthy smokers. Arch. Oral Biol. 2015, 60, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Carta, C.F.L.; Oliveira Alves, M.G.; de Barros, P.P.; Campos, M.S.; Scholz, J.; Jorge, A.O.C.; Nunes, F.D.; Almeida, J.D. Screening methylation of DNA repair genes in the oral mucosa of chronic smokers. Arch. Oral Biol. 2018, 92, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinzon-Reyes, E.; Alvarez, W.A.; Rondon-Villarreal, P.; Hernandez, H.G. Softepigen: Primers Design Web-Based Tool for MS-HRM Technique. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 354–357. [Google Scholar] [CrossRef]
- Flavahan, W.A. Epigenetic plasticity, selection, and tumorigenesis. Biochem. Soc. Trans. 2020, 48, 1609–1621. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| DNA Methylomes from Cancer-Free Subset | |||
|---|---|---|---|
| Heavy Smokers (n = 37) | Controls (n = 31) | p-Values | |
| Age, years (mean ± standard deviation) | 62.73 ± 8.044 | 58.45 ± 12.01 | §: 0.097 |
| Males, n (%) | 24 (64.86%) | 19 (61.29) | †: 0.959 |
| Alcohol consumption | ‡: 0.041 * | ||
| No alcohol consumption, n (%) | 4 (10.81%) | 7 (22.58) | |
| Alcohol low frequency, n (%) | 20 (54.05) | 21 (67.74) | |
| Alcohol high frequency, n (%) | 13(35.14) | 3 (9.68) | |
| DNA methylomes from cancer-diagnosed subset | |||
| Heavy smokers (n = 92) | Controls (n = 47) | p-values | |
| Age, years (mean ± standard deviation) | 57.57 ± 12.55 | 60.77 ± 11.07 | §: 0.144 |
| Males, n (%) | 69(76) | 22(66) | †: 0.286 |
| Alcohol consumption | ‡: 0.002 * | ||
| No alcohol consumption, n (%) | 4 (0.44%) | 6 (13%) | |
| Alcohol low frequency, n (%) | 42 (45.6%) | 31 (67%) | |
| Alcohol high frequency, n (%) | 46 (50%) | 10 (21%) | |
| Probe ID | Gene Symbol | GeneID | logFC | AveExpr | t | p-Value | Adj.P.Val | B | Δβ | Dir |
|---|---|---|---|---|---|---|---|---|---|---|
| cg02599361 | ADAMTS2 | 9509 | 0.133 | 0.664 | 4.93 | 5.8 × 10−6 | 0.026 | 2.45 | 0.10 | ↑ |
| cg04450456 | FAM184B | 27146 | 0.101 | 0.811 | 5.75 | 2.42 × 10−7 | 0.008 | 5.57 | 0.10 | ↑ |
| cg15017067 | FAM184B | 27146 | 0.093 | 0.766 | 4.65 | 1.62 × 10−5 | 0.037 | 1.44 | 0.09 | ↑ |
| Probe ID | Gene | GeneID | AveExpr | t | p-Value | Adj.P.Val | B | Δβ | Dir |
|---|---|---|---|---|---|---|---|---|---|
| cg05575921 | AHRR | 57491 | 0.72 | −5.37 | 3.20 × 10−7 | 2.23 × 10−2 | 4.86 | −0.13 | ↓ |
| cg05951221 | - | - | 0.40 | −7.16 | 4.32 × 10−11 | 9.05 × 10−6 | 13.63 | −0.10 | ↓ |
| cg06126421 | - | 0.50 | −5.22 | 6.55 × 10−7 | 3.92 × 10−2 | 4.21 | −0.11 | ↓ | |
| cg21566642 | - | - | 0.40 | −6.44 | 1.81 × 10−9 | 2.53 × 10−4 | 9.95 | −0.10 | ↓ |
| hg19 Coordinates | Width | Gene(s) | Group | #p | Minpval | Meanpval | Maxbetafc | Mean Dbeta | Dir |
|---|---|---|---|---|---|---|---|---|---|
| chr12:2943902-2944493 | 592 | NRIP2 | 1st exon, 5′UTR, TSS200 | 8 | 2.50 × 10−8 | 3.72 × 10−8 | 0.015 | 0.066 | ↑ |
