Abstract
Cancer drivers play an important role in regulating cell growth, cell cycles, and DNA replication. Identifying these cancer drivers provides cancer researchers with indispensable knowledge that has important implications for clinical decision making. Some methods have been recently proposed to identify coding and non-coding cancer drivers through controllability analysis in network and eigenvector centrality based on community detection. However, the performance of these methods is not satisfactory. In this work, we focus on the strategy of selecting a set of critical nodes in cancer-special network as cancer drivers, and propose a novel approach for identifying coding and non-coding drives via a network-based voting mechanism. We name our approach HWVoteRank. Compared with two recent methods to identify cancer drivers, CBNA and NIBNA, and three algorithms for identifying key nodes on BRCA dataset, our method can achieve the best efficiency. By analyzing the results, it is found that our approach has better ability in identifying miRNA cancer drivers. We also applied our approach to identification of drivers of miRNA during Epithelial–Mesenchymal transition and drivers for cancer subtype. Through literature research, we found that those drivers explored by our approach are of biological significance.
1. Introduction
Cancer is a kind of disease that can affect any part of the human body. It is the second leading cause of death worldwide, killing about 10 million people a year and causing about one in six deaths [1]. The main causes of cancer are genetic disorders and environmental factors [2,3]. Cancer drivers are genes that play an active and crucial role in the evolution of cancer and give tumor cells an advantage in selective growth [4]. Identifying all the genes that drive tumors is a milestone in understanding and overcoming cancer.
Traditional cancer drivers refer to genes that have mutations in their DNA sequences. In order to reduce the cost of biological experiments, many methods based on mutation data and network structure have been proposed to identify cancer drivers. For example, OncodriveFM [5] was proposed to detect cancer drivers based on the function impact of gene mutation. MutSigCV, developed by Lawrence et al. [6], explores cancer genes by calculating the significance of mutations in gene sequence. The structural consequences of gene mutation is used by the models of OncodriveCLUST [7] and ActivaDriver [8]. Hou et al. [9] adopted a ranking framework of pagerank in gene–gene interaction networks to predict cancer drivers. Horn et al. [10] expanded discovery of cancer driver genes from cancer genomes by combining molecular network and tumor genome expression data.
However, genes without mutation and non-coding miRNA also have been proven as causal factors contributing to cancer. For instance, the unmutated gene CDC42 has been shown to promote the formation of invadopodia in breast cancer cells by activating N-WASp [11]. As oncomiR, the dysfunction of miR-423 leads to oncogenesis and is involved in signaling pathways of multiple cancer progression [12]. The high expression of miR-15b-3p promotes tumorigenesis and malignant transformation in gastric cancer cells [13]. Therefore, several models have been proposed to predict both coding and non-coding cancer drivers. Pham et al. [14] first integrated multiple biological data sets into a directed network, and proposed a model CBNA based on controllability analysis in network to predict cancer drivers, including genes with/without mutation and miRNAs. The network control idea is introduced by CBNA and the subset of nodes that guide the complex network can be regarded as driver nodes. Although CBNA is a complement for tradition methods in predicting cancer drivers, only a small percentage of cancer drivers can be identified. In order to improve the performance, a network-based algorithm NIBNA [15] was developed. The importance score is calculated for each node in the constructed undirected network based on community detection and eigenvector centrality. The nodes with higher importance score are more likely to be cancer drivers. NIBNA achieves better performance than CBNA in uncovering coding drivers. As there is no experimentally confirmed miRNA–miRNA association in the network constructed by CBNA and NIBNA, similar to CBNA, NIBNA is weak in detecting cancer driver of miRNA. In order to further improve the performance of the algorithm, it is necessary to design new algorithms to identify cancer drivers.
As cancer drivers have a strong ability to influence other genes, we adopt a common and classical strategy of a vote mechanism to select top-ranked key nodes as drivers. The critical factors of nodes’ importance are weights and degrees of the underlying network. In this work, we design a novel approach to identify key nodes in networks. We name our approach HWVoteRank. We take into account the local topology of nodes, as well as the state of its neighboring nodes in our approach. Then, we apply HWVoteRank in detecting cancer drivers in a special breast cancer network that is built by gene expression data of breast cancer samples and multiple biological datasets. By comparison, HWVoteRank outperforms the state-of-the-art models CBNA and NIBNA. In particular, it has great advantages in predicting miRNA cancer driver. HWVoteRank also achieves better performance than three well-known algorithms, which have excellent performance in identifying key nodes in weighted network. In order to further demonstrate the effectiveness of our method, we use HWVoteRank to identify miRNA drivers during Epithelial–Mesenchymal transition and drivers for cancer subtype. These results demonstrate that our method can identify key drivers under different conditions.
2. Materials and Methods
2.1. Datasets
A total of 747 cancer samples were downloaded from the BRCA dataset in TCGA [16], containing coding genes expression, miRNA expression, and mutation data. According to the list of transcription factors (TFs) provided by Lizio et al. [17], coding genes can be divided into transcription factors and mRNA. Then, we downloaded the protein–protein interactions network (PPIs) [18] and deleted coding genes that are not in PPIs. In addition, we obtained miRNA–mRNA/TF associations from miRTarBase [19], TarBase [20], miRWalk [21], and TargetScan databases [22], and TF–miRNA associations from TransmiR Database [23].
