From Batch to Flow Stripping Analysis with Screen-Printed Electrodes: A Possible Way to Decentralize Trace Inorganic Analysis


Abstract
:1. Introduction
2. Materials and Methods
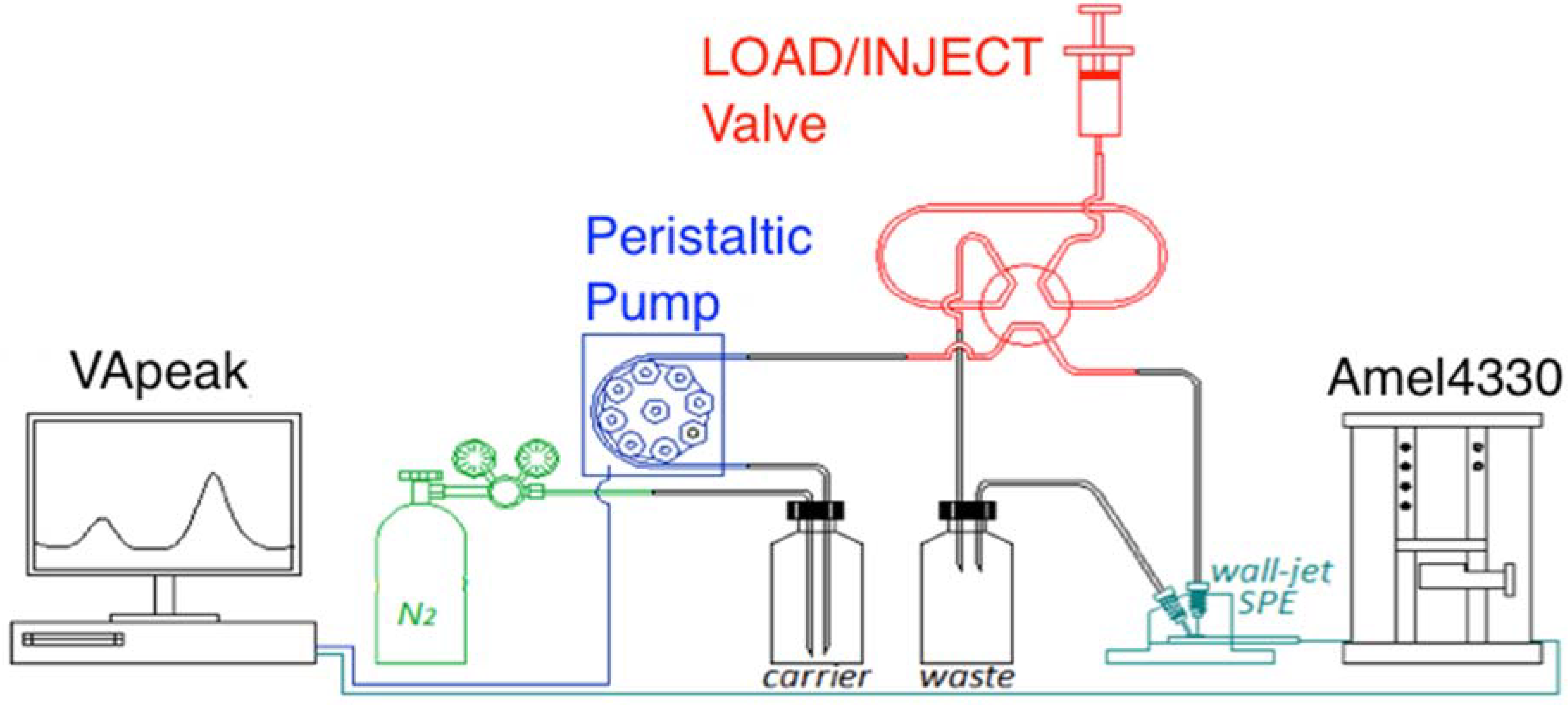
2.1. Instrumental Setup
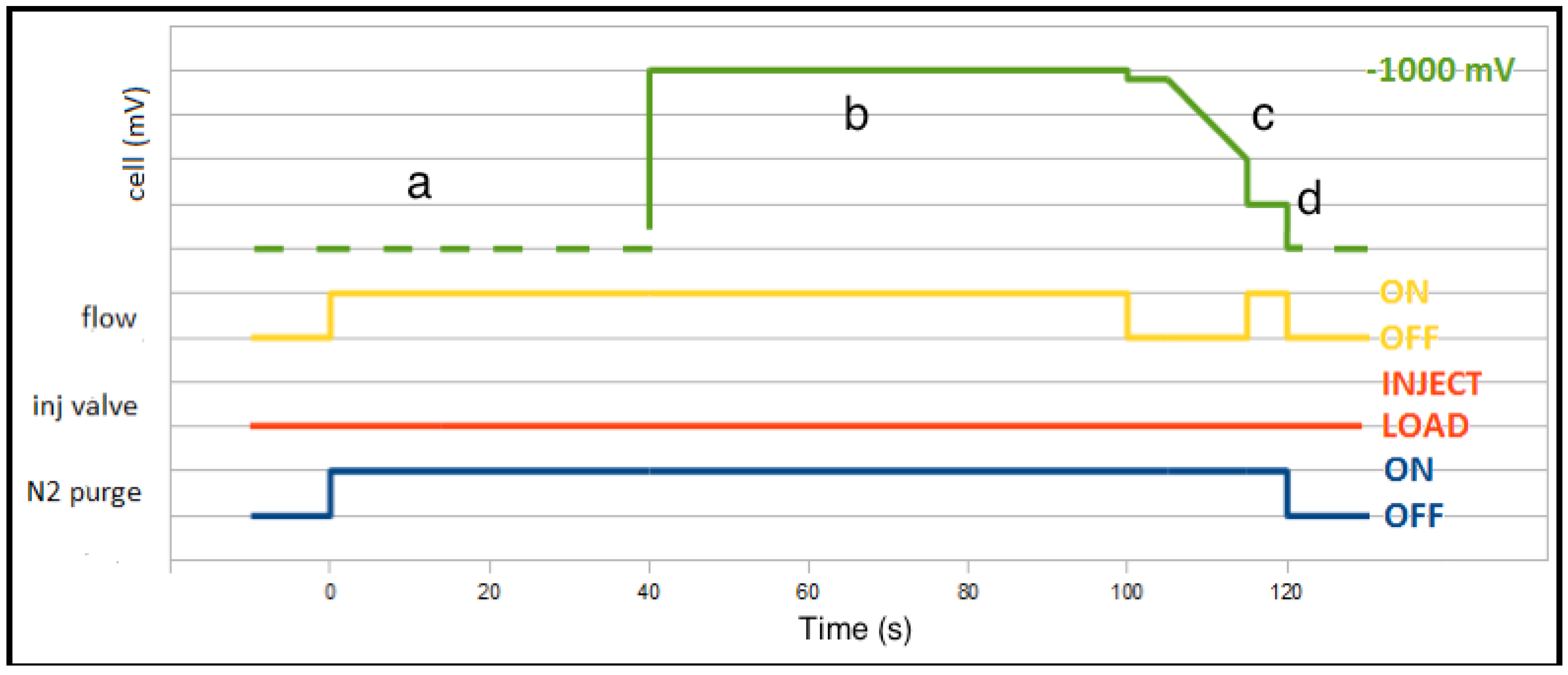
2.2. Cells and Flow System
2.3. Measurement Procedures
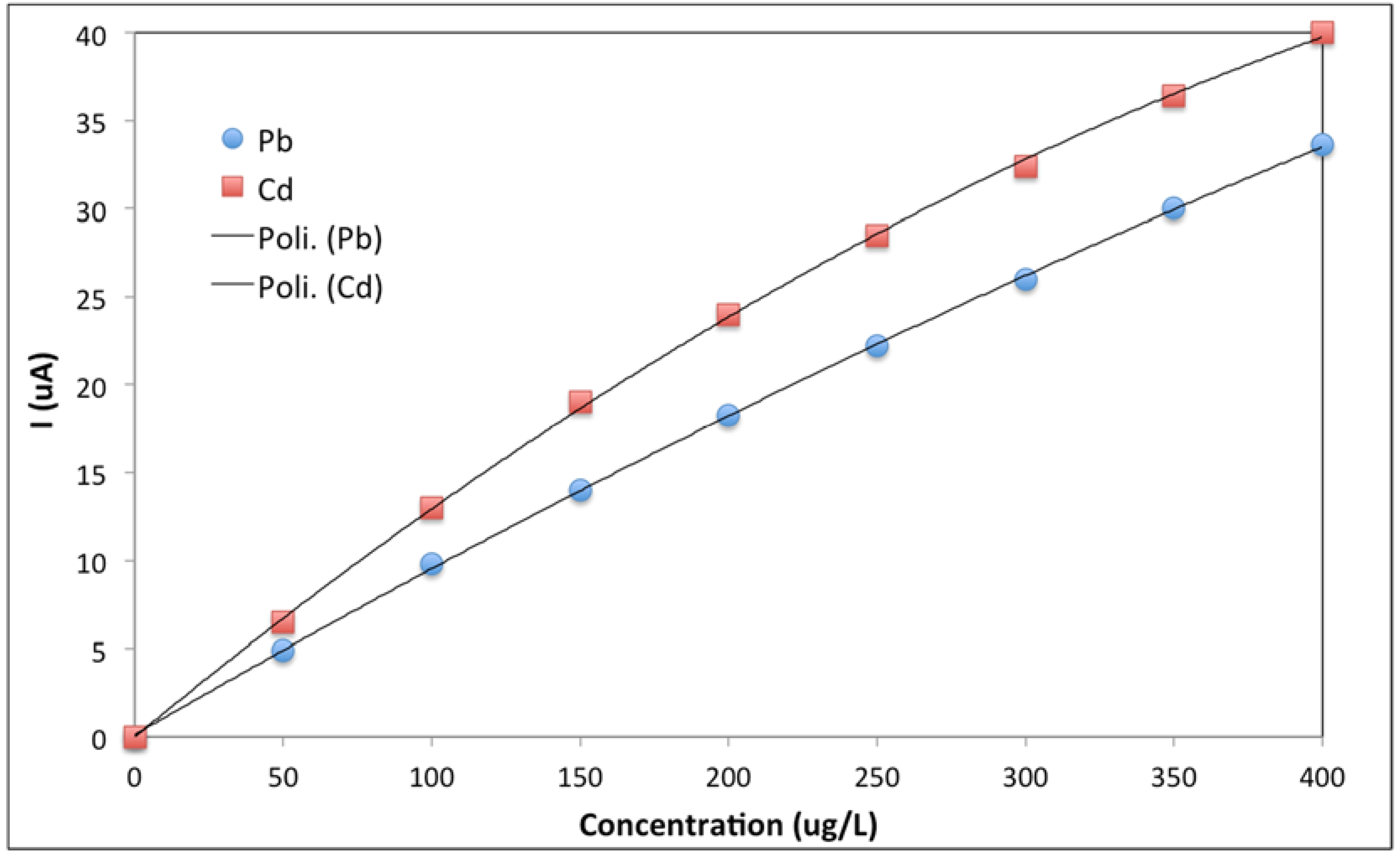
3. Results and Discussion
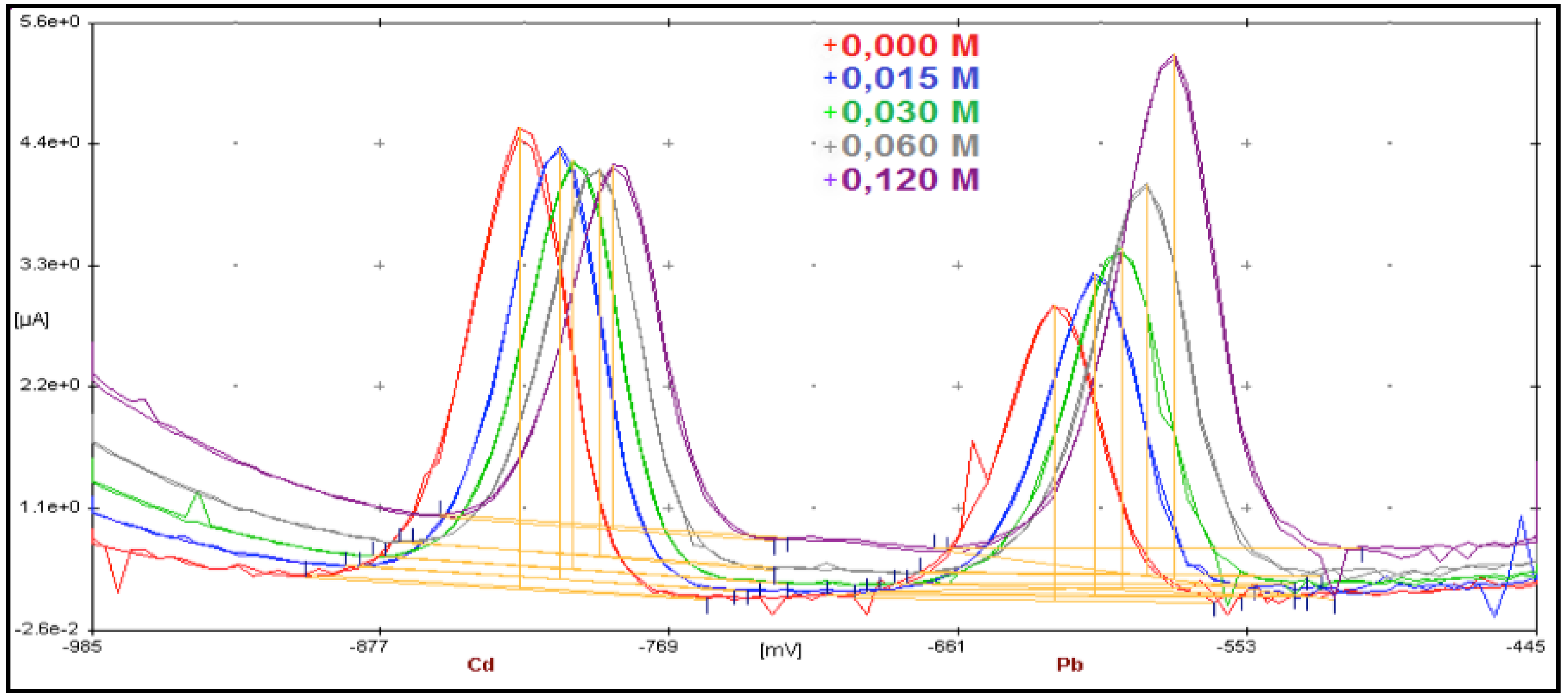
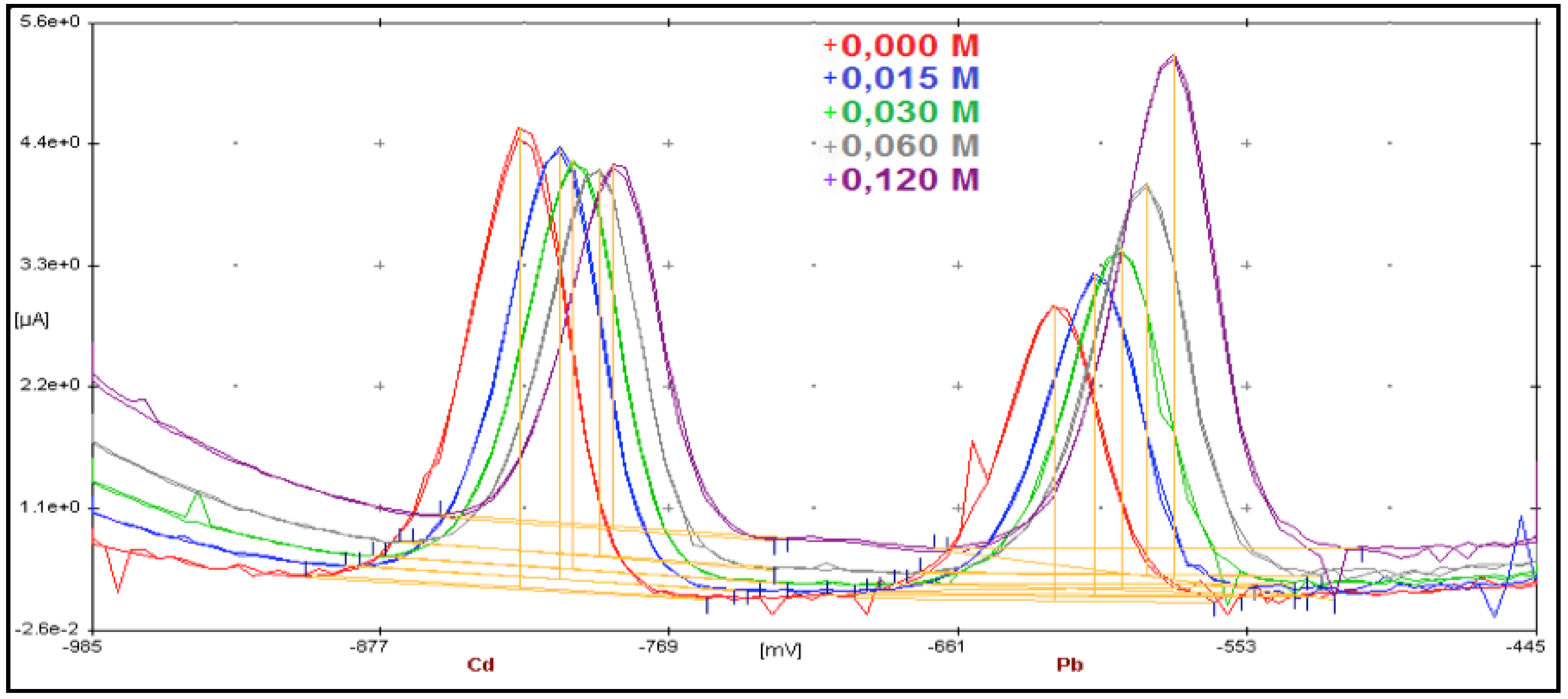
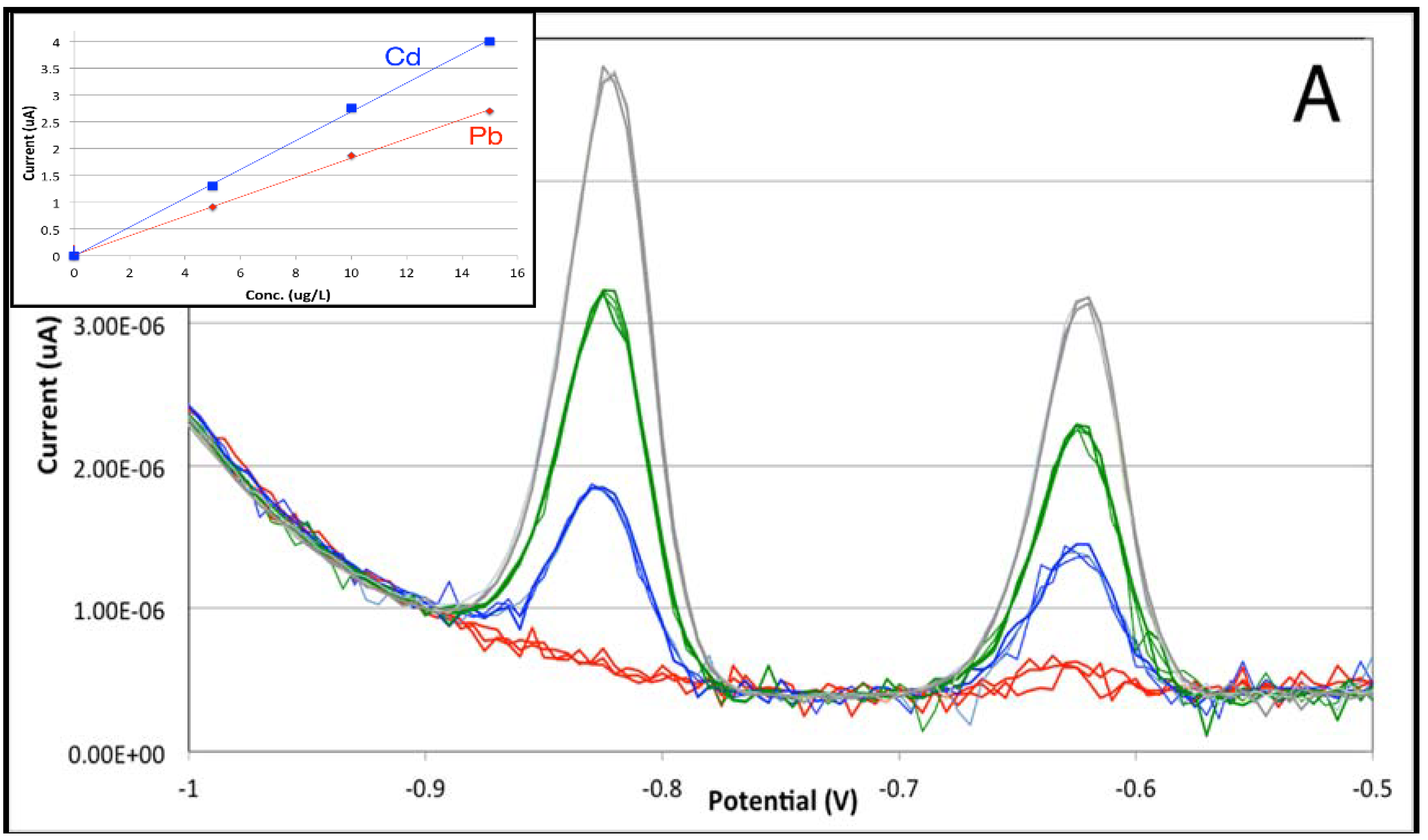
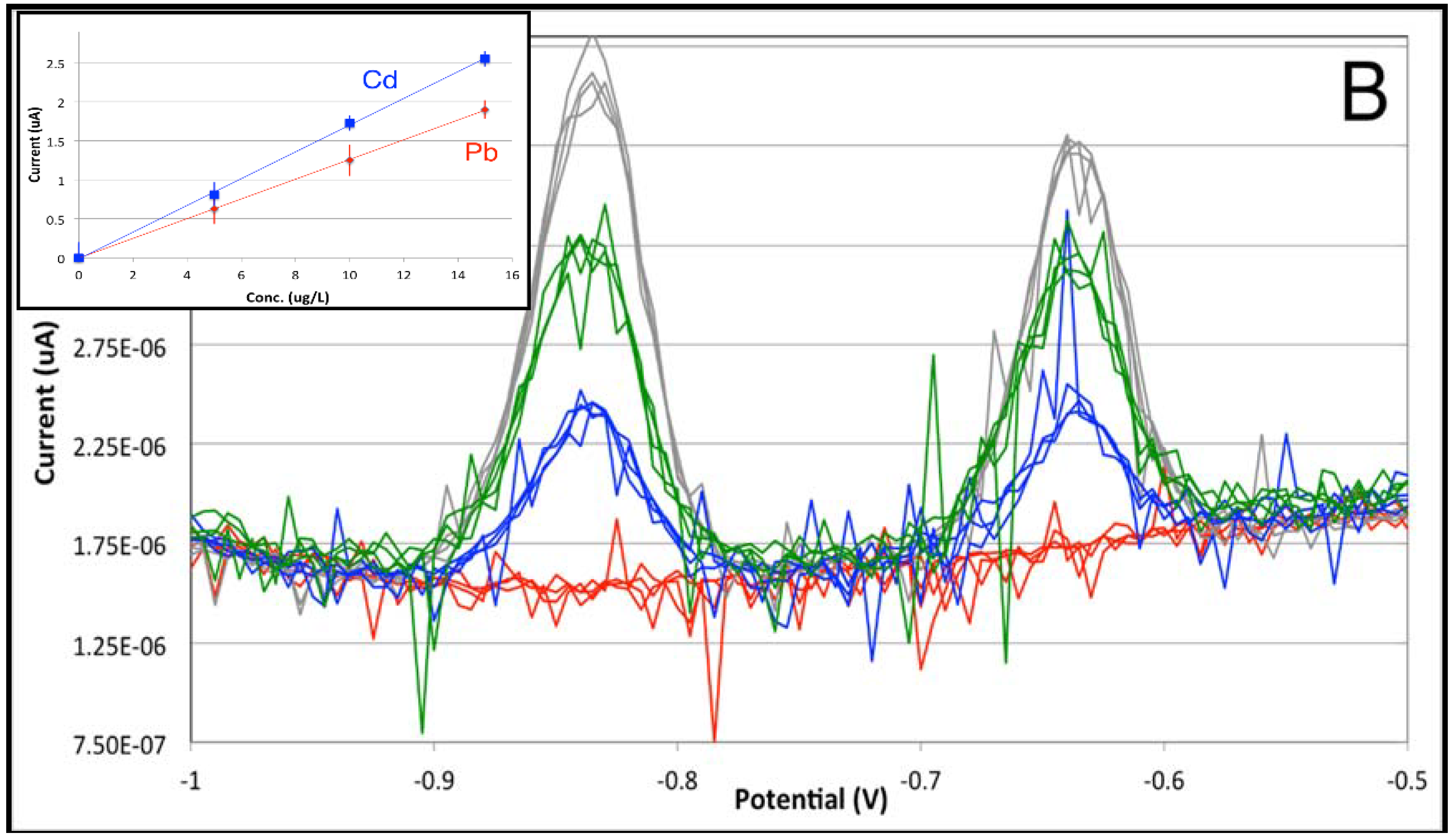
3.1. From Batch to Stopped-Flow Differential Pulse Stripping Voltammetry
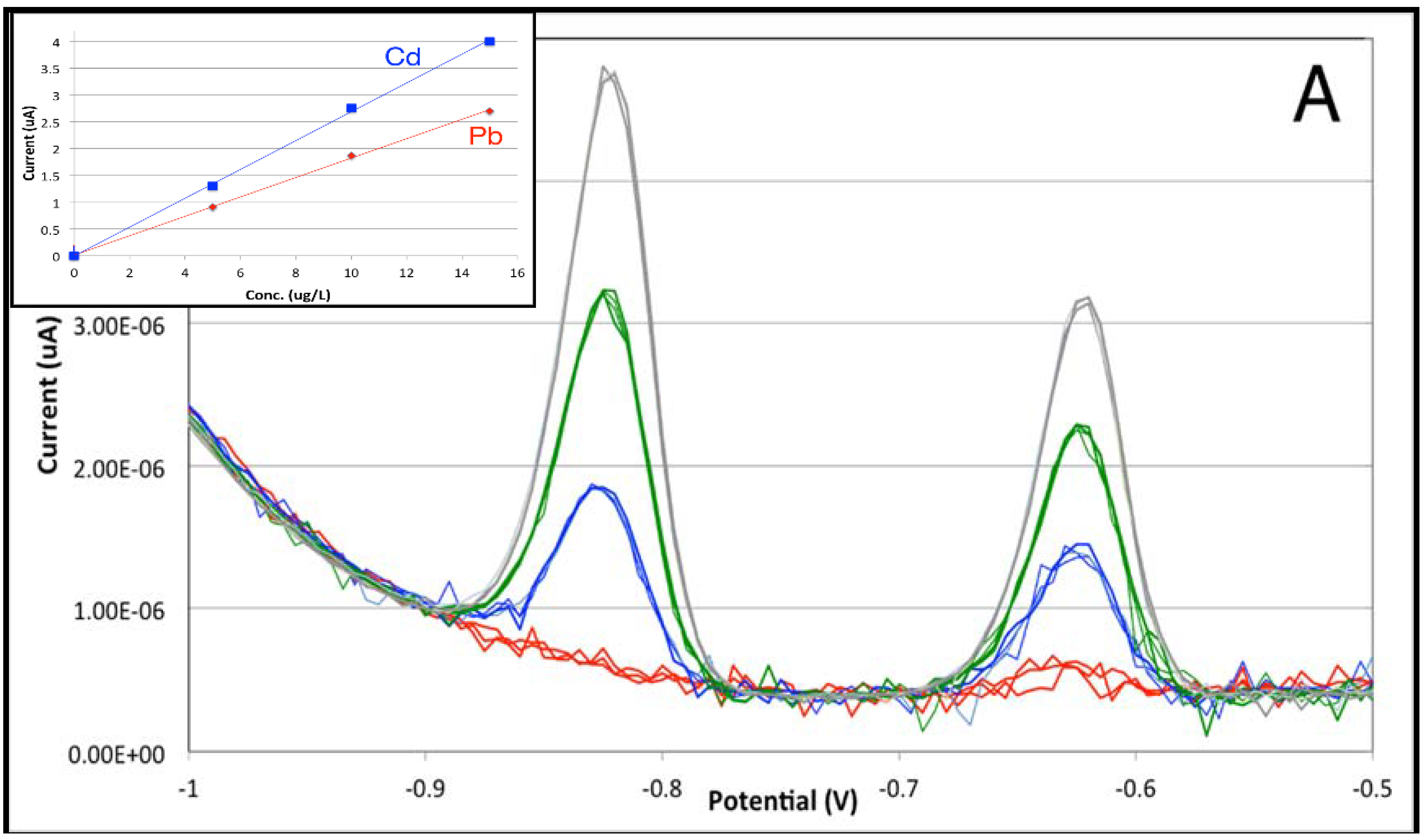
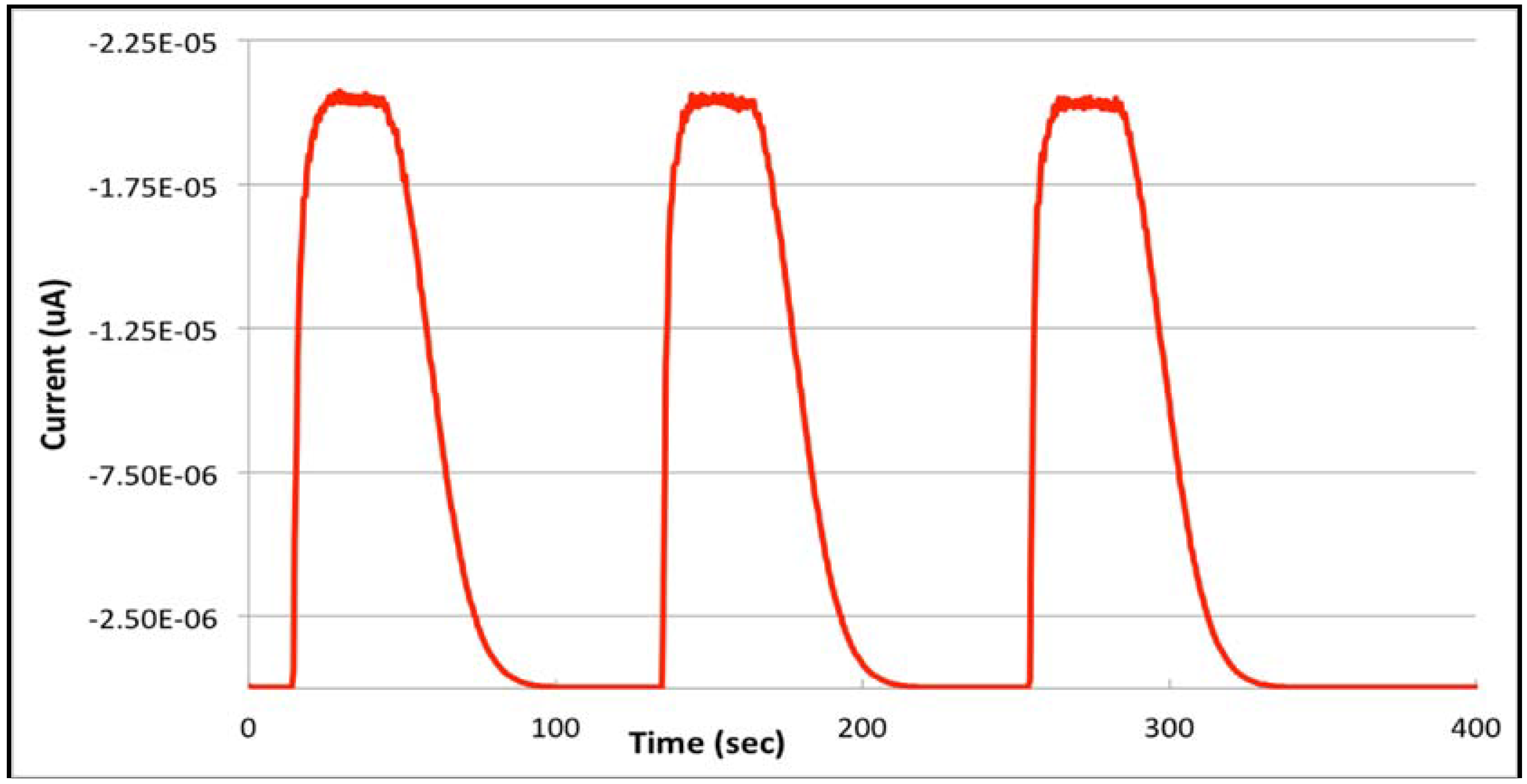
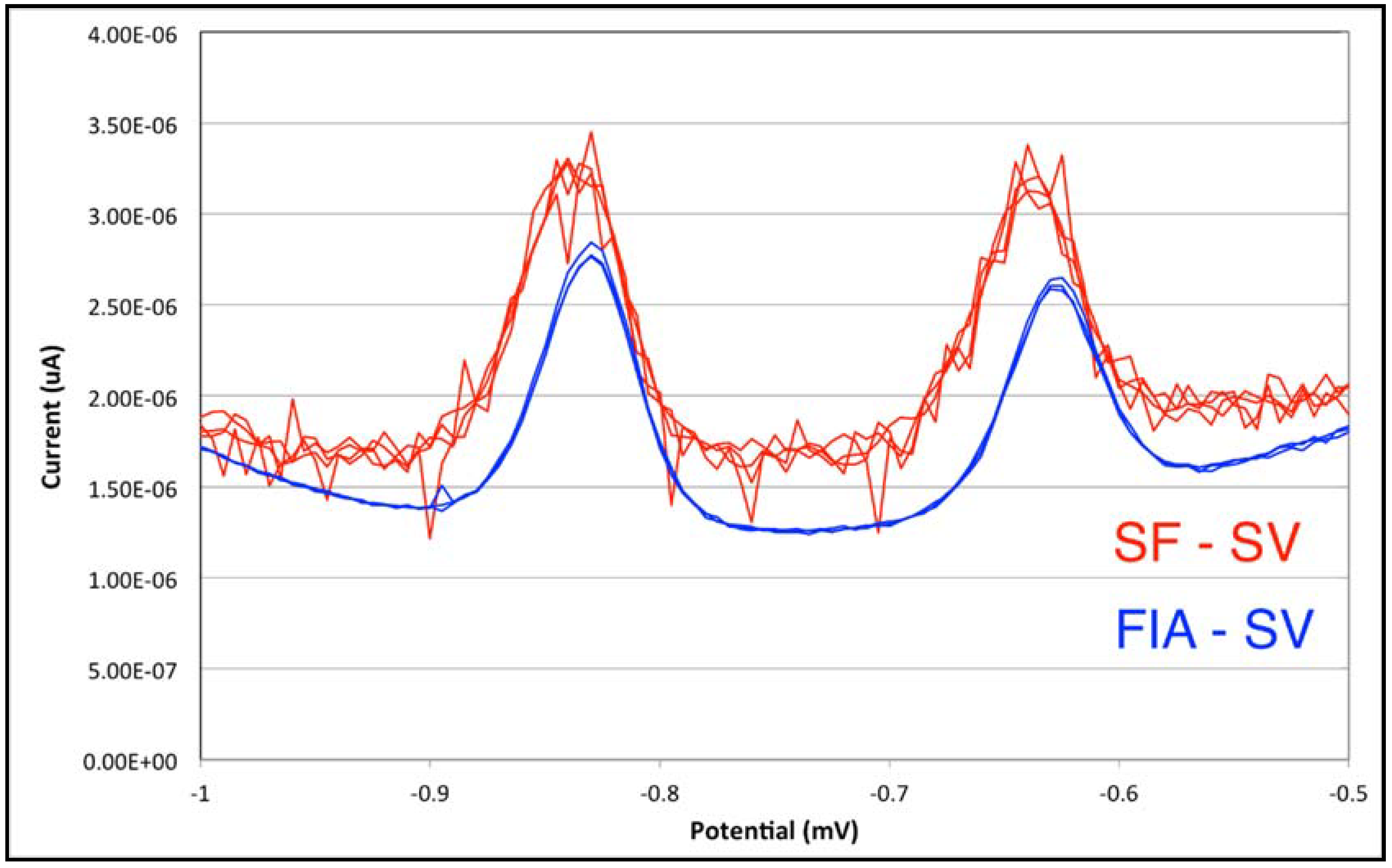
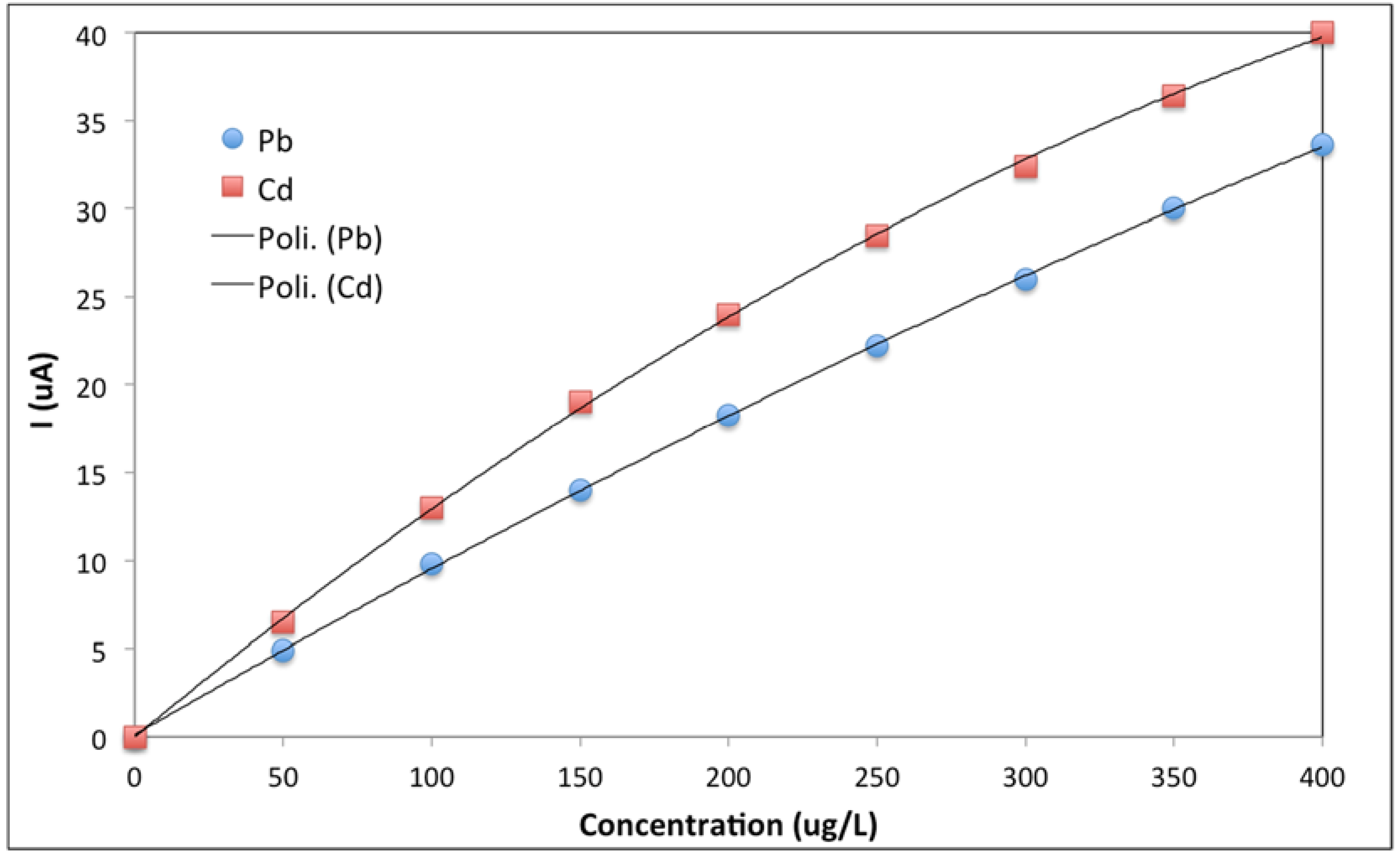
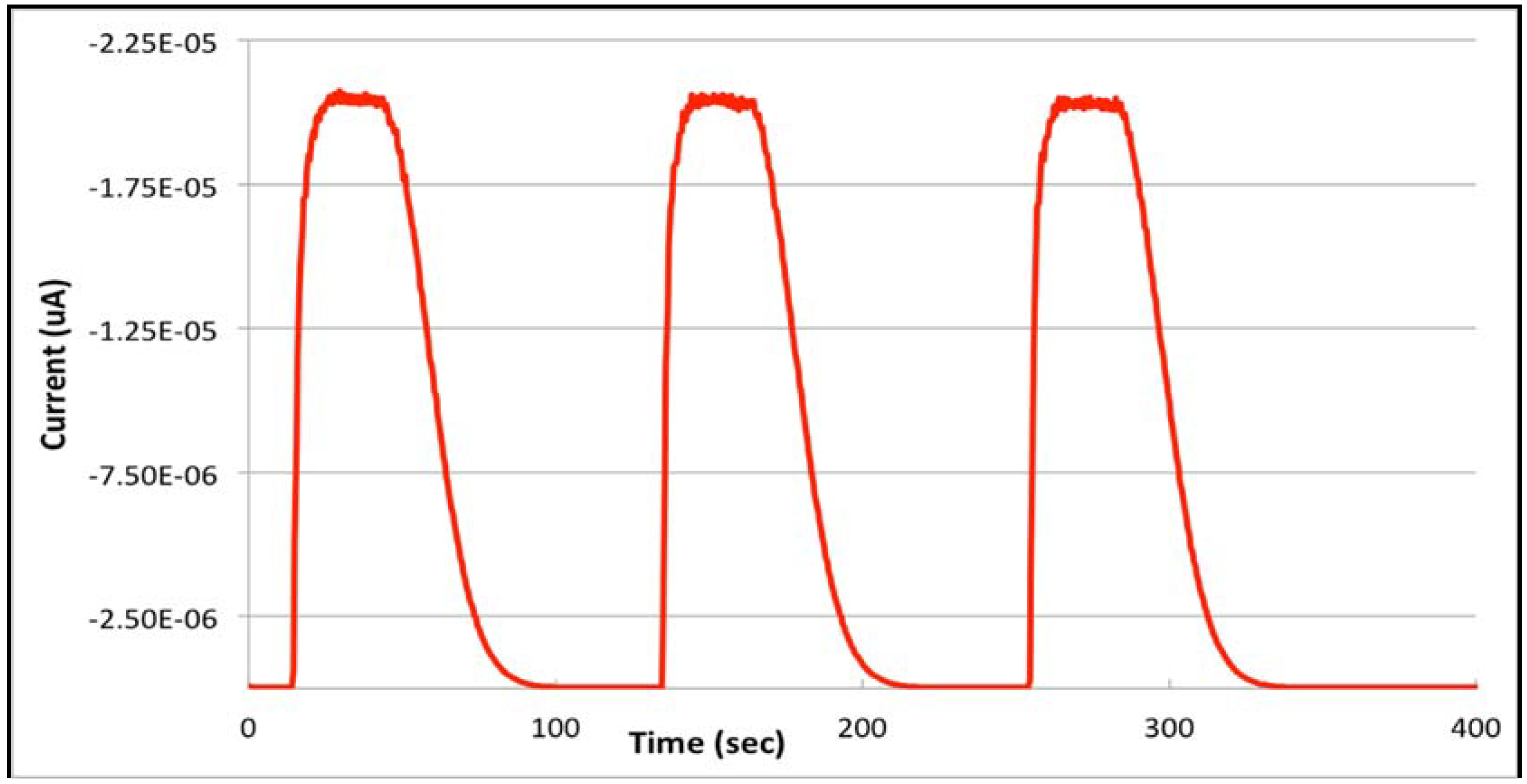
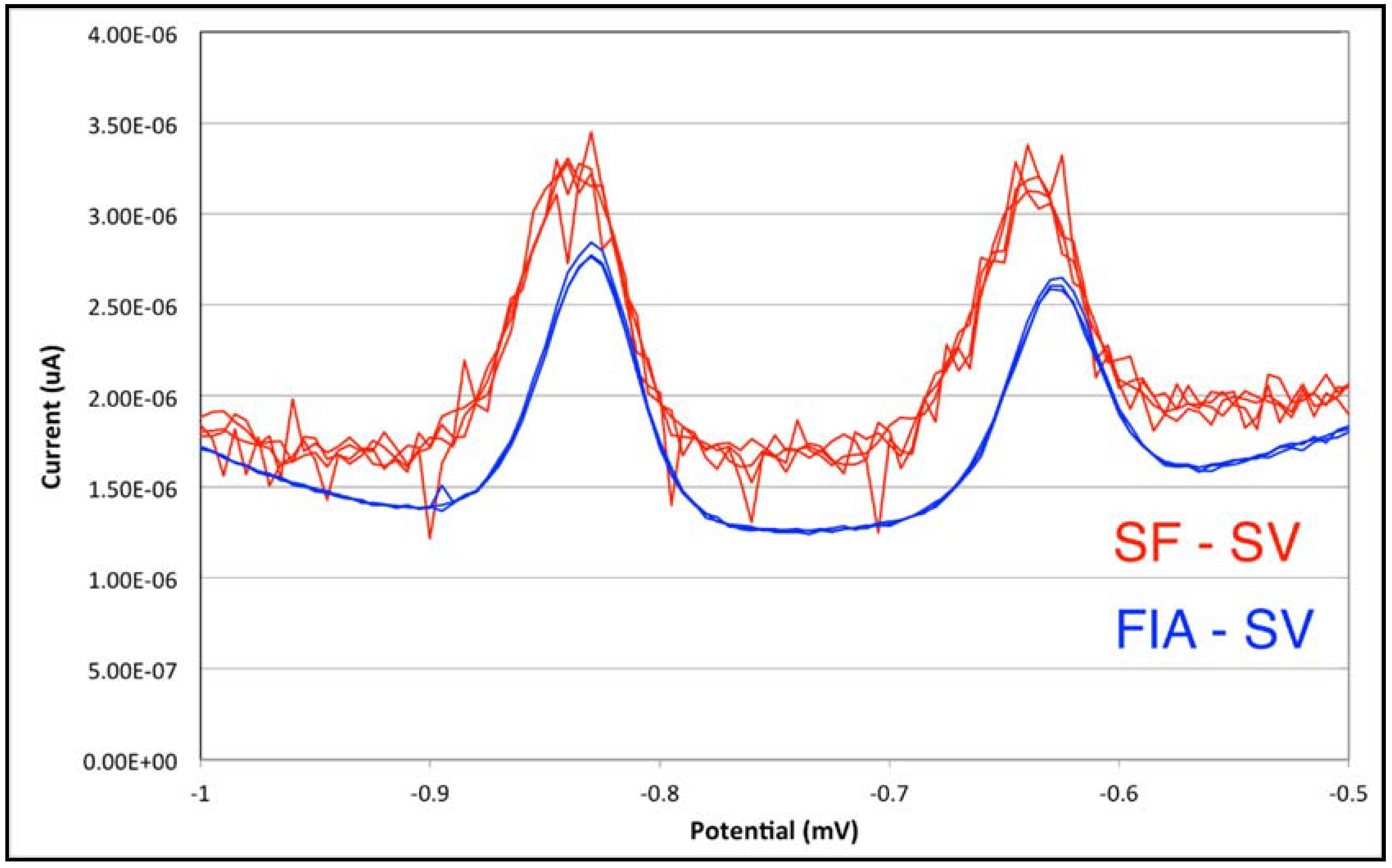
3.2. From Stopped-Flow to Flow Injection Analysis in Differential Pulse Stripping Voltammetry
