
An Overview of Chronic Kidney Disease Pathophysiology: The Impact of Gut Dysbiosis and Oral Disease

 , , , and
, , , and
Abstract
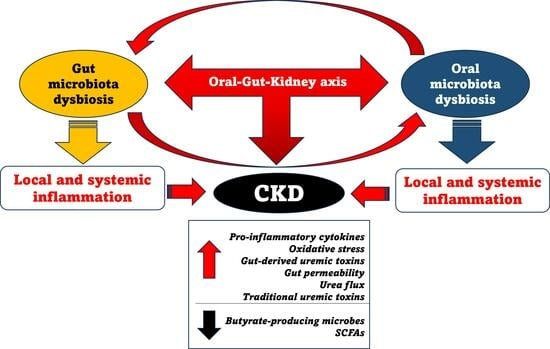
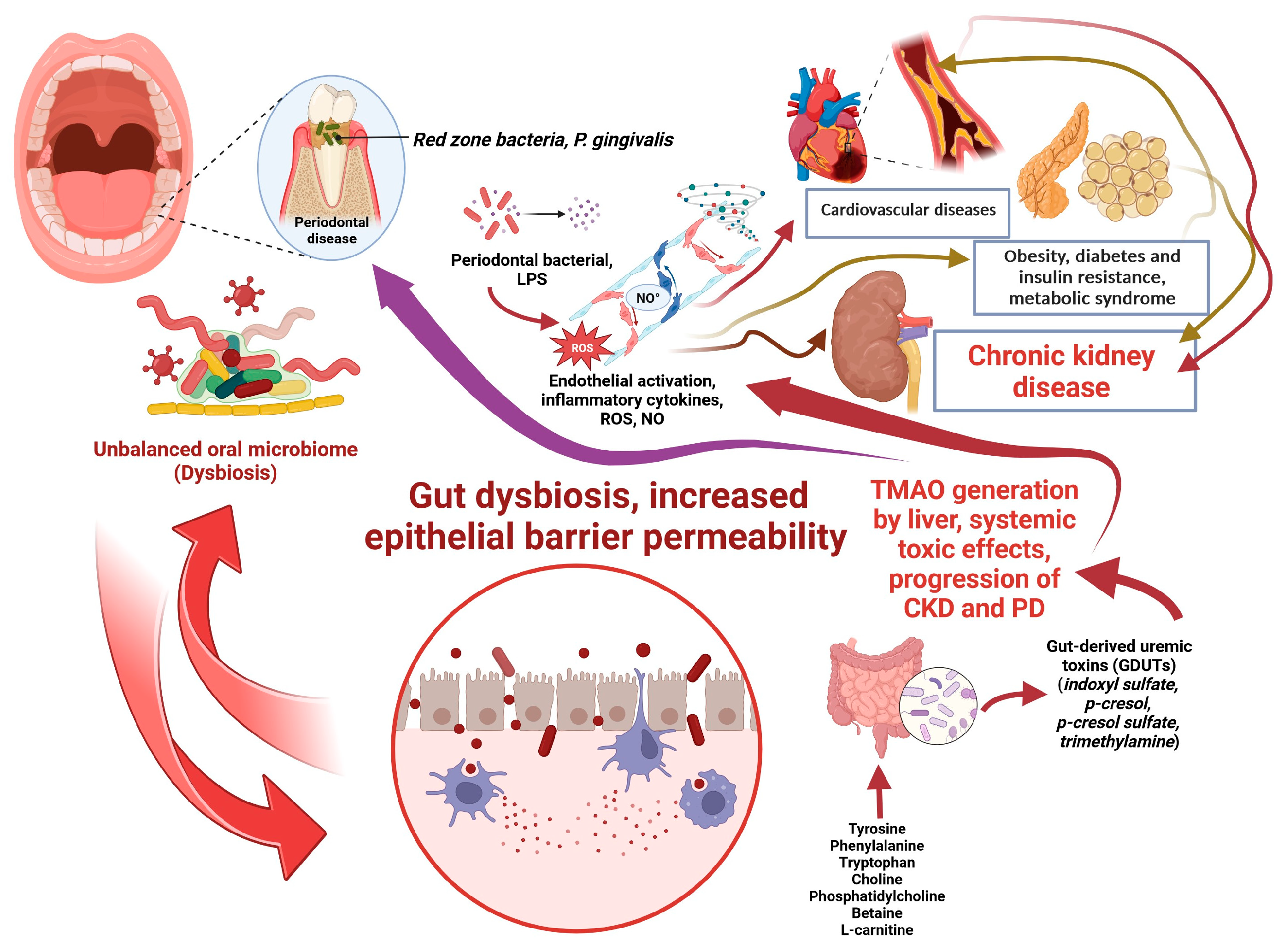
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Inflammation and Metabolic Products in CKD
2.1. Inflammasomes in CKD
2.2. Oxidative Stress (OS) and Endothelial Dysfunction (ED) in CKD
2.3. Uremic Toxins (UTs) and Gut-Derived Uremic Toxins (GDUTs) in CKD
2.4. The Bi-Directional Relationship between Visceral Fat and CKD
3. Gut-Kidney Axis and CKD
3.1. The Impact of Gut Dysbiosis on CKD
3.2. The Impact of CKD on Gut Microbiota: A Vicious Circle
3.3. Unhealthy Eating Habits as Pro-Dysbiosis Factors in CKD
3.4. Can Probiotics Be a Valid Support to Slow the Progression of CKD?
4. Impaired Oral Health and CKD
4.1. The Bidirectional Link between CKD and Periodontal Disease (PD)
4.2. The Association between Oral Dysbiosis and Impaired Kidney Function
5. Impact of Periodontal Treatment on Kidney Function
6. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levey, A.S.; Becker, C.; Inker, L.A. Glomerular filtration rate and albuminuria for detection and staging of acute and chronic kidney disease in adults: A systematic review. JAMA 2015, 313, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Manns, L.; Scott-Douglas, N.; Tonelli, M.; Weaver, R.; Tam-Tham, H.; Chong, C.; Hemmelgarn, B. A Population-Based Analysis of Quality Indicators in CKD. Clin. J. Am. Soc. Nephrol. 2017, 12, 727–733. [Google Scholar] [CrossRef]
- Inker, L.A.; Astor, B.C.; Fox, C.H.; Isakova, T.; Lash, J.P.; Peralta, C.A.; Kurella Tamura, M.; Feldman, H.I. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am. J. Kidney Dis. 2014, 63, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.A.; Zhao, J.; McCullough, K.; Fuller, D.S.; Figueiredo, A.E.; Bieber, B.; Finkelstein, F.O.; Shen, J.; Kanjanabuch, T.; Kawanishi, H.; et al. Burden of Kidney Disease, Health-Related Quality of Life, and Employment among Patients Receiving Peritoneal Dialysis and In-Center Hemodialysis: Findings From the DOPPS Program. Am. J. Kidney Dis. 2021, 78, 489–500.e481. [Google Scholar] [CrossRef]
- Collaboration, G.B.D.C.K.D. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Kofod, D.H.; Carlson, N.; Ballegaard, E.F.; Almdal, T.P.; Torp-Pedersen, C.; Gislason, G.; Svendsen, J.H.; Feldt-Rasmussen, B.; Hornum, M. Cardiovascular mortality in patients with advanced chronic kidney disease with and without diabetes: A nationwide cohort study. Cardiovasc. Diabetol. 2023, 22, 140. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef]
- Nair, N.; Kalra, R.; Chandra Bhatt, G.; Narang, A.; Kumar, G.; Raina, R. The Effect and Prevalence of Comorbidities in Adolescents with CKD and Obesity. Adv. Chronic Kidney Dis. 2022, 29, 251–262. [Google Scholar] [CrossRef]
- Gregg, L.P.; Hedayati, S.S. Management of Traditional Cardiovascular Risk Factors in CKD: What Are the Data? Am. J. Kidney Dis. 2018, 72, 728–744. [Google Scholar] [CrossRef]
- Speer, T.; Dimmeler, S.; Schunk, S.J.; Fliser, D.; Ridker, P.M. Targeting innate immunity-driven inflammation in CKD and cardiovascular disease. Nat. Rev. Nephrol. 2022, 18, 762–778. [Google Scholar] [CrossRef]
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular disease in dialysis patients. Nephrol. Dial. Transplant. 2018, 33, iii28–iii34. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M.; Alberta Kidney Disease, N. Cause of Death in Patients with Reduced Kidney Function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Batra, G.; Ghukasyan Lakic, T.; Lindback, J.; Held, C.; White, H.D.; Stewart, R.A.H.; Koenig, W.; Cannon, C.P.; Budaj, A.; Hagstrom, E.; et al. Interleukin 6 and Cardiovascular Outcomes in Patients with Chronic Kidney Disease and Chronic Coronary Syndrome. JAMA Cardiol. 2021, 6, 1440–1445. [Google Scholar] [CrossRef]
- Mathew, R.O.; Bangalore, S.; Lavelle, M.P.; Pellikka, P.A.; Sidhu, M.S.; Boden, W.E.; Asif, A. Diagnosis and management of atherosclerotic cardiovascular disease in chronic kidney disease: A review. Kidney Int. 2017, 91, 797–807. [Google Scholar] [CrossRef]
- Xu, H.; Matsushita, K.; Su, G.; Trevisan, M.; Arnlov, J.; Barany, P.; Lindholm, B.; Elinder, C.G.; Lambe, M.; Carrero, J.J. Estimated Glomerular Filtration Rate and the Risk of Cancer. Clin. J. Am. Soc. Nephrol. 2019, 14, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Hand, T.W.; Vujkovic-Cvijin, I.; Ridaura, V.K.; Belkaid, Y. Linking the Microbiota, Chronic Disease, and the Immune System. Trends Endocrinol. Metab. 2016, 27, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Lu, P.C.; Hou, C.Y.; Tain, Y.L. Blood Pressure Abnormalities Associated with Gut Microbiota-Derived Short Chain Fatty Acids in Children with Congenital Anomalies of the Kidney and Urinary Tract. J. Clin. Med. 2019, 8, 1090. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lu, P.C.; Lo, M.H.; Lin, I.C.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Gut Microbiota-Dependent Trimethylamine N-Oxide Pathway Associated with Cardiovascular Risk in Children with Early-Stage Chronic Kidney Disease. Int. J. Mol. Sci. 2018, 19, 3699. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bai, M.; Ning, X.; Qin, Y.; Wang, Y.; Yu, Z.; Dong, R.; Zhang, Y.; Sun, S. Expansion of Escherichia-Shigella in Gut Is Associated with the Onset and Response to Immunosuppressive Therapy of IgA Nephropathy. J. Am. Soc. Nephrol. 2022, 33, 2276–2292. [Google Scholar] [CrossRef]
- Stavropoulou, E.; Kantartzi, K.; Tsigalou, C.; Konstantinidis, T.; Romanidou, G.; Voidarou, C.; Bezirtzoglou, E. Focus on the Gut-Kidney Axis in Health and Disease. Front. Med. 2020, 7, 620102. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef]
- Pluznick, J.L. The gut microbiota in kidney disease. Science 2020, 369, 1426–1427. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xin, W.; Xiong, J.; Yao, M.; Zhang, B.; Zhao, J. The Intestinal Microbiota and Metabolites in the Gut-Kidney-Heart Axis of Chronic Kidney Disease. Front. Pharmacol. 2022, 13, 837500. [Google Scholar] [CrossRef]
- Sabatino, A.; Regolisti, G.; Brusasco, I.; Cabassi, A.; Morabito, S.; Fiaccadori, E. Alterations of intestinal barrier and microbiota in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Nouri, Z.; Zhang, X.Y.; Khakisahneh, S.; Degen, A.A.; Wang, D.H. The microbiota-gut-kidney axis mediates host osmoregulation in a small desert mammal. NPJ Biofilm. Microbiomes 2022, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Ebert, T.; Neytchev, O.; Witasp, A.; Kublickiene, K.; Stenvinkel, P.; Shiels, P.G. Inflammation and Oxidative Stress in Chronic Kidney Disease and Dialysis Patients. Antioxid. Redox Signal 2021, 35, 1426–1448. [Google Scholar] [CrossRef] [PubMed]
- Graterol Torres, F.; Molina, M.; Soler-Majoral, J.; Romero-Gonzalez, G.; Rodriguez Chitiva, N.; Troya-Saborido, M.; Socias Rullan, G.; Burgos, E.; Paul Martinez, J.; Urrutia Jou, M.; et al. Evolving Concepts on Inflammatory Biomarkers and Malnutrition in Chronic Kidney Disease. Nutrients 2022, 14, 4297. [Google Scholar] [CrossRef]
- Kadatane, S.P.; Satariano, M.; Massey, M.; Mongan, K.; Raina, R. The Role of Inflammation in CKD. Cells 2023, 12, 1581. [Google Scholar] [CrossRef]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Grabulosa, C.C.; Manfredi, S.R.; Canziani, M.E.; Quinto, B.M.R.; Barbosa, R.B.; Rebello, J.F.; Batista, M.C.; Cendoroglo, M.; Dalboni, M.A. Chronic kidney disease induces inflammation by increasing Toll-like receptor-4, cytokine and cathelicidin expression in neutrophils and monocytes. Exp. Cell Res. 2018, 365, 157–162. [Google Scholar] [CrossRef]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef] [PubMed]
- Munoz Mendoza, J.; Isakova, T.; Cai, X.; Bayes, L.Y.; Faul, C.; Scialla, J.J.; Lash, J.P.; Chen, J.; He, J.; Navaneethan, S.; et al. Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int. 2017, 91, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Schunk, S.J.; Triem, S.; Schmit, D.; Zewinger, S.; Sarakpi, T.; Becker, E.; Hutter, G.; Wrublewsky, S.; Kuting, F.; Hohl, M.; et al. Interleukin-1alpha Is a Central Regulator of Leukocyte-Endothelial Adhesion in Myocardial Infarction and in Chronic Kidney Disease. Circulation 2021, 144, 893–908. [Google Scholar] [CrossRef]