2.2. Constructing Network
Pearson’s correlation coefficient is the commonly used measurement for the strength of the association between a pair of genes as it conforms well to the intuitive biological notion [24]. It is widely used to identify cancer drivers, prognostic genes, and key regulators [15,25,26]. Based on the expression profile of tumor data, Pearson’s correlation coefficients between miRNAs, TFs, and mRNAs were calculated. Then a miRNA–TF–mRNA network can be constructed, in which miRNAs, TFs, and mRNAs are nodes, and the correlation coefficients between nodes are edge weights. Following the suggestion of Pham et al. [15], we constructed a cancer-specific network by integrating multiple databases, including PPIs, miRTarBase, TarBase, miRWalk, TargetScan, and TransmiR. The cancer-specific network was built through removing the edges of miRNA–TF–mRNA network that not exist in the above 6 databases. Finally, there are 7726 nodes and 128,264 edges in this cancer-specific network. The node set includes 1719 miRNAs nodes, 839 TF nodes, and 5168 mRNA nodes. The edge set includes 16,087 miRNA-TF links, 73,347 miRNA-mRNA links, 18,950 TF-miRNA links, 1812 TF-TF links, 1188 TF-mRNA links, and 16,880 mRNA-mRNA links. It is worth noting that the cancer-specific network is an undirected, weighted network without considering the directionality of edges.
2.3. Method
Identifying the key nodes in complex networks is a crucial task in various major fields. The key nodes have very important influence on the whole network, and the influence of nodes in the network is closely related to its location and topological structure of network. At present, a number of methods have been proposed to identify the key nodes based on closeness centrality [27], betweenness centrality [28], degree centrality [28], eigenvector centrality [29], H-index [30], etc. VoteRank algorithm is one popular method. However, most of the methods are only applicable to the unweighted network, and the remaining methods that can deal with the weighted network have a common limitation. Due the phenomenon of rich club, they cannot accurately identify the key nodes in the top ranked ones [31]. In order to overcome this difficulty, Sun et al. [32] extended VoteRank algorithm [33] to the weighted network and proposed the WVoteRank algorithm to find influential nodes. Subsequently, Kumar et al. [34] improved WVoteRank by considering 2-hop neighbors. At the same time, Liu et al. [35] developed the VoteRank++ algorithm by redefining voting mechanism in VoteRank algorithm. Although VoteRank++ cannot be applied to a weighted network, it achieves excellent performance in unweighted networks. In this work, inspired by the WVoteRank and VoteRank++ algorithms, we propose HWVoteRank to identify the key nodes in heterogeneous network.
For a network , V denotes the set of nodes and E denotes the set of edges. HWVoteRank adopts the idea of VoteRank++ and WVoteRank to find key nodes through a voting mechanism. Nodes in the network can be voted as key nodes by their neighbors, and they can vote for their neighbors. We set a state tuple for each node in the network, where represents the voting ability of the node and represents the voting score obtained by voting of neighbor nodes. The voting ability of each node is initialized. We assigned voting ability as 1 for all nodes in G.
Then, a novel voting rule is defined. Liu et al. [35] think if the degree of a neighbor node is larger, it is more likely to become a key node. In VoteRank++, the voting rule is based on the ratio of the degree of the neighbor node to the sum of the degrees of all neighbor nodes. Sun et al. [32] believe that nodes are more likely to vote for neighbor nodes with high correlation. In other words, for node v, it is likely to obtain more voting scores from node u when the edge weight is greater than that from u to its other neighbors. Thus, the voting rule of WVoteRank is set by the weight of edges. In HWVoteRank, we take into account the degree of the neighbor nodes and the edge weights between nodes, and balanced their by a parameter of . For node v, the voting score of u against v is calculated as follows:
Based on the assumption that the topology of the node and the topology of its neighbors are equally important for identifying the key nodes, we set . After a round of voting, the total voting score of node v can be calculated as follows:
where is the number of neighbors of node u. After the voting, the node with the highest voting score is selected as the key node. The voting capability of nodes in a network needs to be updated for the next voting. First, the voting ability of the selected node is set to 0 to prevent it from participating in the next round of voting. In order to avoid the “rich club” phenomenon, both WVoteRank algorithm and VoteRank++ algorithm suppress the voting ability of a key node’s neighbors. To reduce computation, they only update 1-hop neighbors and 2-hop neighbors by a parameter of p. For 1-hop neighbor nodes of the selected key node, , and for 2-hop neighbor nodes of the selected key node, . Their updated rules are defined as follows.
In WVoteRank algorithm,
where is the average degree of network.
In VoteRank++ algorithm,
where is a suppressing factor.
According to the definition of [36,37], a network with multiple types of nodes and/or multiple types of edges can be called a heterogeneous network. For a network , is defined as the node type mapping function of G, where O is the set of node type and the type of each node belongs to O. is defined as the edge type mapping function of G, where R is the set of edge type and the type of each edge belongs to R. If , the network G can be called a heterogeneous network; otherwise, it is a homogeneous network. At present, most key node identification algorithms are applicable to homogeneous networks, including WVoteRank and VoteRank++. In this study, we design a new, updated rule that enables our algorithm WVoteRank to be applied to heterogeneous networks which have multiple types of nodes. We assume that there is only suppression among nodes of the same type. The voting ability of the node v is updated as follows:
where is the average degree of nodes whose type is i. The definition of p is the same as WVoteRank and VoteRank++.
2.4. Identifying Cancer Drivers
Cancer drivers are closely related to the progress of cancer. In this paper, we identify the drivers of cancer by identifying the key nodes in a cancer-specific network. The flow chart of our model is shown in Figure 1. Firstly, we calculate Pearson’s correlation coefficient between genes through the expression profile of cancer, including coding genes of TF/mRNA and non-coding genes of miRNA, and construct a miRNA–TF–mRNA fully connected network. We then use multiple biological databases to calibrate the network and delete coding gene nodes that do not exist in PPIs and edges that do not exist in these databases. A cancer-specific network can be generated. Finally, using the topology of the network, we identify the cancer drivers in the network through a novel voting mechanism in heterogeneous networks.