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ochsenkühn-Petropoulou, M.; Ochsenkühn, K.M. Comparison of inductively coupled plasma-atomic emission spectrometry, anodic stripping voltammetry and instrumental neutron-activation analysis for the determination of heavy metals in airborne particulate matter. Fres. J. Anal. Chem. 2001, 369, 629–632. [Google Scholar] [CrossRef]
- Wang, J. Analytical Electrochemistry, 3rd ed.; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Ozkan, S.A. Principles and Techniques of Electroanalytical Stripping Methods for Pharmaceutically Active Compounds in Dosage Forms and Biological Samples. Curr. Pharm. Anal. 2009, 5, 127–143. [Google Scholar] [CrossRef]
- Economou, A.; Fielden, P.R. Applications, potentialities and limitations of adsorptive stripping analysis on mercury film electrodes. Trends Anal. Chem. 1997, 16, 286–292. [Google Scholar] [CrossRef]
- Wang, J. Decentralized Electrochemical Monitoring of Trace Metals: From Disposable Strips to Remote Electrodes. Analyst 1994, 119, 763–766. [Google Scholar] [CrossRef]
- Arduini, F.; Amine, A.; Moscone, D.; Ricci, F.; Palleschi, G. Fast, sensitive and cost-effective detection of nerve agents in the gas phase using a portable instrument and an electrochemical biosensor. Anal. Bioanal. Chem. 2007, 388, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Arduini, F.; Cassisi, A.; Amine, A.; Ricci, F.; Moscone, D.; Palleschi, G. Electrocatalytic oxidation of thiocholine at chemically modified cobalt hexacyanoferrate screen-printed electrodes. J. Electroanal. Chem. 2009, 626, 66–74. [Google Scholar] [CrossRef]
- Palchetti, I.; Laschi, S.; Mascini, M. Miniaturised stripping-based carbon modified sensor for in field analysis of heavy metals. Anal. Chim. Acta 2005, 530, 61–67. [Google Scholar] [CrossRef]
- Gomez, V.S.; Barcelò, C.; Serrano, N.; Esteban, M. Antimony film screen-printed carbon electrode for stripping analysis of Cd(II), Pb(II), and Cu(II) in natural sample. Anal. Chim. Acta 2015, 855, 34–40. [Google Scholar]
- Hoĉevar, S.B.; Ogorevc, B.; Wang, J.; Pihlar, B. A Study on Operational Parameters for Advanced Use of Bismuth Film Electrode in Anodic Stripping Voltammetry. Electroanalysis 2002, 14, 1707–1712. [Google Scholar] [CrossRef]