- Sun, J.; Axelsson, J.; Machowska, A.; Heimburger, O.; Barany, P.; Lindholm, B.; Lindstrom, K.; Stenvinkel, P.; Qureshi, A.R. Biomarkers of Cardiovascular Disease and Mortality Risk in Patients with Advanced CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Darisipudi, M.N.; Knauf, F. An update on the role of the inflammasomes in the pathogenesis of kidney diseases. Pediatr. Nephrol. 2016, 31, 535–544. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Leemans, J.C.; Kors, L.; Anders, H.J.; Florquin, S. Pattern recognition receptors and the inflammasome in kidney disease. Nat. Rev. Nephrol. 2014, 10, 398–414. [Google Scholar] [CrossRef]
- Zewinger, S.; Schumann, T.; Fliser, D.; Speer, T. Innate immunity in CKD-associated vascular diseases. Nephrol. Dial. Transplant. 2016, 31, 1813–1821. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, Q. Uncoupled pyroptosis and IL-1beta secretion downstream of inflammasome signaling. Front. Immunol. 2023, 14, 1128358. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhang, Z.; Li, Y. Relevance of the Pyroptosis-Related Inflammasome Pathway in the Pathogenesis of Diabetic Kidney Disease. Front. Immunol. 2021, 12, 603416. [Google Scholar] [CrossRef]
- Gregg, L.P.; Tio, M.C.; Li, X.; Adams-Huet, B.; de Lemos, J.A.; Hedayati, S.S. Association of Monocyte Chemoattractant Protein-1 with Death and Atherosclerotic Events in Chronic Kidney Disease. Am. J. Nephrol. 2018, 47, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Chertow, G.M.; Devarajan, P.; Levin, A.; Andreoli, S.P.; Bangalore, S.; Warady, B.A. Chronic Inflammation in Chronic Kidney Disease Progression: Role of Nrf2. Kidney Int. Rep. 2021, 6, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Carlstrom, M.; Montenegro, M.F. Therapeutic value of stimulating the nitrate-nitrite-nitric oxide pathway to attenuate oxidative stress and restore nitric oxide bioavailability in cardiorenal disease. J. Intern. Med. 2019, 285, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Gherghina, M.E.; Peride, I.; Tiglis, M.; Neagu, T.P.; Niculae, A.; Checherita, I.A. Uric Acid and Oxidative Stress-Relationship with Cardiovascular, Metabolic, and Renal Impairment. Int. J. Mol. Sci. 2022, 23, 3188. [Google Scholar] [CrossRef]
- Jung, S.W.; Kim, S.M.; Kim, Y.G.; Lee, S.H.; Moon, J.Y. Uric acid and inflammation in kidney disease. Am. J. Physiol. Renal Physiol. 2020, 318, F1327–F1340. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef]
- Wang, D.; Li, J.; Luo, G.; Zhou, J.; Wang, N.; Wang, S.; Zhao, R.; Cao, X.; Ma, Y.; Liu, G.; et al. Nox4 as a novel therapeutic target for diabetic vascular complications. Redox Biol. 2023, 64, 102781. [Google Scholar] [CrossRef]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef]
- Gorin, Y.; Wauquier, F. Upstream regulators and downstream effectors of NADPH oxidases as novel therapeutic targets for diabetic kidney disease. Mol. Cells 2015, 38, 285–296. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal 2016, 25, 119–146. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Oates, J.C.; Russell, D.L.; Van Beusecum, J.P. Endothelial cells: Potential novel regulators of renal inflammation. Am. J. Physiol. Renal Physiol. 2022, 322, F309–F321. [Google Scholar] [CrossRef]
- Chintam, K.; Chang, A.R. Strategies to Treat Obesity in Patients with CKD. Am. J. Kidney Dis. 2021, 77, 427–439. [Google Scholar] [CrossRef]
- Yan, M.T.; Chao, C.T.; Lin, S.H. Chronic Kidney Disease: Strategies to Retard Progression. Int. J. Mol. Sci. 2021, 22, 10084. [Google Scholar] [CrossRef]
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.A.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Maggo, J.K.; Campbell, K.L.; Craig, J.C.; Johnson, D.W.; Sutanto, B.; Ruospo, M.; Tong, A.; Strippoli, G.F. Dietary interventions for adults with chronic kidney disease. Cochrane Database Syst. Rev. 2017, 4, CD011998. [Google Scholar] [CrossRef]
- Kochan, Z.; Szupryczynska, N.; Malgorzewicz, S.; Karbowska, J. Dietary Lipids and Dyslipidemia in Chronic Kidney Disease. Nutrients 2021, 13, 3138. [Google Scholar] [CrossRef] [PubMed]
- Marsche, G.; Heine, G.H.; Stadler, J.T.; Holzer, M. Current Understanding of the Relationship of HDL Composition, Structure and Function to Their Cardioprotective Properties in Chronic Kidney Disease. Biomolecules 2020, 10, 1348. [Google Scholar] [CrossRef]
- Ceja-Galicia, Z.A.; Aranda-Rivera, A.K.; Amador-Martinez, I.; Aparicio-Trejo, O.E.; Tapia, E.; Trujillo, J.; Ramirez, V.; Pedraza-Chaverri, J. The Development of Dyslipidemia in Chronic Kidney Disease and Associated Cardiovascular Damage, and the Protective Effects of Curcuminoids. Foods 2023, 12, 921. [Google Scholar] [CrossRef]
- Kasikara, C.; Doran, A.C.; Cai, B.; Tabas, I. The role of non-resolving inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 2713–2723. [Google Scholar] [CrossRef]
- Del Pinto, R.; Pagliacci, S.; De Feo, M.; Grassi, D.; Ferri, C.; Italian Society of Hypertension and Federfarma. Prevalence of hypertension and associated cardiovascular risk factors among pharmacies customers: An Italian nationwide epidemiological survey. Eur. J. Prev. Cardiol. 2020, 27, 1228–1230. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef]
- Hamed, S.A. Neurologic conditions and disorders of uremic syndrome of chronic kidney disease: Presentations, causes, and treatment strategies. Expert. Rev. Clin. Pharmacol. 2019, 12, 61–90. [Google Scholar] [CrossRef]
- Espi, M.; Koppe, L.; Fouque, D.; Thaunat, O. Chronic Kidney Disease-Associated Immune Dysfunctions: Impact of Protein-Bound Uremic Retention Solutes on Immune Cells. Toxins 2020, 12, 300. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Cuppari, L. The 2020 Updated KDOQI Clinical Practice Guidelines for Nutrition in Chronic Kidney Disease. Blood Purif. 2021, 50, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Tain, Y.L. Chronic Kidney Disease and Gut Microbiota: What Is Their Connection in Early Life? Int. J. Mol. Sci. 2022, 23, 3954. [Google Scholar] [CrossRef]
- Tanaka, S.; Watanabe, H.; Nakano, T.; Imafuku, T.; Kato, H.; Tokumaru, K.; Arimura, N.; Enoki, Y.; Maeda, H.; Tanaka, M.; et al. Indoxyl Sulfate Contributes to Adipose Tissue Inflammation through the Activation of NADPH Oxidase. Toxins 2020, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.D.; Urquhart, B.L. Cerebrovascular Disease, Cardiovascular Disease, and Chronic Kidney Disease: Interplays and Influences. Curr. Neurol. Neurosci. Rep. 2022, 22, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Zixin, Y.; Lulu, C.; Xiangchang, Z.; Qing, F.; Binjie, Z.; Chunyang, L.; Tai, R.; Dongsheng, O. TMAO as a potential biomarker and therapeutic target for chronic kidney disease: A review. Front. Pharmacol. 2022, 13, 929262. [Google Scholar] [CrossRef] [PubMed]
- Swierczynska-Mroz, K.; Nowicka-Suszko, D.; Fleszar, M.G.; Fortuna, P.; Krajewski, P.K.; Krajewska, M.; Bialynicki-Birula, R.; Szepietowski, J.C. Serum Level of Protein-Bound Uraemic Toxins in Haemodialysis Patients with Chronic Kidney Disease-Associated Pruritus: Myths and Facts. J. Clin. Med. 2023, 12, 2310. [Google Scholar] [CrossRef]
- Adesso, S.; Ruocco, M.; Rapa, S.F.; Piaz, F.D.; Raffaele Di Iorio, B.; Popolo, A.; Autore, G.; Nishijima, F.; Pinto, A.; Marzocco, S. Effect of Indoxyl Sulfate on the Repair and Intactness of Intestinal Epithelial Cells: Role of Reactive Oxygen Species’ Release. Int. J. Mol. Sci. 2019, 20, 2280. [Google Scholar] [CrossRef]
- Ribeiro, A.; Liu, F.; Srebrzynski, M.; Rother, S.; Adamowicz, K.; Wadowska, M.; Steiger, S.; Anders, H.J.; Schmaderer, C.; Koziel, J.; et al. Uremic Toxin Indoxyl Sulfate Promotes Macrophage-Associated Low-Grade Inflammation and Epithelial Cell Senescence. Int. J. Mol. Sci. 2023, 24, 8031. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef]
- Velasquez, M.T.; Centron, P.; Barrows, I.; Dwivedi, R.; Raj, D.S. Gut Microbiota and Cardiovascular Uremic Toxicities. Toxins 2018, 10, 287. [Google Scholar] [CrossRef]