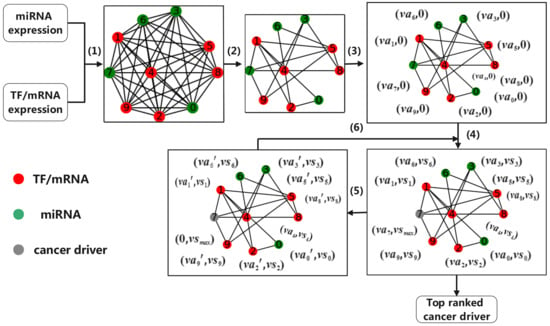
Figure 1.
An illustration of identifying cancer driver. (1) Calculating Pearson’s correlation coefficient between genes; (2) Refining network; (3) Initializing voting ability; (4) Voting according to voting mechanism; (5) Updating voting ability; and (6) Repeating step (4).
3. Results and Discussion
In this work, we proposed a network-based voting approach (HWVoteRank) to identify coding and non-coding cancer drivers. The idea of HWVoteRank is to detect key nodes in the cancer-specific network. We adopted the innovation points of VoteRank++ and WVoteRank and considered the heterogeneous characteristics of biological network. Compared with traditional algorithms for identifying key nodes, HWVoteRank takes into full consideration the local topology of nodes, as well as the state of its neighboring nodes. More importantly, HWVoteRank can be applied to heterogeneous networks, which prevents the rich club phenomenon in the same type nodes through the voting approach. To our knowledge, this is the first work to use network-based voting approach for identifying cancer drivers.
3.1. Experiment Setting
The cancer-specific network contains coding genes and non-coding RNA of miRNA. Based on mutation data from the BRCA dataset, we classified coding genes into mutated and non-mutated. For mutated genes, we verified cancer drivers predicted by our method through the Cancer Gene Census (CGC) of the COSMIC Database [38], a database that collects mutated genes which have a direct causal association with cancer. For genes without mutations, as there were no real ground-truth data, we performed GO enrichment analysis through an online software of DAVID [39] to evaluate the ability of HWVoteRank of identifying meaningful coding non-mutation cancer driver. For non-coding RNA of miRNA, we validated miRNA drivers that result in tumorigenesis of BRCA through OncomiR [40], an online resource for exploring miRNA dysregulation in cancer. In addition, we also applied HWVoteRank algorithm to detect drivers in the process of Epithelial–Mesenchymal transition and identify drivers used for cancer subtypes.
3.2. Comparison Experiments
In order to prove the performance of HWVoteRank in detecting cancer drivers in cancer-specific networks, we compared HWVoteRank with CBNA [14] and NIBNA [15], which are currently efficient methods for identifying both coding and non-coding cancer drivers based on expression data. Some well-known methods for identifying key nodes in weighted networks include weighted degree [41], weighted H-index [42], weighted coreness [43], and weighted betweenness [44]. We also compared HWVoteRank with weighted degree (w_degree), weighted H-index (w_hindex), and improved WVoteRank [34] (called PWVoteRank in this paper) that has excellent performance in identifying key nodes in a weighted network (by voting approach). As weighted coreness and weighted betweenness need to traverse all paths from each node to other nodes in the network, they have very high computational complexity and are not suitable for large biological information networks. In additional, according to the comparison results of [42], weighted degree is better than weighted coreness and weighted betweenness, so we did not compare HWVoteRank with weighted coreness and weighted betweenness. As CBNA cannot rank identified cancer drivers of miRNA (it just ranks genes by mutation frequency), we only compared HWVoteRank, NIBNA, PWVoteRank, w_degree, and w_hindex by the validated number of cancer drivers, precision, recall, and F1 values among the top 200 identified cancer drivers, excluding genes without mutations. The results are shown in Table 1 and Figure 2. Clearly, HWVoteRank, proposed in this study, achieves the highest performance in exploring cancer drivers of coding mutated gene and non-coding miRNA. We found that the performance of w_degree is second only to ours, because as there is no experimentally confirmed miRNA–miRNA association in cancer-specific network, the absence of edges between miRNA nodes reduced the influence of rich club phenomenon on the performance of w_degree.

Table 1.
The comparison of validated number of cancer drivers among PHVoteRank, NIBNA, HWVoteRank, w_hindex, and w_degree in top 200 predicted cancer drivers, excluding genes without mutations.
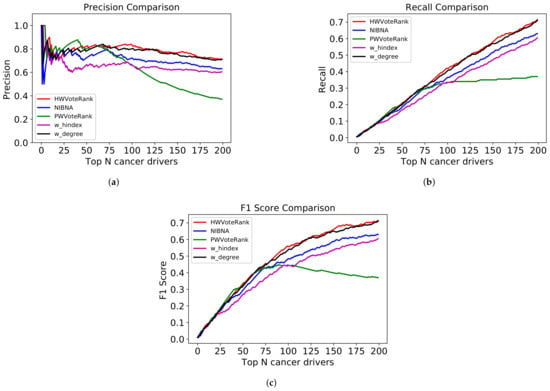
Figure 2.
Performance comparison among PHVoteRank, NIBNA, HWVoteRank, w_hindex, and w_degree in top 200 predicted cancer drivers, excluding genes without mutations. (a) is the precision comparison, (b) is the recall comparison, and (c) is the F1 score comparison.
