PAPE (Prefractionation-Assisted Phosphoprotein Enrichment): A Novel Approach for Phosphoproteomic Analysis of Green Tissues from Plants

Abstract
:
1. Introduction
2. Experimental
2.1. Plant Growth
2.2. Protein Extraction
2.3. Precipitation of Protein Extract
2.4. Phosphoprotein Enrichment (MOAC)
2.5. SDS-PAGE and Phosphoprotein Staining
2.6. In-Solution Digestion
2.7. Mass Spectrometry
2.8. Spectral Data Analysis
3. Results and Discussion
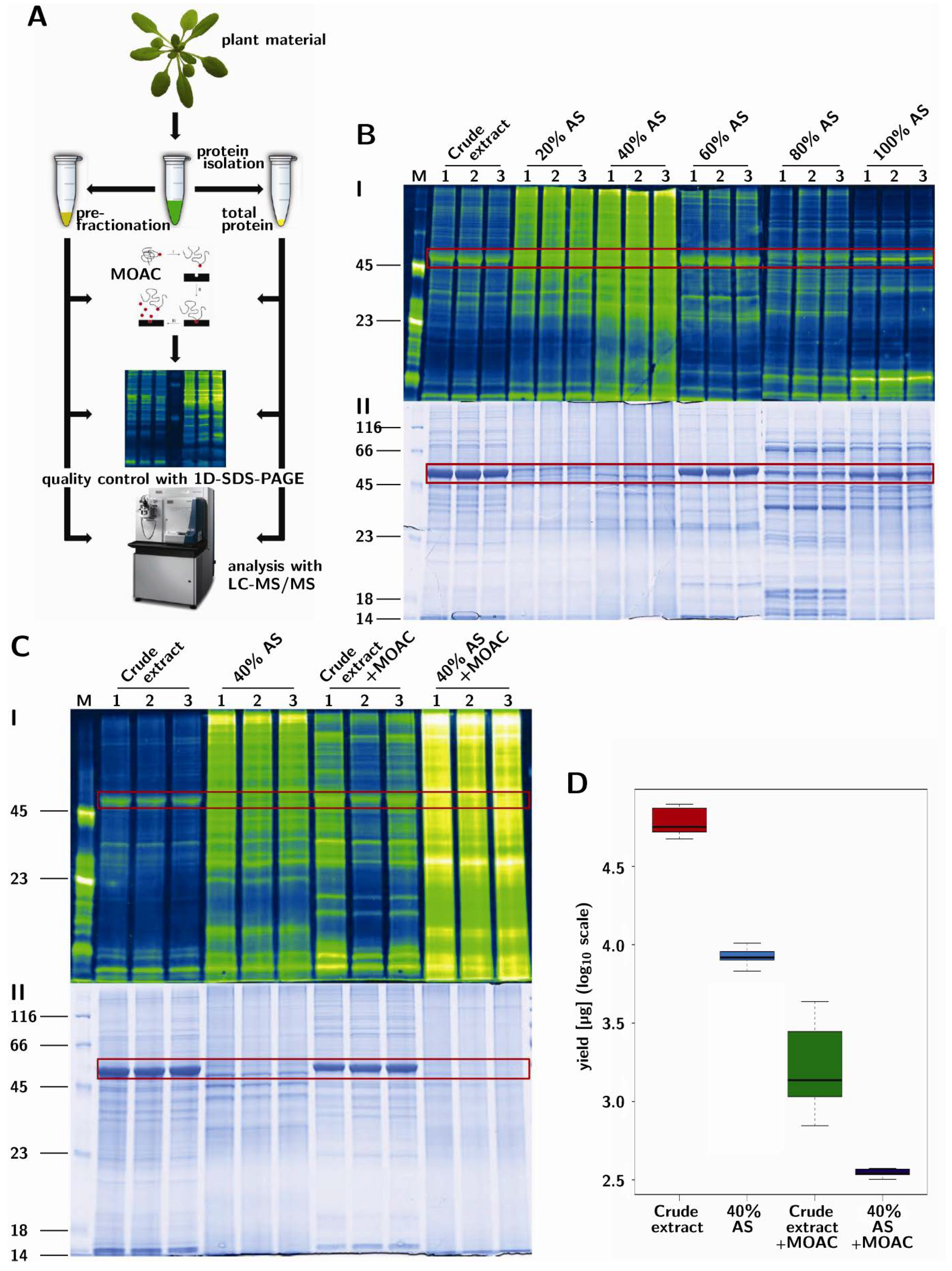
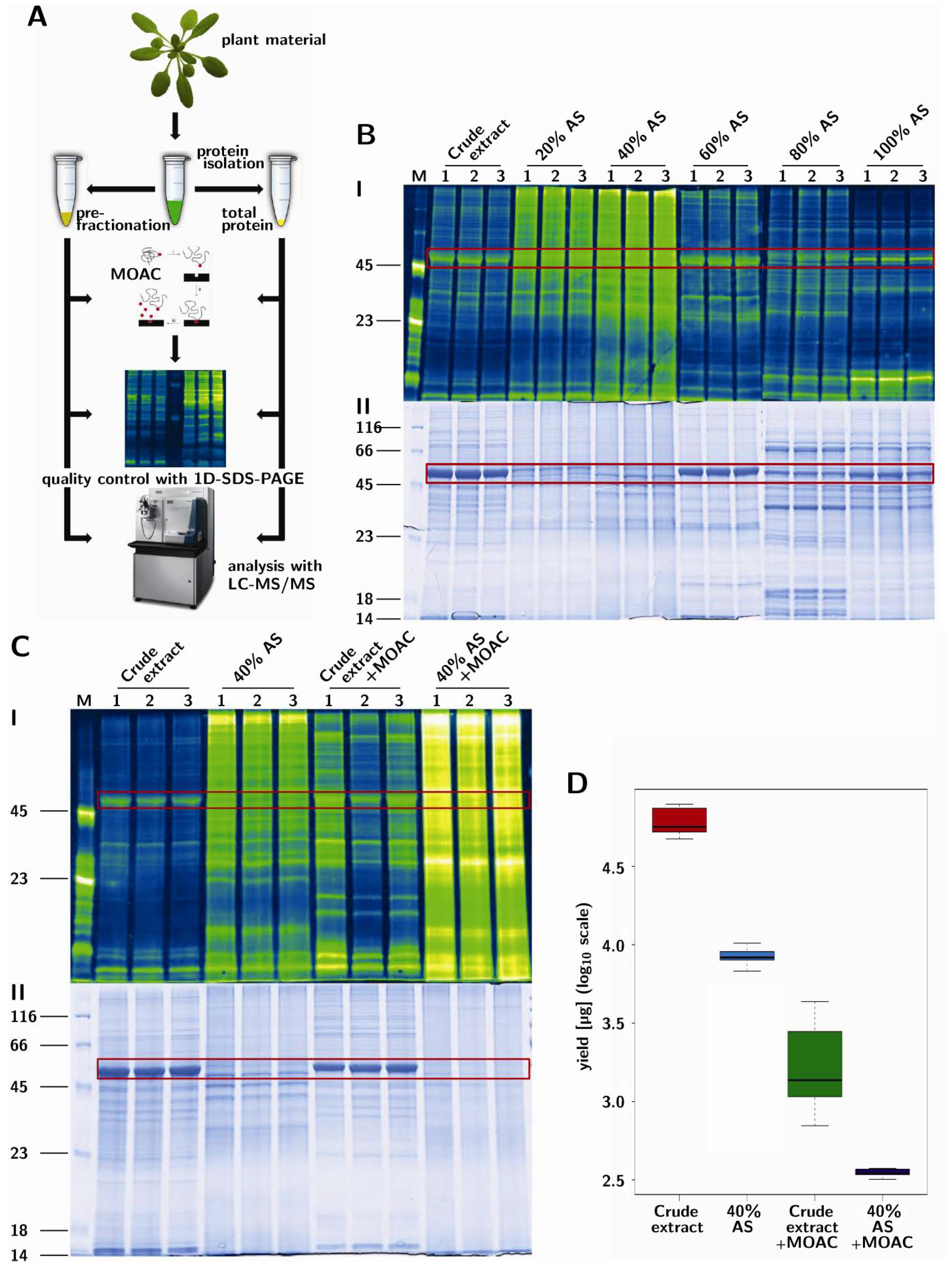