- Yamagami, F.; Tajiri, K.; Yumino, D.; Ieda, M. Uremic Toxins and Atrial Fibrillation: Mechanisms and Therapeutic Implications. Toxins 2019, 11, 597. [Google Scholar] [CrossRef]
- Zaidan, N.; Nazzal, L. The Microbiome and Uremic Solutes. Toxins 2022, 14, 245. [Google Scholar] [CrossRef] [PubMed]
- Addi, T.; Dou, L.; Burtey, S. Tryptophan-Derived Uremic Toxins and Thrombosis in Chronic Kidney Disease. Toxins 2018, 10, 412. [Google Scholar] [CrossRef]
- Oe, Y.; Takahashi, N. Tissue Factor, Thrombosis, and Chronic Kidney Disease. Biomedicines 2022, 10, 2737. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Ballew, S.H.; Wang, A.Y.; Kalyesubula, R.; Schaeffner, E.; Agarwal, R. Epidemiology and risk of cardiovascular disease in populations with chronic kidney disease. Nat. Rev. Nephrol. 2022, 18, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Shi, Y.; Chen, H.; Tao, M.; Zhou, X.; Li, J.; Ma, X.; Wang, Y.; Liu, N. Blockade of Autophagy Prevents the Progression of Hyperuricemic Nephropathy through Inhibiting NLRP3 Inflammasome-Mediated Pyroptosis. Front. Immunol. 2022, 13, 858494. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Camara, N.O.S. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef]
- Kim, S.M.; Lee, S.H.; Kim, Y.G.; Kim, S.Y.; Seo, J.W.; Choi, Y.W.; Kim, D.J.; Jeong, K.H.; Lee, T.W.; Ihm, C.G.; et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2015, 308, F993–F1003. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- Mei, Y.; Dong, B.; Geng, Z.; Xu, L. Excess Uric Acid Induces Gouty Nephropathy through Crystal Formation: A Review of Recent Insights. Front. Endocrinol. 2022, 13, 911968. [Google Scholar] [CrossRef]
- Balakumar, P.; Alqahtani, A.; Khan, N.A.; Mahadevan, N.; Dhanaraj, S.A. Mechanistic insights into hyperuricemia-associated renal abnormalities with special emphasis on epithelial-to-mesenchymal transition: Pathologic implications and putative pharmacologic targets. Pharmacol. Res. 2020, 161, 105209. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Pontremoli, R.; Burnier, M. Management of Hyperuricemia in Patients with Chronic Kidney Disease: A Focus on Renal Protection. Curr. Hypertens. Rep. 2020, 22, 102. [Google Scholar] [CrossRef]
- Del Pinto, R.; Viazzi, F.; Pontremoli, R.; Ferri, C.; Carubbi, F.; Russo, E. The URRAH study. Panminerva Med. 2021, 63, 416–423. [Google Scholar] [CrossRef]
- Johnson, R.J.; Sanchez Lozada, L.G.; Lanaspa, M.A.; Piani, F.; Borghi, C. Uric Acid and Chronic Kidney Disease: Still More to Do. Kidney Int. Rep. 2023, 8, 229–239. [Google Scholar] [CrossRef]
- Aroor, A.R.; McKarns, S.; Demarco, V.G.; Jia, G.; Sowers, J.R. Maladaptive immune and inflammatory pathways lead to cardiovascular insulin resistance. Metabolism 2013, 62, 1543–1552. [Google Scholar] [CrossRef]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am. J. Kidney Dis. 1992, 20, 1–17. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Berenguer-Martinez, J.M.; Bernal-Celestino, R.J.; Leon-Martin, A.A.; Gonzalez-Moro, M.T.R.; Fernandez-Calvo, N.; Arias-Del-Campo, L.; Civera-Miguel, M. Quality of Life and Related Factors in Patients Undergoing Renal Replacement Therapy at the Hospital General Universitario de Ciudad Real: Cross Sectional Descriptive Observational Study. J. Clin. Med. 2023, 12, 2250. [Google Scholar] [CrossRef]
- Rodrigues, F.G.; Ormanji, M.S.; Heilberg, I.P.; Bakker, S.J.L.; de Borst, M.H. Interplay between gut microbiota, bone health and vascular calcification in chronic kidney disease. Eur. J. Clin. Investig. 2021, 51, e13588. [Google Scholar] [CrossRef] [PubMed]
- Dusing, P.; Zietzer, A.; Goody, P.R.; Hosen, M.R.; Kurts, C.; Nickenig, G.; Jansen, F. Vascular pathologies in chronic kidney disease: Pathophysiological mechanisms and novel therapeutic approaches. J. Mol. Med. 2021, 99, 335–348. [Google Scholar] [CrossRef]
- Serrano, E.; Shenoy, P.; Martinez Cantarin, M.P. Adipose tissue metabolic changes in chronic kidney disease. Immunometabolism 2023, 5, e00023. [Google Scholar] [CrossRef] [PubMed]
- Weisinger, J.R.; Kempson, R.L.; Eldridge, F.L.; Swenson, R.S. The nephrotic syndrome: A complication of massive obesity. Ann. Intern. Med. 1974, 81, 440–447. [Google Scholar] [CrossRef]
- Hsu, C.Y.; McCulloch, C.E.; Iribarren, C.; Darbinian, J.; Go, A.S. Body mass index and risk for end-stage renal disease. Ann. Intern. Med. 2006, 144, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Iseki, K.; Ikemiya, Y.; Kinjo, K.; Inoue, T.; Iseki, C.; Takishita, S. Body mass index and the risk of development of end-stage renal disease in a screened cohort. Kidney Int. 2004, 65, 1870–1876. [Google Scholar] [CrossRef]
- de Vries, A.P.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Pongrac Barlovic, D.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Hall, J.E.; Mouton, A.J.; da Silva, A.A.; Omoto, A.C.M.; Wang, Z.; Li, X.; do Carmo, J.M. Obesity, kidney dysfunction, and inflammation: Interactions in hypertension. Cardiovasc. Res. 2021, 117, 1859–1876. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, Y.; Zhao, X.; Cui, H.; Han, M.; Ren, X.; Gang, X.; Wang, G. Obesity and chronic kidney disease. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E24–E41. [Google Scholar] [CrossRef]
- D’Agati, V.D.; Chagnac, A.; de Vries, A.P.; Levi, M.; Porrini, E.; Herman-Edelstein, M.; Praga, M. Obesity-related glomerulopathy: Clinical and pathologic characteristics and pathogenesis. Nat. Rev. Nephrol. 2016, 12, 453–471. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Z.; Chen, Y.; Dong, Y. Kidney Damage Caused by Obesity and Its Feasible Treatment Drugs. Int. J. Mol. Sci. 2022, 23, 747. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Herrington, W.G.; Haynes, R.; Emberson, J.; Landray, M.J.; Sudlow, C.L.M.; Woodward, M.; Baigent, C.; Lewington, S.; Staplin, N. Conventional and Genetic Evidence on the Association between Adiposity and CKD. J. Am. Soc. Nephrol. 2021, 32, 127–137. [Google Scholar] [CrossRef]
- Chang, Y.; Ryu, S.; Choi, Y.; Zhang, Y.; Cho, J.; Kwon, M.J.; Hyun, Y.Y.; Lee, K.B.; Kim, H.; Jung, H.S.; et al. Metabolically Healthy Obesity and Development of Chronic Kidney Disease: A Cohort Study. Ann. Intern. Med. 2016, 164, 305–312. [Google Scholar] [CrossRef]
- Cho, Y.K.; Lee, J.; Kim, H.S.; Park, J.Y.; Lee, W.J.; Kim, Y.J.; Jung, C.H. Impact of Transition in Metabolic Health and Obesity on the Incident Chronic Kidney Disease: A Nationwide Cohort Study. J. Clin. Endocrinol. Metab. 2020, 105, e148–e157. [Google Scholar] [CrossRef]
- Brennan, E.; Kantharidis, P.; Cooper, M.E.; Godson, C. Pro-resolving lipid mediators: Regulators of inflammation, metabolism and kidney function. Nat. Rev. Nephrol. 2021, 17, 725–739. [Google Scholar] [CrossRef]
- Li, C.; Lin, Y.; Luo, R.; Chen, S.; Wang, F.; Zheng, P.; Levi, M.; Yang, T.; Wang, W. Intrarenal renin-angiotensin system mediates fatty acid-induced ER stress in the kidney. Am. J. Physiol. Renal Physiol. 2016, 310, F351–F363. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ge, X.; Li, X.; He, J.; Wei, X.; Du, J.; Sun, J.; Li, X.; Xun, Z.; Liu, W.; et al. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis. 2020, 11, 914. [Google Scholar] [CrossRef]
- Wang, H.; Li, J.; Gai, Z.; Kullak-Ublick, G.A.; Liu, Z. TNF-alpha Deficiency Prevents Renal Inflammation and Oxidative Stress in Obese Mice. Kidney Blood Press. Res. 2017, 42, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Weldegiorgis, M.; Woodward, M. Elevated triglycerides and reduced high-density lipoprotein cholesterol are independently associated with the onset of advanced chronic kidney disease: A cohort study of 911,360 individuals from the United Kingdom. BMC Nephrol. 2022, 23, 312. [Google Scholar] [CrossRef]
- Zhang, Y.B.; Sheng, L.T.; Wei, W.; Guo, H.; Yang, H.; Min, X.; Guo, K.; Yang, K.; Zhang, X.; He, M.; et al. Association of blood lipid profile with incident chronic kidney disease: A Mendelian randomization study. Atherosclerosis 2020, 300, 19–25. [Google Scholar] [CrossRef]
- Hammoud, S.H.; AlZaim, I.; Al-Dhaheri, Y.; Eid, A.H.; El-Yazbi, A.F. Perirenal Adipose Tissue Inflammation: Novel Insights Linking Metabolic Dysfunction to Renal Diseases. Front. Endocrinol. 2021, 12, 707126. [Google Scholar] [CrossRef]