We analyzed non-coding drivers of miRNA in the top 200 predicted cancer drivers. The comparison results of number of validated miRNA are shown in Figure 3. In addition, CBNA predicted 17 miRNA drivers, 11 of which were confirmed by OncomiR associated with BRCA tumorigenesis. Obviously, HWVoteRank has ability to explore higher number of non-coding drivers of miRNA. The reason of HWVoteRank proves superior to other methods is that HWVoteRank takes full advantage of the topology structural of cancer-special network.
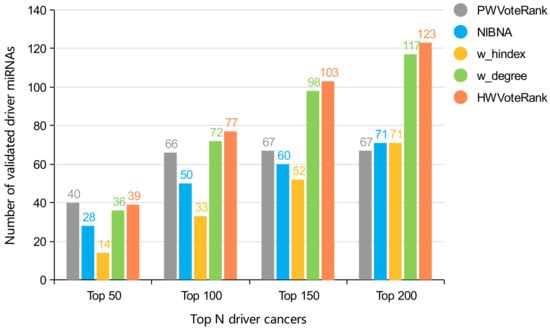
Figure 3.
Comparison of the number of confirmed miRNA drivers in the top 200 predicted cancer drivers, excluding genes without mutations.
We compared HWVoteRank with NIBNA and CBNA, two of the most popular methods for identifying coding and non-coding cancer drivers, in the number of predicted cancer driver of gene with mutations in the top 50 genes. The comparison results are shown in Figure 4. It is demonstrated that the proposed HWVoteRank approach outperforms the other two methods in top 50. Therefore, methods based on key node identification can effectively identify cancer drivers of genes with mutations. It is a novel development direction to identify cancer drivers in biological network by the algorithms for identifying the key nodes in the network.
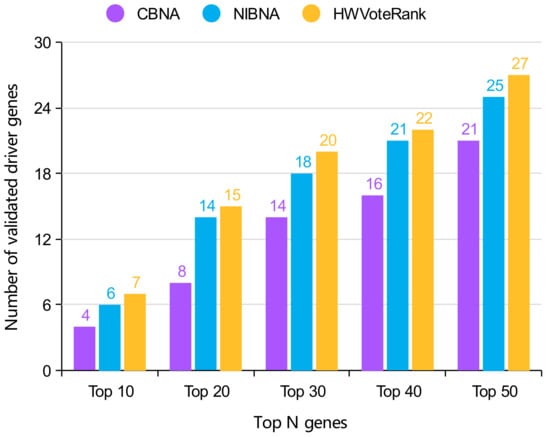
Figure 4.
Comparison of the number of confirmed drivers in the top 50 predicted cancer driver of gene with mutations.
3.3. Identifying Cancer Drivers of Coding Gene without Mutations
To evaluate the ability of HWVoteRank in discovering cancer drivers of coding genes without mutations, Go enrichment analysis of biological processes (BP) and molecular functions (MF) were performed by online software DAVID. We selected the top 50 predicted candidate cancer drivers of coding genes without mutations. The top 10 enriched terms of GO BP and GO MF involved with the highest number of predicted coding genes without mutations, and the top 20 coding genes without mutations with the highest frequency in the top 10 enriched terms, were selected. The results of the enrichment analysis are presented as heatmaps in Figure 5, and the corresponding terms of biological processes and molecular functions are provided in Table A1 and Table A2 of Appendix A. Two heatmaps show that there is a significant association between coding genes without mutations and the enrichment terms of BP and MF. We conducted further analysis through literature research, and there is much evidence demonstrating those cancer-drivers of genes without mutations have biological significance. For example, CEBPB, involved both in BP and MF, promotes migration and invasion of breast cancer cell by deregulating the expression of CLDN4 [45]. Chen et al. [46] found that JunD activates proximal aromatase associated with breast cancer by integrating prostaglandin E2. FGF2 has been shown to promote breast cancer growth through independent ligand activation and interaction with MYC regulatory sequence [47]. RBPJ was identified as a medullary breast carcinoma autoantigens, which can block serological characteristics in breast cancer [48]. The expression of CEBPD is reduced by promoter methylation in the sum-52pe human breast cancer cell line and primary breast cancer [49], and the interaction between FOXP2 and FOXA2 can prevent metastasis of breast cancer [50].
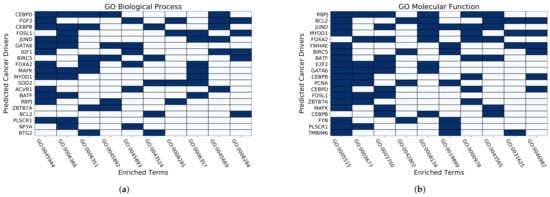
Figure 5.
The heatmaps of (a,b) show enrichment profiles for GO biological processes and GO molecular functions, respectively.
3.4. Identifying Drivers during Epithelial–Mesenchymal Transition
Epithelial–Mesenchymal transition (EMT) is a complex process that drives cells to become invasive and motile during cancer progression [51]. Non-coding RNA of miRNA can regulate EMT via interacting with critical molecules involved in EMT engineering [52]. For example, miRNA-21 induces EMT by the AKT/PTEN pathway in breast cancer [53]. MiRNA-520f reverses EMT by targeting two targets of TGFBR2 and ADAM9 [54]. Considering that HWVoteRank has excellent performance in exploring cancer drivers of miRNA, we applied it in identifying drivers during Epithelial–Mesenchymal transition. The BRCA dataset used in this study was categorized into four phenotypes according to EMT score [55], including epithelial, mesenchymal, intermediately epithelial, and intermediately mesenchymal. HWVoteRank was performed on the mesenchymal class with the largest sample size to detect EMT drivers of miRNA. We selected the top 100 EMT drivers, of which 19 predicted EMT drivers of miRNAs were confirmed by the list of pro-mesenchymal miRNAs provided by [56]. The top 20 predicted EMT drivers of miRNAs are given in Table 2, which can be prioritized for the study of their biological function in EMT.