| chr4:17643702-17643749 | 48 | FAM184B | Body | 2 | 3.11 × 10−6 | 3.11 × 10−6 | 0.015 | 0.098 | ↑ |
| chr2:233215939-233217079 | 1141 | 6 | 2.89 × 10−3 | 6.95 × 10−3 | 0.015 | 0.071 | ↑ | ||
| chr1:102312608-102312671 | 64 | OLFM3 | Body | 3 | 8.87 × 10−3 | 8.89 × 10−3 | 0.013 | 0.078 | ↑ |
| chr19:49001890-49002477 | 588 | LMTK3 | Body | 3 | 9.16 × 10−3 | 2.16 × 10−2 | 0.012 | 0.071 | ↑ |
| chr17:80708279-80708513 | 235 | FN3K, TBCD | Body, TSS1500 | 3 | 1.25 × 10−2 | 1.26 × 10−2 | 0.018 | 0.093 | ↑ |
| chr19:18888799-18889003 | 205 | CRTC1 | 3′UTR | 2 | 1.41 × 10−2 | 1.84 × 10−2 | 0.011 | 0.075 | ↑ |
| chr5:373299373887 | 589 | AHRR | Body | 3 | 1.61 × 10−2 | 2.34 × 10−2 | −0.019 | −0.069 | ↓ |
| chr1:19110734-19110978 | 245 | 3 | 2.51 × 10−2 | 2.55 × 10−2 | 0.029 | 0.132 | ↑ | ||
| chr7:52341648-52342124 | 477 | 3 | 3.43 × 10−2 | 4.09 × 10−2 | 0.010 | 0.062 | ↑ | ||
| chr21:37437505-37437565 | 61 | SETD4 | TSS1500 | 2 | 4.17 × 10−2 | 4.24 × 10−2 | −0.025 | −0.062 | ↓ |
| chr4:100242862-100242957 | 96 | ADH1B | TSS1500 | 2 | 4.23 × 10−2 | 4.50 × 10−2 | 0.013 | 0.074 | ↑ |
| hg19 Coordinates | Width | Gene(s) | Group | #p | Minpval | Meanpval | Maxbetafc | Mean Dbeta | Dir |
|---|---|---|---|---|---|---|---|---|---|
| chr5:373299-373887 | 589 | AHRR | Body | 3 | 7.58 × 10−5 | 3.75 × 10−3 | −0.02 | −0.07 | ↓ |
| chr19:17000585-17000585 | 1 | F2RL3 | Body | 1 | 4.50 × 10−2 | 4.50 × 10−2 | −0.01 | −0.08 | ↓ |
| Gene | ProbeID | logFC | r | t | p-Value | Adj.P.Val | B | Δβ | Dir |
|---|---|---|---|---|---|---|---|---|---|
| Cancer diagnosed subset | |||||||||
| FAM184B | cg04450456 | 0.10 | 0.81 | 5.37 | 1.16 × 10−6 | 9.24 × 10−3 | 3.98 | 0.10 | ↑ |
| ADAM2 | cg02599361 | 0.13 | 0.66 | 5.34 | 1.27 × 10−6 | 9.31 × 10−3 | 3.89 | 0.10 | ↑ |
| Cancer-free subset | |||||||||
| AHRR | cg05575921 | −0.10 | 0.72 | −5.66 | 8.89 × 10−8 | 5.31 × 10−3 | 6.07 | −0.13 | ↓ |
| Intergenic | cg21566642 | −0.10 | 0.40 | −6.65 | 6.62 × 10−10 | 1.38 × 10−4 | 10.91 | −0.10 | ↓ |
| Intergenic | cg05951221 | −0.10 | 0.40 | −6.12 | 9.69 × 10−9 | 1.35 × 10−3 | 8.24 | −0.10 | ↓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández, H.G.; Aranzazu-Moya, G.C.; Pinzón-Reyes, E.H. Aberrant AHRR, ADAMTS2 and FAM184 DNA Methylation: Candidate Biomarkers in the Oral Rinse of Heavy Smokers. Biomedicines 2023, 11, 1797. https://doi.org/10.3390/biomedicines11071797
Hernández HG, Aranzazu-Moya GC, Pinzón-Reyes EH. Aberrant AHRR, ADAMTS2 and FAM184 DNA Methylation: Candidate Biomarkers in the Oral Rinse of Heavy Smokers. Biomedicines. 2023; 11(7):1797. https://doi.org/10.3390/biomedicines11071797
Chicago/Turabian StyleHernández, Hernán Guillermo, Gloria Cristina Aranzazu-Moya, and Efraín Hernando Pinzón-Reyes. 2023. "Aberrant AHRR, ADAMTS2 and FAM184 DNA Methylation: Candidate Biomarkers in the Oral Rinse of Heavy Smokers" Biomedicines 11, no. 7: 1797. https://doi.org/10.3390/biomedicines11071797