- Huang, N.; Mao, E.W.; Hou, N.N.; Liu, Y.P.; Han, F.; Sun, X.D. Novel insight into perirenal adipose tissue: A neglected adipose depot linking cardiovascular and chronic kidney disease. World J. Diabetes 2020, 11, 115–125. [Google Scholar] [CrossRef]
- D’Marco, L.; Salazar, J.; Cortez, M.; Salazar, M.; Wettel, M.; Lima-Martinez, M.; Rojas, E.; Roque, W.; Bermudez, V. Perirenal fat thickness is associated with metabolic risk factors in patients with chronic kidney disease. Kidney Res. Clin. Pract. 2019, 38, 365–372. [Google Scholar] [CrossRef]
- Liu, B.X.; Sun, W.; Kong, X.Q. Perirenal Fat: A Unique Fat Pad and Potential Target for Cardiovascular Disease. Angiology 2019, 70, 584–593. [Google Scholar] [CrossRef]
- Ma, S.; Zhu, X.Y.; Eirin, A.; Woollard, J.R.; Jordan, K.L.; Tang, H.; Lerman, A.; Lerman, L.O. Perirenal Fat Promotes Renal Arterial Endothelial Dysfunction in Obese Swine through Tumor Necrosis Factor-alpha. J. Urol. 2016, 195, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Scherer, P.E. Immunologic and endocrine functions of adipose tissue: Implications for kidney disease. Nat. Rev. Nephrol. 2018, 14, 105–120. [Google Scholar] [CrossRef]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-17. [Google Scholar] [CrossRef]
- Belkaid, Y.; Harrison, O.J. Homeostatic Immunity and the Microbiota. Immunity 2017, 46, 562–576. [Google Scholar] [CrossRef]
- Briskey, D.; Tucker, P.; Johnson, D.W.; Coombes, J.S. The role of the gastrointestinal tract and microbiota on uremic toxins and chronic kidney disease development. Clin. Exp. Nephrol. 2017, 21, 7–15. [Google Scholar] [CrossRef]
- Chen, L.; Liu, B.; Ren, L.; Du, H.; Fei, C.; Qian, C.; Li, B.; Zhang, R.; Liu, H.; Li, Z.; et al. High-fiber diet ameliorates gut microbiota, serum metabolism and emotional mood in type 2 diabetes patients. Front. Cell Infect. Microbiol. 2023, 13, 1069954. [Google Scholar] [CrossRef] [PubMed]
- Pham, V.T.; Calatayud, M.; Rotsaert, C.; Seifert, N.; Richard, N.; Van den Abbeele, P.; Marzorati, M.; Steinert, R.E. Antioxidant Vitamins and Prebiotic FOS and XOS Differentially Shift Microbiota Composition and Function and Improve Intestinal Epithelial Barrier In Vitro. Nutrients 2021, 13, 1125. [Google Scholar] [CrossRef] [PubMed]
- Cosola, C.; Rocchetti, M.T.; Sabatino, A.; Fiaccadori, E.; Di Iorio, B.R.; Gesualdo, L. Microbiota issue in CKD: How promising are gut-targeted approaches? J. Nephrol. 2019, 32, 27–37. [Google Scholar] [CrossRef]
- Rysz, J.; Franczyk, B.; Lawinski, J.; Olszewski, R.; Cialkowska-Rysz, A.; Gluba-Brzozka, A. The Impact of CKD on Uremic Toxins and Gut Microbiota. Toxins 2021, 13, 252. [Google Scholar] [CrossRef]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 69, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Barros, A.F.; Borges, N.A.; Ferreira, D.C.; Carmo, F.L.; Rosado, A.S.; Fouque, D.; Mafra, D. Is there interaction between gut microbial profile and cardiovascular risk in chronic kidney disease patients? Future Microbiol. 2015, 10, 517–526. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Voroneanu, L.; Burlacu, A.; Brinza, C.; Covic, A.; Balan, G.G.; Nistor, I.; Popa, C.; Hogas, S.; Covic, A. Gut Microbiota in Chronic Kidney Disease: From Composition to Modulation towards Better Outcomes-A Systematic Review. J. Clin. Med. 2023, 12, 1948. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, M.; Wang, J.; Li, R.; Zhang, Y. Alterations to the Gut Microbiota and Their Correlation with Inflammatory Factors in Chronic Kidney Disease. Front. Cell Infect. Microbiol. 2019, 9, 206. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, H.; Guo, J.; Zhang, M.; Zheng, H.; Liu, Y.; Liu, W. Gut microbiota-derived trimethylamine N-oxide is associated with the risk of all-cause and cardiovascular mortality in patients with chronic kidney disease: A systematic review and dose-response meta-analysis. Ann. Med. 2023, 55, 2215542. [Google Scholar] [CrossRef] [PubMed]
- Kaysen, G.A.; Johansen, K.L.; Chertow, G.M.; Dalrymple, L.S.; Kornak, J.; Grimes, B.; Dwyer, T.; Chassy, A.W.; Fiehn, O. Associations of Trimethylamine N-Oxide with Nutritional and Inflammatory Biomarkers and Cardiovascular Outcomes in Patients New to Dialysis. J. Ren. Nutr. 2015, 25, 351–356. [Google Scholar] [CrossRef]
- Kim, R.B.; Morse, B.L.; Djurdjev, O.; Tang, M.; Muirhead, N.; Barrett, B.; Holmes, D.T.; Madore, F.; Clase, C.M.; Rigatto, C.; et al. Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int. 2016, 89, 1144–1152. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef]
- Xu, K.Y.; Xia, G.H.; Lu, J.Q.; Chen, M.X.; Zhen, X.; Wang, S.; You, C.; Nie, J.; Zhou, H.W.; Yin, J. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci. Rep. 2017, 7, 1445. [Google Scholar] [CrossRef] [PubMed]
- Aquilani, R.; Bolasco, P.; Murtas, S.; Maestri, R.; Iadarola, P.; Testa, C.; Deiana, M.L.; Esposito, M.P.; Contu, R.; Cadeddu, M.; et al. Effects of a Metabolic Mixture on Gut Inflammation and Permeability in Elderly Patients with Chronic Kidney Disease: A Proof-of-Concept Study. Metabolites 2022, 12, 987. [Google Scholar] [CrossRef] [PubMed]
- Ticinesi, A.; Milani, C.; Guerra, A.; Allegri, F.; Lauretani, F.; Nouvenne, A.; Mancabelli, L.; Lugli, G.A.; Turroni, F.; Duranti, S.; et al. Understanding the gut-kidney axis in nephrolithiasis: An analysis of the gut microbiota composition and functionality of stone formers. Gut 2018, 67, 2097–2106. [Google Scholar] [CrossRef]
- Anders, H.J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef]
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Vesey, D.A.; Coombes, J.S.; Weston, K.S.; Hawley, C.M.; McWhinney, B.C.; Ungerer, J.P.; et al. Protein-bound uremic toxins, inflammation and oxidative stress: A cross-sectional study in stage 3–4 chronic kidney disease. Arch. Med. Res. 2014, 45, 309–317. [Google Scholar] [CrossRef]
- Donadei, C.; Angeletti, A.; Pizzuti, V.; Zappulo, F.; Conte, D.; Cappuccilli, M.; Chiocchini, A.L.; Scrivo, A.; Apuzzo, D.; Mariggio, M.A.; et al. Impact of Single Hemodialysis Treatment on immune Cell Subpopulations. J. Clin. Med. 2023, 12, 3107. [Google Scholar] [CrossRef]
- Kurashima, Y.; Kiyono, H. Mucosal Ecological Network of Epithelium and Immune Cells for Gut Homeostasis and Tissue Healing. Annu. Rev. Immunol. 2017, 35, 119–147. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Jian, Y.; Wang, F.; Yu, S.; Guo, J.; Kan, J.; Guo, W. NLRP3 and Gut Microbiota Homeostasis: Progress in Research. Cells 2022, 11, 3758. [Google Scholar] [CrossRef]
- Lohia, S.; Vlahou, A.; Zoidakis, J. Microbiome in Chronic Kidney Disease (CKD): An Omics Perspective. Toxins 2022, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.; Kesper, M.S.; Marschner, J.A.; Konrad, L.; Ryu, M.; Kumar Vr, S.; Kulkarni, O.P.; Mulay, S.R.; Romoli, S.; Demleitner, J.; et al. Intestinal Dysbiosis, Barrier Dysfunction, and Bacterial Translocation Account for CKD-Related Systemic Inflammation. J. Am. Soc. Nephrol. 2017, 28, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, S.; Merckelbach, E.; Noels, H.; Vohra, A.; Jankowski, J. Homeostasis in the Gut Microbiota in Chronic Kidney Disease. Toxins 2022, 14, 648. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Melekoglu, E.; Samur, F.G. Dietary strategies for gut-derived protein-bound uremic toxins and cardio-metabolic risk factors in chronic kidney disease: A focus on dietary fibers. Crit. Rev. Food Sci. Nutr. 2023, 63, 3994–4008. [Google Scholar] [CrossRef]
- Mahmoodpoor, F.; Rahbar Saadat, Y.; Barzegari, A.; Ardalan, M.; Zununi Vahed, S. The impact of gut microbiota on kidney function and pathogenesis. Biomed. Pharmacother. 2017, 93, 412–419. [Google Scholar] [CrossRef]
- Lau, W.L.; Vaziri, N.D. The Leaky Gut and Altered Microbiome in Chronic Kidney Disease. J. Ren. Nutr. 2017, 27, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Burnier, M.; Damianaki, A. Hypertension as Cardiovascular Risk Factor in Chronic Kidney Disease. Circ. Res. 2023, 132, 1050–1063. [Google Scholar] [CrossRef]
- Spoto, B.; Pisano, A.; Zoccali, C. Insulin resistance in chronic kidney disease: A systematic review. Am. J. Physiol. Renal Physiol. 2016, 311, F1087–F1108. [Google Scholar] [CrossRef]