3.1. Prefractionation of Arabidopsis Leaf Proteins
3.2. PAPE: Prefractionation-Assisted Phosphoprotein Enrichment
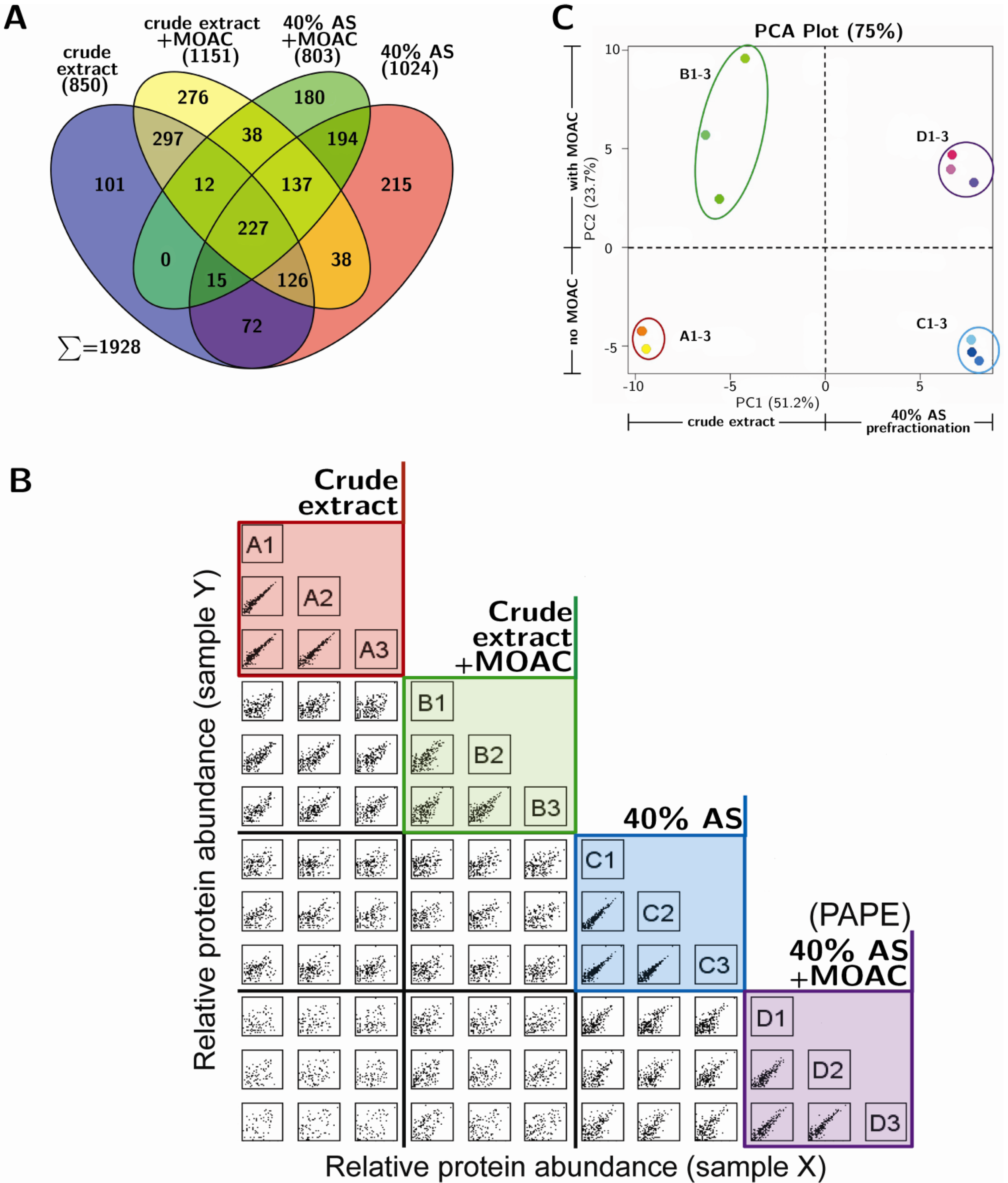