- Romanus, A.; Mueller, H.; Kirsch, D. Application of adsorptive stripping voltammetry (AdSV) for the analysis of trace metals in brine. Fresenius J. Anal. Chem. 1991, 340, 371–376. [Google Scholar] [CrossRef]
- Economou, A. Recent developments in on-line electrochemical stripping analysis—An overview of the last 12 years. Anal. Chim. Acta 2010, 683, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, K.C. Screen-printed electrochemical sensors and biosensors for monitoring metal pollutants. Sci. J. 2012, 2, 1–51. [Google Scholar] [CrossRef]
- Guell, R.; Fontas, C.; Aragay, G.; Merkoci, A.; Antico, E. Screen-printed electrodes incorporated in a flow system for the decentralized monitoring of lead, cadmium and copper in natural and wastewater samples. Int. J. Environ. Anal. Chem. 2012, 93, 1–12. [Google Scholar] [CrossRef]
- Mozo, J.D.; Carbajo, J.; Sturm, J.C.; Nunez-Vergara, L.J.; Salgado, P.; Squella, J.A. Determination of Nifuroxazide by flow injection linear adsorptive stripping voltammetry on a screen-printed carbon nanofiber modified electrode. Electroanalysis 2010, 24, 676–682. [Google Scholar] [CrossRef]