- Yang, J.; Lim, S.Y.; Ko, Y.S.; Lee, H.Y.; Oh, S.W.; Kim, M.G.; Cho, W.Y.; Jo, S.K. Intestinal barrier disruption and dysregulated mucosal immunity contribute to kidney fibrosis in chronic kidney disease. Nephrol. Dial. Transplant. 2019, 34, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Jiang, S.; Persson, P.B.; Persson, E.A.G.; Lai, E.Y.; Patzak, A. Reactive oxygen species in renal vascular function. Acta Physiol. 2020, 229, e13477. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jin, H.; Ju, H.; Sun, M.; Chen, H.; Li, L. Circulating Trimethylamine-N-Oxide and Risk of All-Cause and Cardiovascular Mortality in Patients with Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Front. Med. 2022, 9, 828343. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef]
- Lin, X.; Liang, W.; Li, L.; Xiong, Q.; He, S.; Zhao, J.; Guo, X.; Xiang, S.; Zhang, P.; Wang, H.; et al. The Accumulation of Gut Microbiome-derived Indoxyl Sulfate and P-Cresyl Sulfate in Patients with End-stage Renal Disease. J. Ren. Nutr. 2022, 32, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. CKD impairs barrier function and alters microbial flora of the intestine: A major link to inflammation and uremic toxicity. Curr. Opin. Nephrol. Hypertens. 2012, 21, 587–592. [Google Scholar] [CrossRef]
- Lau, W.L.; Vaziri, N.D. Urea, a true uremic toxin: The empire strikes back. Clin. Sci. 2017, 131, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Ikee, R.; Sasaki, N.; Yasuda, T.; Fukazawa, S. Chronic Kidney Disease, Gut Dysbiosis, and Constipation: A Burdensome Triplet. Microorganisms 2020, 8, 1862. [Google Scholar] [CrossRef] [PubMed]
- Onal, E.M.; Afsar, B.; Covic, A.; Vaziri, N.D.; Kanbay, M. Gut microbiota and inflammation in chronic kidney disease and their roles in the development of cardiovascular disease. Hypertens. Res. 2019, 42, 123–140. [Google Scholar] [CrossRef]
- Feng, Z.; Wang, T.; Dong, S.; Jiang, H.; Zhang, J.; Raza, H.K.; Lei, G. Association between gut dysbiosis and chronic kidney disease: A narrative review of the literature. J. Int. Med. Res. 2021, 49, 3000605211053276. [Google Scholar] [CrossRef]
- Fu, C.; Yan, D.; Wang, L.; Duan, F.; Gu, D.; Yao, N.; Sun, M.; Wang, D.; Lin, X.; Wu, Y.; et al. High prevalence of sarcopenia and myosteatosis in patients undergoing hemodialysis. Front. Endocrinol. 2023, 14, 1117438. [Google Scholar] [CrossRef] [PubMed]
- Noor, H.; Reid, J.; Slee, A. Resistance exercise and nutritional interventions for augmenting sarcopenia outcomes in chronic kidney disease: A narrative review. J. Cachexia Sarcopenia Muscle 2021, 12, 1621–1640. [Google Scholar] [CrossRef]
- Raphael, K.L. Metabolic Acidosis and Subclinical Metabolic Acidosis in CKD. J. Am. Soc. Nephrol. 2018, 29, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Zheng, S.; Zhang, W.; Hu, G. Progress in the study of nutritional status and selenium in dialysis patients. Ann. Med. 2023, 55, 2197296. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, B.; Hassounah, F.; Price, S.R.; Klein, J.; Mohamed, T.M.A.; Wang, Y.; Park, J.; Cai, H.; Zhang, X.; et al. The impact of senescence on muscle wasting in chronic kidney disease. J. Cachexia Sarcopenia Muscle 2023, 14, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Martin-Del-Campo, F.; Avesani, C.M.; Stenvinkel, P.; Lindholm, B.; Cueto-Manzano, A.M.; Cortes-Sanabria, L. Gut microbiota disturbances and protein-energy wasting in chronic kidney disease: A narrative review. J. Nephrol. 2023, 36, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Aycart, D.F.; Acevedo, S.; Eguiguren-Jimenez, L.; Andrade, J.M. Influence of Plant and Animal Proteins on Inflammation Markers among Adults with Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Nutrients 2021, 13, 1660. [Google Scholar] [CrossRef]
- Ko, G.J.; Rhee, C.M.; Kalantar-Zadeh, K.; Joshi, S. The Effects of High-Protein Diets on Kidney Health and Longevity. J. Am. Soc. Nephrol. 2020, 31, 1667–1679. [Google Scholar] [CrossRef]
- Fang, Y.; Lee, H.; Son, S.; Oh, S.; Jo, S.K.; Cho, W.; Kim, M.G. Association between Consumption of Dietary Supplements and Chronic Kidney Disease Prevalence: Results of the Korean Nationwide Population-Based Survey. Nutrients 2023, 15, 822. [Google Scholar] [CrossRef]
- Goraya, N.; Wesson, D.E. Management of the Metabolic Acidosis of Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2017, 24, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, Y.; Kaimori, J.Y.; Isaka, Y. Plant-Dominant Low Protein Diet: A Potential Alternative Dietary Practice for Patients with Chronic Kidney Disease. Nutrients 2023, 15, 1002. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Buysse, J.M.; Bushinsky, D.A. Mechanisms of Metabolic Acidosis-Induced Kidney Injury in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2020, 31, 469–482. [Google Scholar] [CrossRef]
- Asahina, Y.; Sakaguchi, Y.; Kajimoto, S.; Hattori, K.; Doi, Y.; Oka, T.; Kaimori, J.Y.; Isaka, Y. Time-updated anion gap and cardiovascular events in advanced chronic kidney disease: A cohort study. Clin. Kidney J. 2022, 15, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Asahina, Y.; Sakaguchi, Y.; Kajimoto, S.; Hattori, K.; Doi, Y.; Oka, T.; Kaimori, J.Y.; Isaka, Y. Association of Time-Updated Anion Gap with Risk of Kidney Failure in Advanced CKD: A Cohort Study. Am. J. Kidney Dis. 2022, 79, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, J.; Yu, D.; Liu, M. Plant or Animal-Based or PLADO Diets: Which Should Chronic Kidney Disease Patients Choose? J. Ren. Nutr. 2023, 33, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Gonzalez-Ortiz, A.; Avesani, C.M.; Bakker, S.J.L.; Bellizzi, V.; Chauveau, P.; Clase, C.M.; Cupisti, A.; Espinosa-Cuevas, A.; Molina, P.; et al. Plant-based diets to manage the risks and complications of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Mafra, D.; Borges, N.; Alvarenga, L.; Esgalhado, M.; Cardozo, L.; Lindholm, B.; Stenvinkel, P. Dietary Components That May Influence the Disturbed Gut Microbiota in Chronic Kidney Disease. Nutrients 2019, 11, 496. [Google Scholar] [CrossRef]
- Khosroshahi, H.T.; Abedi, B.; Ghojazadeh, M.; Samadi, A.; Jouyban, A. Effects of fermentable high fiber diet supplementation on gut derived and conventional nitrogenous product in patients on maintenance hemodialysis: A randomized controlled trial. Nutr. Metab. 2019, 16, 18. [Google Scholar] [CrossRef]
- Borrelli, S.; Matarazzo, I.; Lembo, E.; Peccarino, L.; Annoiato, C.; Scognamiglio, M.R.; Foderini, A.; Ruotolo, C.; Franculli, A.; Capozzi, F.; et al. Chronic Hyperkaliemia in Chronic Kidney Disease: An Old Concern with New Answers. Int. J. Mol. Sci. 2022, 23, 6378. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Backhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Magliocca, G.; Mone, P.; Di Iorio, B.R.; Heidland, A.; Marzocco, S. Short-Chain Fatty Acids in Chronic Kidney Disease: Focus on Inflammation and Oxidative Stress Regulation. Int. J. Mol. Sci. 2022, 23, 5354. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lv, D.; Jiang, S.; Jiang, J.; Liang, M.; Hou, F.; Chen, Y. Quantitative reduction in short-chain fatty acids, especially butyrate, contributes to the progression of chronic kidney disease. Clin. Sci. 2019, 133, 1857–1870. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Kaul, R.; Chaari, A. Renal Health Improvement in Diabetes through Microbiome Modulation of the Gut-Kidney Axis with Biotics: A Systematic and Narrative Review of Randomized Controlled Trials. Int. J. Mol. Sci. 2022, 23, 14838. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214. [Google Scholar] [CrossRef]
- Rios-Covian, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de Los Reyes-Gavilan, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Fazeli, G.; Di Micco, L.; Autore, G.; Adesso, S.; Dal Piaz, F.; Heidland, A.; Di Iorio, B. Supplementation of Short-Chain Fatty Acid, Sodium Propionate, in Patients on Maintenance Hemodialysis: Beneficial Effects on Inflammatory Parameters and Gut-Derived Uremic Toxins, A Pilot Study (PLAN Study). J. Clin. Med. 2018, 7, 315. [Google Scholar] [CrossRef]