Table 2.
Top 20 miRNA EMT drivers, with the names of predicted drivers validated by [56] in bold.
3.5. Identifying Drivers for Cancer Subtype
Breast cancer with the same pathologic morphology is highly heterogeneous at the molecular level. The subtype of breast cancer can reflect the biological behavior of tumors more accurately, judge the prognosis, and facilitate the selection and research of more targeted personalized treatment methods. We performed HWVoteRank to detect candidate drivers that are capable of typing breast cancer. Following the suggestions of Pham et al. [14,15], we classified the BRCA dataset used in this study into five subtypes by Pam50 [57,58]. We obtained 221 samples of Luminal A, 165 samples of Luminal B, 158 samples of Basal, 108 samples of Her2, and 95 samples of Normal-like. For each type, the drivers of coding gene with mutation, coding gene without mutation, and miRNA were predicted and are listed in Table A3 of Appendix A. Coding genes with mutation are considered as drivers if they mutate more frequently than any other cancer subtype, and the top 10 of each type was selected. For coding genes without mutation, and miRNA, we selected the top 10 candidate drivers for each breast cancer subtype ranked in the top 500 and top 200 predicted drivers, respectively, by removing common candidates between at least two subtypes.
Among the predicted drivers, FN1 is a hub node of the protein–protein interactions networks constructed by differentially expressed mRNAs in luminal subtype B breast cancer tumor tissues and adjacent tissues [59]. STAT1 has been observed to have a statistically significant correlation with IDO1, which is found in stromal cells and tumor-associated macrophages, and has a higher incidence with luminal B samples [60]. The expression of FOXA1 is closely related to luminal A breast cancer, and it can be used as a significant predictor of survival in patients tumors [61]. Several miRNAs associated with breast cancer subtype predicted by Pham et al. [15] were also identified by us, for example, the expression of hsa-miR-590-5p, hsa-miR-10a-5p, and hsa-miR-877-5p in subtype of Basal and the expression of hsa-miR-215-5p in subtype of Her2 are abnormal.
4. Conclusions
Cancer is one of the most common diseases that endangers human health. Cancer drivers play an important role in the initiation, progression, and metastasis of cancer. Successful identification of cancer drivers is helpful to understand the pathology of cancer and to design accurate treatment and prognosis plans for cancer. In this work, we proposed an efficient approach, HWVoteRank, to detect cancer drivers, including coding genes with mutation, coding genes without mutation, and non-coding RNA of miRNA. In our method, we first constructed a cancer-specific network by integrating several biological datasets. Each gene in the network is endowed with a voting ability according to its degree, and it also can be voted for by its neighbor genes through a voting mechanism, which considers both the local topology of genes and its neighbors. After a round of voting, the node with the highest voting score is considered a driver, and the voting ability of each node is updated. With multiple votes, we can iterate to identify top cancer drivers in a cancer-specific network. In order to prove the effectiveness of our method, we compared it with other popular methods, CBNA, NIBNA, and three algorithms for identifying key nodes. The results show that our method has the best performance, and especially has powerful capability in identifying cancer drivers of miRNA. An experiment based on identifying drivers of miRNA during the Epithelial–Mesenchymal transition, and identifying drivers for cancer subtypes, was also conducted in this work. Some drivers identified in different condition by our method do have biological significance, found through literature research. HWVoteRank is proven to be a reliable potential method for identifying cancer drivers, and it contributes to the improvement for the understanding of cancer pathology.
Author Contributions
D.Y. and Z.Y. conceived the experiment, D.Y. conducted the experiment, and D.Y. and Z.Y. analysed the results. All authors have read and agreed to the published version of the manuscript.
Funding
The authors thank the anonymous reviewers for their valuable suggestions. This work was supported by funds from the National Natural Science Foundation of China (grant number: 11871061); The National Key Research and Development Program of China (grant number: 2020YFC0832405); Hunan Provincial Innovation Foundation for Postgraduate (grant number: CX20200611).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available in a publicly accessible repository. The data presented in this study are openly available in reference number [16,17,18,19,20,21,22,23,38,40,56]. All source codes and data sets used in our paper are provided in Github, and can be downloaded from https://github.com/Ydongling/HWVoteRank.git/ anytime.
Acknowledgments
The authors thank the anonymous reviewers for their valuable suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| BRCA | breast cancer |
| TF | transcription factors |
| PPIs | protein-protein interactions network |
| BP | biological processes |
| MF | molecular functions |
Appendix A
Table A1.
Lists of top 10 enriched terms and the corresponding biological processes.
Table A1.
Lists of top 10 enriched terms and the corresponding biological processes.
| Enriched-Terms | Term |
|---|---|
| GO:0045944 | positive regulation of transcription from RNA polymerase II promoter |
| GO:0006366 | transcription from RNA polymerase II promoter |
| GO:0006351 | transcription, DNA-templated |
| GO:0045892 | negative regulation of transcription, DNA-templated |
| GO:0045893 | positive regulation of transcription, DNA-templated |
| GO:0043524 | negative regulation of neuron apoptotic process |
| GO:0008285 | negative regulation of cell proliferation |
| GO:0006357 | regulation of transcription from RNA polymerase II promoter |
| GO:0045669 | positive regulation of osteoblast differentiation |
| GO:0008284 | positive regulation of cell proliferation |
Table A2.