- Dossi, C.; Stropeni, F.; Monticelli, D. Screen Printed Electrode-flow stripping voltammetry for inorganic analysis. In Sensors-Lecture Notes in Electrical Engineering; Compagnone, D., Baldini, F., Di Natale, C., Betta, G., Siciliano, P., Eds.; Springer: Berlin, Germany, 2015; Chapter 25; Volume 319. [Google Scholar]
- Pizeta, I.; Omanovic, D.; Branica, M. The influence of data treatment on the interpretation of experimental results in voltammetry. Anal. Chim. Acta 1999, 401, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Mocak, J.; Bond, A.M.; Mitchell, S.; Schollary, G. A statistical overview of standard (IUPAC and ACS) and new procedures for determining the limits of detection and quantification: Application to voltammetric and stripping techniques. IUPAC Technical Report 1997. Pure Appl. Chem. 1997, 69, 297–328. [Google Scholar] [CrossRef]
- Jakubowska, M.; Piech, R.; Dzierwa, T.; Wcislo, J.; Kubiak, W.W. The evaluation method of smoothing algorithms in voltammetry. Electroanalysis 2003, 15, 1729–1736. [Google Scholar] [CrossRef]
- Margoshes, M.; Burns, D.A. Treatise on Analytical Chemistry, Part 1-Theory and Practice, 2nd ed.; Elving, P.J., Mossotti, E.V.G., Kolthoff, I.K., Eds.; Wiley: New York, NY, USA, 1984; Volume 4, p. 413. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Figure of Merit | Cd | Pb |
|---|---|---|---|
| Batch | Sensitivity (μA × L × μg−1) | 0.2831 | 0.1681 |
| Intercept (μA) | −0.306 | 0.098 | |
| Limit of Detection (μg/L) | 0.17 | 0.19 | |
| Limit of Quantitation (μg/L) | 0.58 | 0.64 | |
| Stopped-Flow | Sensitivity (μA × L × μg−1) | 0.1635 | 0.1202 |
| Intercept (μA) | 0.189 | 0.279 | |
| Limit of Detection (μg/L) | 0.58 | 0.62 | |
| Limit of Quantitation (μg/L) | 1.98 | 2.06 |
| Analyte | Peak Height (μA) | Calculated Concentration (μg/L) | Expected Concentration (μg/L) | % Recovery |
|---|---|---|---|---|
| Cd | 2.4449 | 15.28 | 15 | 102% |
| 0.8463 | 5.11 | 5 | 105% | |
| 4.3753 | 27.56 | 25 | 110% | |
| 3.3041 | 20.75 | 20 | 103% | |
| 1.5945 | 9.87 | 10 | 99% | |