- Vaziri, N.D. Effect of Synbiotic Therapy on Gut-Derived Uremic Toxins and the Intestinal Microbiome in Patients with CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 199–201. [Google Scholar] [CrossRef]
- Anjana; Tiwari, S.K. Bacteriocin-Producing Probiotic Lactic Acid Bacteria in Controlling Dysbiosis of the Gut Microbiota. Front. Cell Infect. Microbiol. 2022, 12, 851140. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Zhou, H.; Deng, J.; Sun, J.; Zhou, X.; Tang, Y.; Qin, W. Effectiveness of Microecological Preparations for Improving Renal Function and Metabolic Profiles in Patients with Chronic Kidney Disease. Front. Nutr. 2022, 9, 850014. [Google Scholar] [CrossRef]
- Tian, N.; Li, L.; Ng, J.K.; Li, P.K. The Potential Benefits and Controversies of Probiotics Use in Patients at Different Stages of Chronic Kidney Disease. Nutrients 2022, 14, 4044. [Google Scholar] [CrossRef]
- Joshi, S.; McMacken, M.; Kalantar-Zadeh, K. Plant-Based Diets for Kidney Disease: A Guide for Clinicians. Am. J. Kidney Dis. 2021, 77, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Beker, B.M.; Colombo, I.; Gonzalez-Torres, H.; Musso, C.G. Decreasing microbiota-derived uremic toxins to improve CKD outcomes. Clin. Kidney J. 2022, 15, 2214–2219. [Google Scholar] [CrossRef]
- Jean, G.; Souberbielle, J.C.; Chazot, C. Vitamin D in Chronic Kidney Disease and Dialysis Patients. Nutrients 2017, 9, 328. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Chen, D.Q.; Chen, L.; Liu, J.R.; Vaziri, N.D.; Guo, Y.; Zhao, Y.Y. Microbiome-metabolome reveals the contribution of gut-kidney axis on kidney disease. J. Transl. Med. 2019, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Xu, W.; Zhang, L.; Li, X.; Wang, R.; Wu, S. Impact of Gut Microbiota and Microbiota-Related Metabolites on Hyperlipidemia. Front. Cell Infect. Microbiol. 2021, 11, 634780. [Google Scholar] [CrossRef] [PubMed]
- Khalesi, S.; Sun, J.; Buys, N.; Jayasinghe, R. Effect of probiotics on blood pressure: A systematic review and meta-analysis of randomized, controlled trials. Hypertension 2014, 64, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Sabico, S.; Al-Mashharawi, A.; Al-Daghri, N.M.; Wani, K.; Amer, O.E.; Hussain, D.S.; Ahmed Ansari, M.G.; Masoud, M.S.; Alokail, M.S.; McTernan, P.G. Effects of a 6-month multi-strain probiotics supplementation in endotoxemic, inflammatory and cardiometabolic status of T2DM patients: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2019, 38, 1561–1569. [Google Scholar] [CrossRef]
- Vitetta, L.; Llewellyn, H.; Oldfield, D. Gut Dysbiosis and the Intestinal Microbiome: Streptococcus thermophilus a Key Probiotic for Reducing Uremia. Microorganisms 2019, 7, 228. [Google Scholar] [CrossRef]
- de Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut microbiome and health: Mechanistic insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef]
- Resta-Lenert, S.; Barrett, K.E. Probiotics and commensals reverse TNF-alpha- and IFN-gamma-induced dysfunction in human intestinal epithelial cells. Gastroenterology 2006, 130, 731–746. [Google Scholar] [CrossRef]
- Wang, I.K.; Wu, Y.Y.; Yang, Y.F.; Ting, I.W.; Lin, C.C.; Yen, T.H.; Chen, J.H.; Wang, C.H.; Huang, C.C.; Lin, H.C. The effect of probiotics on serum levels of cytokine and endotoxin in peritoneal dialysis patients: A randomised, double-blind, placebo-controlled trial. Benef. Microbes 2015, 6, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Y.; Wang, Y.; Xu, H.; Mei, X.; Yu, D.; Wang, Y.; Li, W. Antioxidant Properties of Probiotic Bacteria. Nutrients 2017, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Amini Khiabani, S.; Asgharzadeh, M.; Samadi Kafil, H. Chronic kidney disease and gut microbiota. Heliyon 2023, 9, e18991. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, R.A.B.; Soder, T.F.; Grokoski, K.C.; Benetti, F.; Mendes, R.H. Probiotics in the treatment of chronic kidney disease: A systematic review. J. Bras. Nefrol. 2018, 40, 278–286. [Google Scholar] [CrossRef]
- Ranganathan, N.; Ranganathan, P.; Friedman, E.A.; Joseph, A.; Delano, B.; Goldfarb, D.S.; Tam, P.; Rao, A.V.; Anteyi, E.; Musso, C.G. Pilot study of probiotic dietary supplementation for promoting healthy kidney function in patients with chronic kidney disease. Adv. Ther. 2010, 27, 634–647. [Google Scholar] [CrossRef]
- Yoshifuji, A.; Wakino, S.; Irie, J.; Tajima, T.; Hasegawa, K.; Kanda, T.; Tokuyama, H.; Hayashi, K.; Itoh, H. Gut Lactobacillus protects against the progression of renal damage by modulating the gut environment in rats. Nephrol. Dial. Transplant. 2016, 31, 401–412. [Google Scholar] [CrossRef]
- Stavropoulou, E.; Bezirtzoglou, E. Probiotics in Medicine: A Long Debate. Front. Immunol. 2020, 11, 2192. [Google Scholar] [CrossRef]
- Zhu, H.; Cao, C.; Wu, Z.; Zhang, H.; Sun, Z.; Wang, M.; Xu, H.; Zhao, Z.; Wang, Y.; Pei, G.; et al. The probiotic L. casei Zhang slows the progression of acute and chronic kidney disease. Cell Metab. 2021, 33, 1926–1942.e1928. [Google Scholar] [CrossRef]
- Borges, N.A.; Carmo, F.L.; Stockler-Pinto, M.B.; de Brito, J.S.; Dolenga, C.J.; Ferreira, D.C.; Nakao, L.S.; Rosado, A.; Fouque, D.; Mafra, D. Probiotic Supplementation in Chronic Kidney Disease: A Double-blind, Randomized, Placebo-controlled Trial. J. Ren. Nutr. 2018, 28, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Haghighat, N.; Mohammadshahi, M.; Shayanpour, S.; Haghighizadeh, M.H. Effects of Synbiotics and Probiotics Supplementation on Serum Levels of Endotoxin, Heat Shock Protein 70 Antibodies and Inflammatory Markers in Hemodialysis Patients: A Randomized Double-Blinded Controlled Trial. Probiot. Antimicrob. Proteins 2020, 12, 144–151. [Google Scholar] [CrossRef]
- Ruospo, M.; Palmer, S.C.; Craig, J.C.; Gentile, G.; Johnson, D.W.; Ford, P.J.; Tonelli, M.; Petruzzi, M.; De Benedittis, M.; Strippoli, G.F. Prevalence and severity of oral disease in adults with chronic kidney disease: A systematic review of observational studies. Nephrol. Dial. Transplant. 2014, 29, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Fenton, A.; Dias, I.H.K.; Heaton, B.; Brown, C.L.R.; Sidhu, A.; Rahman, M.; Griffiths, H.R.; Cockwell, P.; Ferro, C.J.; et al. Oxidative stress links periodontal inflammation and renal function. J. Clin. Periodontol. 2021, 48, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Laiho, S.; Vaittinen, O.; Halonen, L.; Ortiz, F.; Forsblom, C.; Groop, P.H.; Lehto, M.; Metsala, M. Biochemical pathways of breath ammonia (NH3) generation in patients with end-stage renal disease undergoing hemodialysis. J. Breath. Res. 2016, 10, 036011. [Google Scholar] [CrossRef]
- Rodrigues, R.; Vidigal, M.T.C.; Vieira, W.A.; Nascimento, G.G.; Sabino-Silva, R.; Blumenberg, C.; Siqueira, M.F.; Siqueira, W.L.; Paranhos, L.R. Salivary changes in chronic kidney disease and in patients undergoing hemodialysis: A systematic review and meta-analysis. J. Nephrol. 2022, 35, 1339–1367. [Google Scholar] [CrossRef]
- Hanna, R.M.; Ghobry, L.; Wassef, O.; Rhee, C.M.; Kalantar-Zadeh, K. A Practical Approach to Nutrition, Protein-Energy Wasting, Sarcopenia, and Cachexia in Patients with Chronic Kidney Disease. Blood Purif. 2020, 49, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Inoue, T.; Ogawa, M.; Shirado, K.; Shirai, N.; Yagi, T.; Momosaki, R.; Kokura, Y. Rehabilitation Nutrition in Patients with Chronic Kidney Disease and Cachexia. Nutrients 2022, 14, 4722. [Google Scholar] [CrossRef]
- Piccoli, G.B.; Cederholm, T.; Avesani, C.M.; Bakker, S.J.L.; Bellizzi, V.; Cuerda, C.; Cupisti, A.; Sabatino, A.; Schneider, S.; Torreggiani, M.; et al. Nutritional status and the risk of malnutrition in older adults with chronic kidney disease—Implications for low protein intake and nutritional care: A critical review endorsed by ERN-ERA and ESPEN. Clin. Nutr. 2023, 42, 443–457. [Google Scholar] [CrossRef]
- Guo, S.; Wu, G.; Liu, W.; Fan, Y.; Song, W.; Wu, J.; Gao, D.; Gu, X.; Jing, S.; Shen, Q.; et al. Characteristics of human oral microbiome and its non-invasive diagnostic value in chronic kidney disease. Biosci. Rep. 2022, 42, BSR20210694. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.L.; Liu, X.Y.; Meng, X.; Zhao, R.Q.; Ou, L.L.; Li, B.Z.; Xing, T. Periodontitis Exacerbates and Promotes the Progression of Chronic Kidney Disease through Oral Flora, Cytokines, and Oxidative Stress. Front. Microbiol. 2021, 12, 656372. [Google Scholar] [CrossRef]