Lists of top-10 enriched terms and the corresponding molecular function.
Table A2.
Lists of top-10 enriched terms and the corresponding molecular function.
| Enriched-Terms | Term |
|---|---|
| GO:0005515 | protein binding |
| GO:0003677 | DNA binding |
| GO:0003700 | transcription factor activity, sequence-specific DNA binding |
| GO:0042802 | identical protein binding |
| GO:0008134 | transcription factor binding |
| GO:0019899 | enzyme binding |
| GO:0000978 | RNA polymerase II core promoter proximal region sequence-specific DNA binding |
| GO:0043565 | sequence-specific DNA binding |
| GO:0031625 | ubiquitin protein ligase binding |
| GO:0046982 | protein heterodimerization activity |
Table A3.
Lists of drivers for breast cancer subtype.
Table A3.
Lists of drivers for breast cancer subtype.
| Subtype | Coding Gene with Mutation | Coding Gene without Mutation | miRNA |
|---|---|---|---|
| Luminal A | AR, TK1, MBD3, CXXC1, FOXA1, CREB1, TCF12, E2F1, RUNX1, FLI1 | GATA6, FOSL1, CEBPD, PPP1CA | hsa-miR-128-1-5p, hsa-miR-296-5p, |
| hsa-miR-484, hsa-miR-491-5p, | |||
| hsa-miR-497-5p, hsa-miR-33a-5p, | |||
| hsa-miR-543, hsa-miR-665, | |||
| hsa-miR-153-3p, hsa-miR-708-5p | |||
| Luminal B | YWHAG, STAT1, FN1, ATXN1, SRC, DLG1, TAF1, TCF3, CTCF, RUNX1T1, | ZBTB7A, TNFSF11, VDR | hsa-miR-615-5p, hsa-miR-122-5p, |
| hsa-miR-766-5p, hsa-miR-134-5p, | |||
| hsa-miR-647, hsa-miR-623 | |||
| Basal | EP300, MAPK1, ERG, SPI1, PRKCA, CREBBP, SP1, VCL, MYC, CTNNB1 | SUMO2, SNRPD1, CNOT7, SNTB2, KLF1, IGF2, TOMM20 | hsa-miR-19b-1-5p, hsa-miR-590-5p, |
| hsa-miR-98-5p, hsa-miR-219a-5p, | |||
| hsa-miR-877-5p, hsa-miR-10a-5p, | |||
| hsa-miR-191-5p, hsa-miR-342-5p, | |||
| hsa-miR-34a-5p, hsa-miR-137 | |||
| Her2 | MAPK6, LCK, TGFBR1, LYN, EGFR, CDK2, ATF7IP, PDPK1, PTPN11, RASA1 | USF2, SET, MYOD1, APOE, RAB11B, RGS19 | hsa-miR-215-5p, hsa-miR-193a-5p, hsa-miR-224-5p, hsa-miR-4728-5p |
| Normal-like | KPNB1, COL1A1, CCND1, C8orf33, ETS1, GOLGA2, PPP3CA, COL6A1, HIPK2, BMPR1A | SUMO4, RASSF1, SSSCA1, PXN, GTF2B, CAPNS1, CHMP1B, ADAM17, TBXA2R, ACP1 | hsa-miR-29a-5p |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Golemis, E.A.; Scheet, P.; Beck, T.N.; Scolnick, E.M.; Hunter, D.J.; Hawk, E.; Hopkins, N. Molecular mechanisms of the preventable causes of cancer in the United States. Genes Dev. 2018, 32, 868–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteman, D.C.; Wilson, L.F. The fractions of cancer attributable to modifiable factors: A global review. Cancer Epidemiol. 2016, 44, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Waks, Z.; Weissbrod, O.; Carmeli, B.; Norel, R.; Utro, F.; Goldschmidt, Y. Driver gene classification reveals a substantial overrepresentation of tumor suppressors among very large chromatin-regulating proteins. Sci. Rep. 2016, 6, 38988. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012, 40, e169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef]
- Reimand, J.; Bader, G.D. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol. Syst. Biol. 2013, 9, 637. [Google Scholar] [CrossRef]
- Hou, J.P.; Ma, J. DawnRank: Discovering personalized driver genes in cancer. Genome Med. 2014, 6, 56. [Google Scholar] [CrossRef]
- Horn, H.; Lawrence, M.S.; Chouinard, C.R.; Shrestha, Y.; Hu, J.X.; Worstell, E.; Shea, E.; Ilic, N.; Kim, E.; Kamburov, A.; et al. NetSig: Network-based discovery from cancer genomes. Nat. Methods 2018, 15, 61–66. [Google Scholar] [CrossRef]
- Pichot, C.S.; Arvanitis, C.; Hartig, S.M.; Jensen, S.A.; Bechill, J.; Marzouk, S.; Yu, J.; Frost, J.A.; Corey, S.J. Cdc42-interacting protein 4 promotes breast cancer cell invasion and formation of invadopodia through activation of N-WASp. Cancer Res. 2010, 70, 8347–8356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, R.; Lv, L.; Zhang, S.; Zhang, F.; Jiang, Y. Functional mechanism and clinical implications of MicroRNA-423 in human cancers. Cancer Med. 2020, 9, 9036–9051. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Peng, L.; Yang, J.; Sang, H.; Jin, D.; Li, X.; Chen, M.; Zhang, W.; Dang, Y.; Zhang, G. Exosomal transfer of miR-15b-3p enhances tumorigenesis and malignant transformation through the DYNLT1/Caspase-3/Caspase-9 signaling pathway in gastric cancer. J. Exp. Clin. Cancer Res. 2020, 39, 32. [Google Scholar] [CrossRef] [Green Version]