| Pb | 1.9857 | 14.49 | 15 | 96% |
| 0.6657 | 4.57 | 5 | 91% | |
| 3.4794 | 25.72 | 25 | 103% | |
| 2.6807 | 19.72 | 20 | 98% | |
| 1.3532 | 9.74 | 10 | 97% |
| Measure Type | Figure of Merit | Cd | Pb |
|---|---|---|---|
| Electrode 1 | Sensitivity (μA × L × μg−1) | 0.1382 | 0.09095 |
| Intercept (μA) | −0.1854 | −0.0936 | |
| Electrode 2 | Sensitivity (μA × L × μg−1) | 0.1363 | 0.08795 |
| Intercept (μA) | −0.3284 | −0.1461 | |
| Electrode 3 | Sensitivity (μA × L × μg−1) | 0.1335 | 0.09007 |
| Intercept (μA) | −0.450 | −0.1820 | |
| Mean result of three electrodes | Limit of Detection (μg/L) | 0.32 | 0.35 |
| Limit of Quantitation (μg/L) | 1.06 | 1.08 |
| Cd | Pb | |||
|---|---|---|---|---|
| Found (μg/L) | %Recovery | Found (μg/L) | %Recovery | |
| Sample 1 | 4.80 | 96% | 4.99 | 100% |
| Sample 2 | 4.51 | 90% | 4.60 | 92% |
| Sample 3 | 4.88 | 97% | 4.81 | 96% |
| Batch | Stopped Flow | Flow Injection Analysis | ||||
|---|---|---|---|---|---|---|
| Cd | Pb | Cd | Pb | Cd | Pb | |
| Sensitivity (μA × L × μg−1) | 0.2831 | 0.1681 | 0.1635 | 0.1202 | 0.1381 | 0.0909 |