- Palmeira, E.; de Liz Perez-Losada, F.; Diaz-Flores-Garcia, V.; Segura-Sampedro, J.J.; Segura-Egea, J.J.; Lopez-Lopez, J. Prevalence of oral infections in chronic kidney disease patients: A cross-sectional study. Oral. Dis. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Serni, L.; Caroti, L.; Barbato, L.; Nieri, M.; Serni, S.; Cirami, C.L.; Cairo, F. Association between chronic kidney disease and periodontitis. A systematic review and metanalysis. Oral Dis. 2023, 29, 40–50. [Google Scholar] [CrossRef]
- Sharma, P.; Dietrich, T.; Ferro, C.J.; Cockwell, P.; Chapple, I.L. Association between periodontitis and mortality in stages 3-5 chronic kidney disease: NHANES III and linked mortality study. J. Clin. Periodontol. 2016, 43, 104–113. [Google Scholar] [CrossRef]
- Gewin, L.; Zent, R.; Pozzi, A. Progression of chronic kidney disease: Too much cellular talk causes damage. Kidney Int. 2017, 91, 552–560. [Google Scholar] [CrossRef]
- Plemmenos, G.; Evangeliou, E.; Polizogopoulos, N.; Chalazias, A.; Deligianni, M.; Piperi, C. Central Regulatory Role of Cytokines in Periodontitis and Targeting Options. Curr. Med. Chem. 2021, 28, 3032–3058. [Google Scholar] [CrossRef]
- Zoccali, C.; Mallamaci, F. Innate Immunity System in Patients with Cardiovascular and Kidney Disease. Circ. Res. 2023, 132, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Altamura, S.; Del Pinto, R.; Pietropaoli, D.; Ferri, C. Oral health as a modifiable risk factor for cardiovascular diseases. Trends Cardiovasc. Med. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Del Pinto, R.; Pietropaoli, D.; Munoz-Aguilera, E.; D’Aiuto, F.; Czesnikiewicz-Guzik, M.; Monaco, A.; Guzik, T.J.; Ferri, C. Periodontitis and Hypertension: Is the Association Causal? High Blood Press. Cardiovasc. Prev. 2020, 27, 281–289. [Google Scholar] [CrossRef]
- Macedo Paizan, M.L.; Vilela-Martin, J.F. Is there an association between periodontitis and hypertension? Curr. Cardiol. Rev. 2014, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, M.; Mochizuki, Y.; Miyata, Y.; Obata, Y.; Mitsunari, K.; Matsuo, T.; Ohba, K.; Mukae, H.; Yoshimura, A.; Nishino, T.; et al. Pathological Characteristics of Periodontal Disease in Patients with Chronic Kidney Disease and Kidney Transplantation. Int. J. Mol. Sci. 2019, 20, 3413. [Google Scholar] [CrossRef] [PubMed]
- Tavares, L.T.R.; Saavedra-Silva, M.; Lopez-Marcos, J.F.; Veiga, N.J.; Castilho, R.M.; Fernandes, G.V.O. Blood and Salivary Inflammatory Biomarkers Profile in Patients with Chronic Kidney Disease and Periodontal Disease: A Systematic Review. Diseases 2022, 10, 12. [Google Scholar] [CrossRef]
- Nylund, K.M.; Meurman, J.H.; Heikkinen, A.M.; Honkanen, E.; Vesterinen, M.; Furuholm, J.O.; Tervahartiala, T.; Sorsa, T.; Ruokonen, H.M. Periodontal Inflammatory Burden and Salivary Matrix Metalloproteinase-8 Concentration among Patients with Chronic Kidney Disease at the Predialysis Stage. J. Periodontol. 2015, 86, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, Y. Matrix Metalloproteinase-10 in Kidney Injury Repair and Disease. Int. J. Mol. Sci. 2022, 23, 2131. [Google Scholar] [CrossRef] [PubMed]
- Gebrayel, P.; Nicco, C.; Al Khodor, S.; Bilinski, J.; Caselli, E.; Comelli, E.M.; Egert, M.; Giaroni, C.; Karpinski, T.M.; Loniewski, I.; et al. Microbiota medicine: Towards clinical revolution. J. Transl. Med. 2022, 20, 111. [Google Scholar] [CrossRef] [PubMed]
- Nallu, A.; Sharma, S.; Ramezani, A.; Muralidharan, J.; Raj, D. Gut microbiome in chronic kidney disease: Challenges and opportunities. Transl. Res. 2017, 179, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Wehedy, E.; Shatat, I.F.; Al Khodor, S. The Human Microbiome in Chronic Kidney Disease: A Double-Edged Sword. Front. Med. 2021, 8, 790783. [Google Scholar] [CrossRef]
- Mahendra, J.; Palathingal, P.; Mahendra, L.; Alzahrani, K.J.; Banjer, H.J.; Alsharif, K.F.; Halawani, I.F.; Muralidharan, J.; Annamalai, P.T.; Verma, S.S.; et al. Impact of Red Complex Bacteria and TNF-alpha Levels on the Diabetic and Renal Status of Chronic Kidney Disease Patients in the Presence and Absence of Periodontitis. Biology 2022, 11, 451. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, Y.; Misaki, T.; Ito, S.; Naka, S.; Wato, K.; Nomura, R.; Matsumoto-Nakano, M.; Nakano, K. Title IgA Nephropathy and Oral Bacterial Species Related to Dental Caries and Periodontitis. Int. J. Mol. Sci. 2022, 23, 725. [Google Scholar] [CrossRef] [PubMed]
- Costacurta, M.; Basilicata, M.; Marrone, G.; Di Lauro, M.; Campolattano, V.; Bollero, P.; Docimo, R.; Di Daniele, N.; Noce, A. The Impact of Chronic Kidney Disease on Nutritional Status and Its Possible Relation with Oral Diseases. Nutrients 2022, 14, 2002. [Google Scholar] [CrossRef] [PubMed]
- Hickey, N.A.; Shalamanova, L.; Whitehead, K.A.; Dempsey-Hibbert, N.; van der Gast, C.; Taylor, R.L. Exploring the putative interactions between chronic kidney disease and chronic periodontitis. Crit. Rev. Microbiol. 2020, 46, 61–77. [Google Scholar] [CrossRef]
- Kim, Y.J.; Moura, L.M.; Caldas, C.P.; Perozini, C.; Ruivo, G.F.; Pallos, D. Evaluation of periodontal condition and risk in patients with chronic kidney disease on hemodialysis. Einstein 2017, 15, 173–177. [Google Scholar] [CrossRef]
- Baciu, S.F.; Mesaros, A.S.; Kacso, I.M. Chronic Kidney Disease and Periodontitis Interplay-A Narrative Review. Int. J. Environ. Res. Public Health 2023, 20, 1298. [Google Scholar] [CrossRef]
- Lam, G.A.; Albarrak, H.; McColl, C.J.; Pizarro, A.; Sanaka, H.; Gomez-Nguyen, A.; Cominelli, F.; Paes Batista da Silva, A. The Oral-Gut Axis: Periodontal Diseases and Gastrointestinal Disorders. Inflamm. Bowel Dis. 2023, 29, 1153–1164. [Google Scholar] [CrossRef]
- de Jongh, C.A.; de Vries, T.J.; Bikker, F.J.; Gibbs, S.; Krom, B.P. Mechanisms of Porphyromonas gingivalis to translocate over the oral mucosa and other tissue barriers. J. OralMicrobiol. 2023, 15, 2205291. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.B.; Ismail, G.; Dumitriu, A.S.; Baston, C.; Berbecar, V.; Jurubita, R.; Andronesi, A.; Dumitriu, H.T.; Sinescu, I. Identification of subgingival periodontal pathogens and association with the severity of periodontitis in patients with chronic kidney diseases: A cross-sectional study. Biomed. Res. Int. 2015, 2015, 370314. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Asai, Y.; Hashimoto, M.; Takeuchi, O.; Kurita, T.; Yoshikai, Y.; Miyake, K.; Akira, S. Cell activation by Porphyromonas gingivalis lipid A molecule through Toll-like receptor 4- and myeloid differentiation factor 88-dependent signaling pathway. Int. Immunol. 2002, 14, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Darveau, R.P.; Pham, T.T.; Lemley, K.; Reife, R.A.; Bainbridge, B.W.; Coats, S.R.; Howald, W.N.; Way, S.S.; Hajjar, A.M. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect. Immun. 2004, 72, 5041–5051. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Krauss, J.L.; Domon, H.; Hosur, K.B.; Liang, S.; Magotti, P.; Triantafilou, M.; Triantafilou, K.; Lambris, J.D.; Hajishengallis, G. Microbial hijacking of complement-toll-like receptor crosstalk. Sci. Signal 2010, 3, ra11. [Google Scholar] [CrossRef]
- Herrera, B.S.; Martins-Porto, R.; Campi, P.; Holzhausen, M.; Teixeira, S.A.; Mendes, G.D.; Costa, S.K.; Gyurko, R.; Van Dyke, T.E.; Spolidorio, L.C.; et al. Local and cardiorenal effects of periodontitis in nitric oxide-deficient hypertensive rats. Arch. Oral Biol. 2011, 56, 41–47. [Google Scholar] [CrossRef]
- Girndt, M.; Sester, M.; Sester, U.; Kaul, H.; Kohler, H. Defective expression of B7-2 (CD86) on monocytes of dialysis patients correlates to the uremia-associated immune defect. Kidney Int. 2001, 59, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Chmielewski, M.; Honda, H.; Pecoits-Filho, R.; Matsuo, S.; Yuzawa, Y.; Tranaeus, A.; Stenvinkel, P.; Lindholm, B. Aspects of immune dysfunction in end-stage renal disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1526–1533. [Google Scholar] [CrossRef]