- Pham, V.V.H.; Liu, L.; Bracken, C.P.; Goodall, G.J.; Long, Q.; Li, J.; Le, T.D. CBNA: A control theory based method for identifying coding and non-coding cancer drivers. PLoS Comput. Biol. 2019, 15, e1007538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, M.S.; Pham, V.V.H.; Le, T.D. NIBNA: A network-based node importance approach for identifying breast cancer drivers. Bioinformatics 2021, 37, 2521–2528. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Lizio, M.; Harshbarger, J.; Abugessaisa, I.; Noguchi, S.; Kondo, A.; Severin, J.; Mungall, C.; Arenillas, D.; Mathelier, A.; Medvedeva, Y.A.; et al. Update of the FANTOM web resource: High resolution transcriptome of diverse cell types in mammals. Nucleic Acids Res. 2016, 45, D737–D743. [Google Scholar] [CrossRef] [PubMed]
- Vinayagam, A.; Vinayagam, A.; Stelzl, U.; Foulle, R.; Plassmann, S.; Zenkner, M.; Timm, J.; Assmus, H.E.; Andrade-Navarro, M.A.; Wanker, E.E. A directed protein interaction network for investigating intracellular signal transduction. Sci. Signal. 2011, 4, rs8. [Google Scholar] [CrossRef]
- Chou, C.H.; Chang, N.W.; Shrestha, S.; Hsu, S.D.; Lin, Y.L.; Lee, W.H.; Yang, C.D.; Hong, H.C.; Wei, T.Y.; Tu, S.J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44, D239–D247. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Paraskevopoulou, M.D.; Karagkouni, D.; Georgakilas, G.; Vergoulis, T.; Kanellos, I.; Anastasopoulos, I.L.; Maniou, S.; Karathanou, K.; Kalfakakou, D.; et al. DIANA-TarBase v7.0: Indexing more than half a million experimentally supported miRNA: MRNA interactions. Nucleic Acids Res. 2015, 43, D153–D159. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, M.; Qiu, C.; Cui, Q. TransmiR: A transcription factor–microRNA regulation database. Nucleic Acids Res. 2010, 38, D119–D122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckmann, N.D.; Comella, P.H.; Cheng, E.; Lepow, L.; Beckmann, A.G.; Tyler, S.R.; Mouskas, K.; Simons, N.W.; Hoffman, G.E.; Francoeur, N.J.; et al. Downregulation of exhausted cytotoxic T cells in gene expression networks of multisystem inflammatory syndrome in children. Nat. Commun. 2021, 12, 4854. [Google Scholar] [CrossRef] [PubMed]
- Sabidussi, G. The centrality index of a graph. Psychometrika 1966, 31, 581–603. [Google Scholar] [CrossRef]
- Freeman, L.C. Centrality in social networks conceptual clarification. Soc. Netw. 1978, 1, 215–239. [Google Scholar] [CrossRef] [Green Version]
- Bonacich, P.; Lloyd, P. Eigenvector-like measures of centrality for asymmet- ric relations. Soc. Netw. 2001, 23, 191–201. [Google Scholar] [CrossRef]
- Lü, L.; Zhou, T.; Zhang, Q.M.; Stanley, H.E. The H-index of a network node and its relation to degree and coreness. Nat. Commun. 2016, 7, 10168. [Google Scholar] [CrossRef] [Green Version]
- Colizza, V.; Flammini, A.; Serrano, M.A.; Vespignani, A. Detecting rich-club ordering in complex networks. Nat. Phys. 2006, 364, 110–115. [Google Scholar] [CrossRef]
- Sun, H.L.; Chen, D.B.; He, J.L.; Ch’ng, E. A voting approach to uncover multiple influential spreaders on weighted networks. Phys. A 2019, 519, 303–312. [Google Scholar] [CrossRef]
- Zhang, J.X.; Chen, D.B.; Dong, Q.; Zhao, Z.D. Identifying a set of influential spreaders in complex networks. Sci. Rep. 2016, 6, 27823. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Panda, A. Identifying influential nodes in weighted complex networks using an improved WVoteRank approach. Appl. Intell. 2022, 52, 1838–1852. [Google Scholar] [CrossRef]
- Liu, P.F.; Li, L.J.; Fang, S.Y.; Yao, K.Y. Identifying influential nodes in social networks: A voting approach. Chaos Solitons Fractals 2021, 152, 111309. [Google Scholar] [CrossRef]
- Sun, Y.Z.; Norick, B.; Han, J.W.; Yan, X.F.; Yu, P.S.; Yu, X. Integrating meta-path selection with user-guided object clustering in heterogeneous information networks. In Proceedings of the 18th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD ’12), Beijing, China, 12–16 August 2012. [Google Scholar]
- Chang, S.; Han, W.; Tang, J.; Qi, G.; Aggarwal, C.C.; Huang, T.S. Heterogeneous Network Embedding via Deep Architectures. In Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Hilton, Sydney, 10–13 August 2015. [Google Scholar]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. Cosmic: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.W.; Chen, Y.; Chen, S.; Wang, X. OncomiR: An online resource for exploring pan-cancer microRNA dysregulation. Bioinformatics 2018, 34, 713–715. [Google Scholar] [CrossRef] [Green Version]
- Opsahl, T.; Colizza, V.; Panzarasa, P.; Ramasco, J.J. Prominence and control: The weighted rich-club effect. Phys. Rev. Lett. 2008, 101, 168702. [Google Scholar] [CrossRef] [Green Version]