| Limit of Detection (μg/L) | 0.17 | 0.19 | 0.58 | 0.62 | 0.32 | 0.35 |
| Analytical Errors | <5% | <5% | <10% | <10% | <10% | <10% |
| Analysis Time a | 175 s | 175 s | 220 s | |||
| Sample Volume | 10–25 mL | 1–2 mL | 0.5 mL (loop volume) | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dossi, C.; Monticelli, D.; Pozzi, A.; Recchia, S. From Batch to Flow Stripping Analysis with Screen-Printed Electrodes: A Possible Way to Decentralize Trace Inorganic Analysis. Chemosensors 2018, 6, 37. https://doi.org/10.3390/chemosensors6030037
Dossi C, Monticelli D, Pozzi A, Recchia S. From Batch to Flow Stripping Analysis with Screen-Printed Electrodes: A Possible Way to Decentralize Trace Inorganic Analysis. Chemosensors. 2018; 6(3):37. https://doi.org/10.3390/chemosensors6030037
Chicago/Turabian StyleDossi, Carlo, Damiano Monticelli, Andrea Pozzi, and Sandro Recchia. 2018. "From Batch to Flow Stripping Analysis with Screen-Printed Electrodes: A Possible Way to Decentralize Trace Inorganic Analysis" Chemosensors 6, no. 3: 37. https://doi.org/10.3390/chemosensors6030037