- Ando, M.; Shibuya, A.; Tsuchiya, K.; Akiba, T.; Nitta, K. Reduced expression of Toll-like receptor 4 contributes to impaired cytokine response of monocytes in uremic patients. Kidney Int. 2006, 70, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, Y.; Tsuchida, K.; Go, I.; Aoyama, M.; Naganuma, T.; Takemoto, Y.; Nakatani, T. A study of innate immunity in patients with end-stage renal disease: Special reference to toll-like receptor-2 and -4 expression in peripheral blood monocytes of hemodialysis patients. Int. J. Mol. Med. 2007, 19, 783–790. [Google Scholar] [CrossRef] [PubMed]
- D’Aiuto, F.; Nibali, L.; Parkar, M.; Patel, K.; Suvan, J.; Donos, N. Oxidative stress, systemic inflammation, and severe periodontitis. J. Dent. Res. 2010, 89, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Onder, C.; Kurgan, S.; Altingoz, S.M.; Bagis, N.; Uyanik, M.; Serdar, M.A.; Kantarci, A.; Gunhan, M. Impact of non-surgical periodontal therapy on saliva and serum levels of markers of oxidative stress. Clin. Oral Investig. 2017, 21, 1961–1969. [Google Scholar] [CrossRef]
- Kanzaki, H.; Wada, S.; Narimiya, T.; Yamaguchi, Y.; Katsumata, Y.; Itohiya, K.; Fukaya, S.; Miyamoto, Y.; Nakamura, Y. Pathways that Regulate ROS Scavenging Enzymes, and Their Role in Defense Against Tissue Destruction in Periodontitis. Front. Physiol. 2017, 8, 351. [Google Scholar] [CrossRef]
- Chaudhry, A.; Kassim, N.K.; Zainuddin, S.L.A.; Taib, H.; Ibrahim, H.A.; Ahmad, B.; Hanafi, M.H.; Adnan, A.S. Potential Effects of Non-Surgical Periodontal Therapy on Periodontal Parameters, Inflammatory Markers, and Kidney Function Indicators in Chronic Kidney Disease Patients with Chronic Periodontitis. Biomedicines 2022, 10, 2752. [Google Scholar] [CrossRef]
- Delbove, T.; Gueyffier, F.; Juillard, L.; Kalbacher, E.; Maucort-Boulch, D.; Nony, P.; Grosgogeat, B.; Gritsch, K. Effect of periodontal treatment on the glomerular filtration rate, reduction of inflammatory markers and mortality in patients with chronic kidney disease: A systematic review. PLoS ONE 2021, 16, e0245619. [Google Scholar] [CrossRef]
- Almeida, S.; Figueredo, C.M.; Lemos, C.; Bregman, R.; Fischer, R.G. Periodontal treatment in patients with chronic kidney disease: A pilot study. J. Periodontal Res. 2017, 52, 262–267. [Google Scholar] [CrossRef]
- Deschamps-Lenhardt, S.; Martin-Cabezas, R.; Hannedouche, T.; Huck, O. Association between periodontitis and chronic kidney disease: Systematic review and meta-analysis. Oral Dis. 2019, 25, 385–402. [Google Scholar] [CrossRef]
- Tasdemir, Z.; Ozsari Tasdemir, F.; Gurgan, C.; Eroglu, E.; Gunturk, I.; Kocyigit, I. The effect of periodontal disease treatment in patients with continuous ambulatory peritoneal dialysis. Int. Urol. Nephrol. 2018, 50, 1519–1528. [Google Scholar] [CrossRef]
- Siribamrungwong, M.; Yothasamutr, K.; Puangpanngam, K. Periodontal treatment reduces chronic systemic inflammation in peritoneal dialysis patients. Ther. Apher. Dial. 2014, 18, 305–308. [Google Scholar] [CrossRef]
- Fang, F.; Wu, B.; Qu, Q.; Gao, J.; Yan, W.; Huang, X.; Ma, D.; Yue, J.; Chen, T.; Liu, F.; et al. The clinical response and systemic effects of non-surgical periodontal therapy in end-stage renal disease patients: A 6-month randomized controlled clinical trial. J. Clin. Periodontol. 2015, 42, 537–546. [Google Scholar] [CrossRef]
- Grubbs, V.; Garcia, F.; Vittinghoff, E.; Jue, B.L.; Ryder, M.; Lovett, D.H.; Offenbacher, S.; Taylor, G.; Ganz, P.; Bibbins-Domingo, K.; et al. Nonsurgical Periodontal Therapy in CKD: Findings of the Kidney and Periodontal Disease (KAPD) Pilot Randomized Controlled Trial. Kidney Med. 2020, 2, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Kuo, H.C.; Liu, H.Y.; Wu, M.Y.; Chang, W.J.; Chen, J.T.; Cherng, Y.G.; Chen, T.J.; Dai, Y.X.; Wu, H.L.; et al. Association between Dental Scaling and Reduced Risk of End-Stage Renal Disease: A Nationwide Matched Cohort Study. Int. J. Environ. Res. Public Health 2021, 18, 8910. [Google Scholar] [CrossRef]
- Chang, Y.; Lee, J.S.; Woo, H.G.; Ryu, D.R.; Kim, J.W.; Song, T.J. Improved oral hygiene care and chronic kidney disease occurrence: A nationwide population-based retrospective cohort study. Medicine 2021, 100, e27845. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Shimbo, T.; Komatsu, Y.; Kobayashi, D. Frequency of tooth brushing as a predictive factor for future kidney function decline. J. Nephrol. 2022, 35, 191–199. [Google Scholar] [CrossRef]
- Hofer, K.; Turnowsky, A.; Ehren, R.; Taylan, C.; Plum, G.; Witte, H.; Noack, M.J.; Weber, L.T. The impact of a needs-oriented dental prophylaxis program on bacteremia after toothbrushing and systemic inflammation in children, adolescents, and young adults with chronic kidney disease. Pediatr. Nephrol. 2022, 37, 403–414. [Google Scholar] [CrossRef]
- Stevens, P.E.; Levin, A.; Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: Synopsis of the kidney disease: Improving global outcomes 2012 clinical practice guideline. Ann. Intern. Med. 2013, 158, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Herrera, D.; Kebschull, M.; Chapple, I.; Jepsen, S.; Beglundh, T.; Sculean, A.; Tonetti, M.S.; Participants, E.F.P.W.; Methodological, C. Treatment of stage I-III periodontitis-The EFP S3 level clinical practice guideline. J. Clin. Periodontol. 2020, 47 (Suppl. S22), 4–60. [Google Scholar] [CrossRef]
- Miyata, Y.; Obata, Y.; Mochizuki, Y.; Kitamura, M.; Mitsunari, K.; Matsuo, T.; Ohba, K.; Mukae, H.; Nishino, T.; Yoshimura, A.; et al. Periodontal Disease in Patients Receiving Dialysis. Int. J. Mol. Sci. 2019, 20, 3805. [Google Scholar] [CrossRef] [PubMed]
- Tonetti, M.S.; D’Aiuto, F.; Nibali, L.; Donald, A.; Storry, C.; Parkar, M.; Suvan, J.; Hingorani, A.D.; Vallance, P.; Deanfield, J. Treatment of periodontitis and endothelial function. N. Engl. J. Med. 2007, 356, 911–920. [Google Scholar] [CrossRef]
- D’Aiuto, F.; Gkranias, N.; Bhowruth, D.; Khan, T.; Orlandi, M.; Suvan, J.; Masi, S.; Tsakos, G.; Hurel, S.; Hingorani, A.D.; et al. Systemic effects of periodontitis treatment in patients with type 2 diabetes: A 12 month, single-centre, investigator-masked, randomised trial. Lancet Diabetes Endocrinol. 2018, 6, 954–965. [Google Scholar] [CrossRef]
- D’Aiuto, F.; Parkar, M.; Nibali, L.; Suvan, J.; Lessem, J.; Tonetti, M.S. Periodontal infections cause changes in traditional and novel cardiovascular risk factors: Results from a randomized controlled clinical trial. Am. Heart J. 2006, 151, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, S.; Chahal, G.S.; Grover, V.; Rathi, M.; Sharma, R.; Sharma, R.; Jain, A. Impact of periodontal treatment on inflammatory oxidative stress in chronic kidney disease subjects: An interventional clinical trial. Am. J. Dent. 2023, 36, 15–20. [Google Scholar] [PubMed]
- Montero, E.; Lopez, M.; Vidal, H.; Martinez, M.; Virto, L.; Marrero, J.; Herrera, D.; Zapatero, A.; Sanz, M. Impact of periodontal therapy on systemic markers of inflammation in patients with metabolic syndrome: A randomized clinical trial. Diabetes Obes. Metab. 2020, 22, 2120–2132. [Google Scholar] [CrossRef] [PubMed]
- Schutz, J.D.S.; de Azambuja, C.B.; Cunha, G.R.; Cavagni, J.; Rosing, C.K.; Haas, A.N.; Thome, F.S.; Fiorini, T. Association between severe periodontitis and chronic kidney disease severity in predialytic patients: A cross-sectional study. Oral Dis. 2020, 26, 447–456. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altamura, S.; Pietropaoli, D.; Lombardi, F.; Del Pinto, R.; Ferri, C. An Overview of Chronic Kidney Disease Pathophysiology: The Impact of Gut Dysbiosis and Oral Disease. Biomedicines 2023, 11, 3033. https://doi.org/10.3390/biomedicines11113033
Altamura S, Pietropaoli D, Lombardi F, Del Pinto R, Ferri C. An Overview of Chronic Kidney Disease Pathophysiology: The Impact of Gut Dysbiosis and Oral Disease. Biomedicines. 2023; 11(11):3033. https://doi.org/10.3390/biomedicines11113033
Chicago/Turabian StyleAltamura, Serena, Davide Pietropaoli, Francesca Lombardi, Rita Del Pinto, and Claudio Ferri. 2023. "An Overview of Chronic Kidney Disease Pathophysiology: The Impact of Gut Dysbiosis and Oral Disease" Biomedicines 11, no. 11: 3033. https://doi.org/10.3390/biomedicines11113033