- Lü, L.Y.; Chen, D.B.; Ren, X.L.; Zhang, Q.M.; Zhou, T. Vital nodes identification in complex networks. Phys. Rev. 2016, 650, 1–63. [Google Scholar]
- Newman, M.E.J. Scientific collaboration networks. ii. Shortest paths, weighted networks, and centrality. Phys. Rev. E 2001, 64, 016132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, U. A faster algorithm for betweenness centrality. J. Math. Sociol. 2001, 25, 163–177. [Google Scholar] [CrossRef]
- Wang, F.; Gao, Y.; Tang, L.; Ning, K.; Geng, N.; Zhang, H.; Li, Y.; Li, Y.; Liu, F.; Li, F. A novel PAK4-CEBPB-CLDN4 axis involving in breast cancer cell migration and invasion. Biochem. Biophys. Res. Commun. 2019, 511, 404–408. [Google Scholar] [CrossRef]
- Chen, D.; Reierstad, S.; Fang, F.; Bulun, S.E. JunD and JunB integrate prostaglandin E2 activation of breast cancer-associated proximal aromatase promoters. Mol. Endocrinol. 2011, 25, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Giulianelli, S.; Riggio, M.; Guillardoy, T.; Pérez Piñero, C.; Gorostiaga, M.A.; Sequeira, G.; Pataccini, G.; Abascal, M.F.; Toledo, M.F.; Jacobsen, B.M.; et al. FGF2 induces breast cancer growth through ligand-independent activation and recruitment of ERα and PRBΔ4 isoform to MYC regulatory sequences. Int. J. Cancer 2019, 145, 1874–1888. [Google Scholar] [PubMed]
- Kostianets, O.; Antoniuk, S.; Filonenko, V.; Kiyamova, R. Immunohistochemical analysis of medullary breast carcinoma autoantigens in different histological types of breast carcinomas. Diagn. Pathol. 2012, 7, 161. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Sivko, G.S.; DeWille, J.W. Promoter methylation reduces C/EBPdelta (CEBPD) gene expression in the SUM-52PE human breast cancer cell line and in primary breast tumors. Breast Cancer Res. Treat. 2006, 95, 161–170. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, T.; Guo, M.; Li, Y.; Zhang, Q.; Tan, G.; Yu, L.; Tan, Y. FOXA2-Interacting FOXP2 Prevents Epithelial-Mesenchymal Transition of Breast Cancer Cells by Stimulating E-Cadherin and PHF2 Transcription. Front. Oncol. 2021, 11, 605025. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Behbahani, G.D.; Ghahhari, N.M.; Javidi, M.A.; Molan, A.F.; Feizi, N.; Babashah, S. MicroRNA-Mediated Post-Transcriptional Regulation of Epithelial to Mesenchymal Transition in Cancer. Pathol. Oncol. Res. 2017, 23, 1–12. [Google Scholar] [CrossRef]
- Wu, Z.H.; Tao, Z.H.; Zhang, J.; Li, T.; Ni, C.; Xie, J.; Zhang, J.F.; Hu, X.C. MiRNA-21 induces epithelial to mesenchymal transition and gemcitabine resistance via the PTEN/AKT pathway in breast cancer. Tumour Biol. 2016, 37, 7245–7254. [Google Scholar] [CrossRef] [PubMed]
- Van Kampen, J.G.M.; van Hooij, O.; Jansen, C.F.; Smit, F.P.; van Noort, P.I.; Schultz, I.; Schaapveld, R.; Schalken, J.A.; Verhaegh, G.W. miRNA-520f Reverses Epithelial-to-Mesenchymal Transition by Targeting ADAM9 and TGFBR2. Cancer Res. 2017, 77, 2008–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef]
- Cursons, J.; Pillman, K.A.; Scheer, K.G.; Gregory, P.A.; Foroutan, M.; Hediyeh-Zadeh, S.; Toubia, J.; Crampin, E.J.; Goodall, G.J.; Bracken, C.P.; et al. Combinatorial Targeting by MicroRNAs Co-ordinates Post-transcriptional Control of EMT. Cell Syst. 2018, 7, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Pitcher, B.N.; Mardis, E.R.; Davies, S.R.; Friedman, P.N.; Snider, J.E.; Vickery, T.L.; Reed, J.P.; DeSchryver, K.; Singh, B.; et al. PAM50 gene signatures and breast cancer prognosis with adjuvant anthracycline- and taxane-based chemotherapy: Correlative analysis of C9741 (Alliance). NPJ Breast Cancer 2016, 2, 15023. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.L.; Jiang, X.M.; Yi, Y.; E, J.F.; Zhang, N.D.; Luo, X.; Zou, N.; Wei, W.; Liu, Y.Y. Identification of differentially expressed lncRNAs and mRNAs in luminal-B breast cancer by RNA-sequencing. BMC Cancer 2019, 19, 1171. [Google Scholar] [CrossRef]
- Anurag, M.; Zhu, M.; Huang, C.; Vasaikar, S.; Wang, J.; Hoog, J.; Burugu, S.; Gao, D.; Suman, V.; Zhang, X.H.; et al. Immune Checkpoint Profiles in Luminal B Breast Cancer (Alliance). J. Natl. Cancer Inst. 2020, 112, 737–746. [Google Scholar] [CrossRef]
- Badve, S.; Turbin, D.; Thorat, M.A.; Morimiya, A.; Nielsen, T.O.; Perou, C.M.; Dunn, S.; Huntsman, D.G.; Nakshatri, H. FOXA1 expression in breast cancer–correlation with luminal subtype A and survival. Clin. Cancer Res. 2007, 13, 4415–4421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).