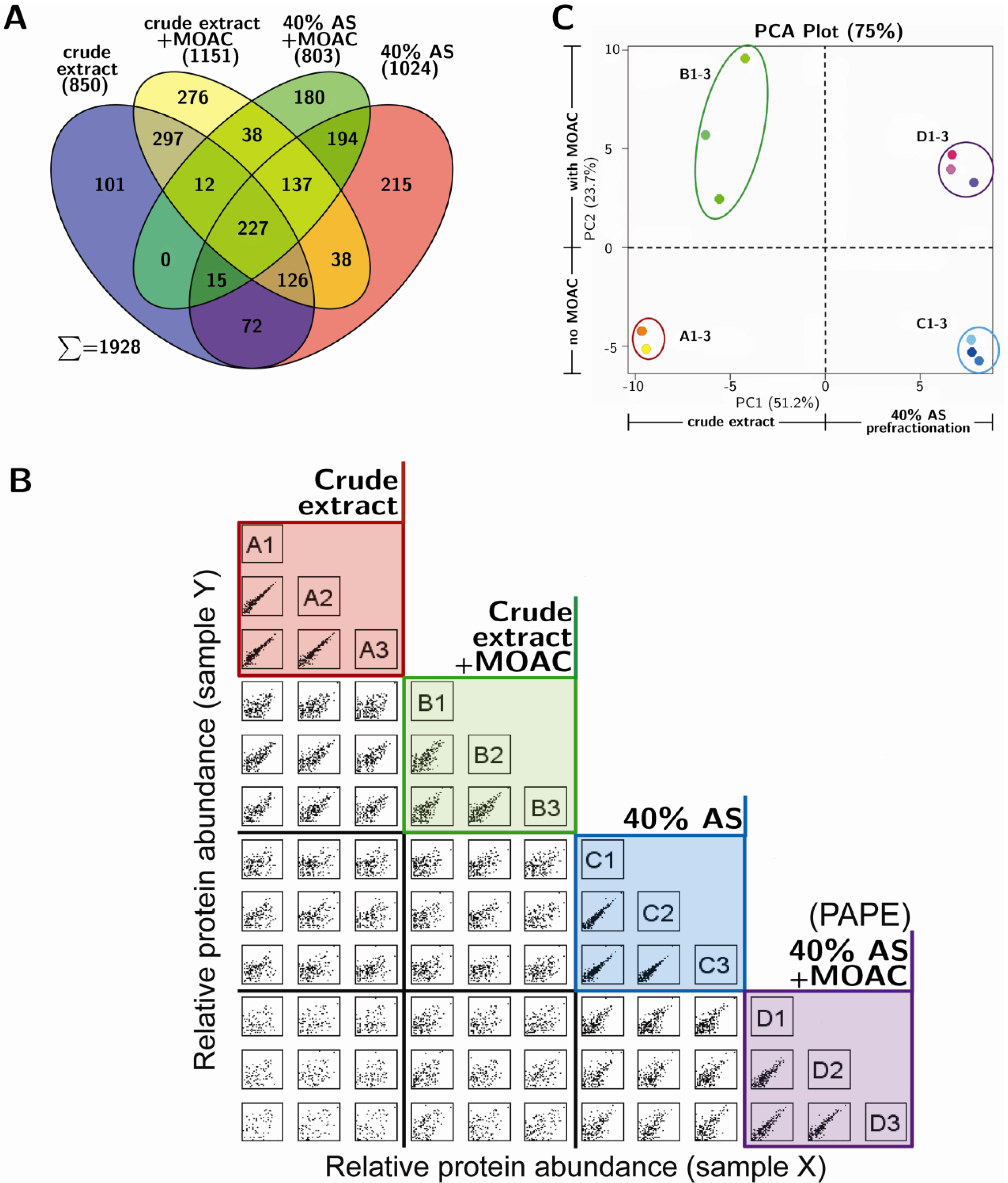
3.3. Reproducibility and Robustness of PAPE on the Basis of Mass Spectrometry Analysis
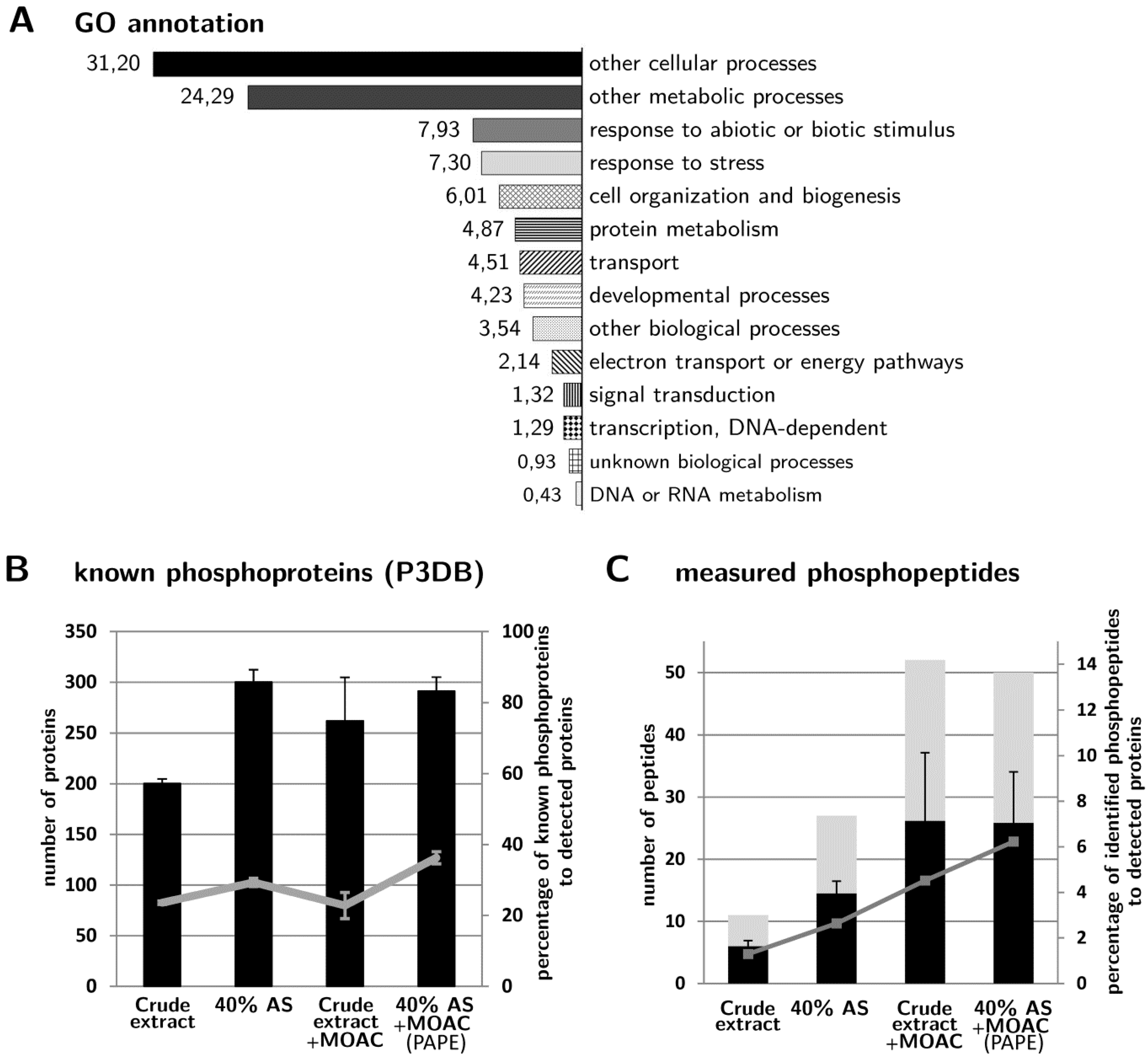
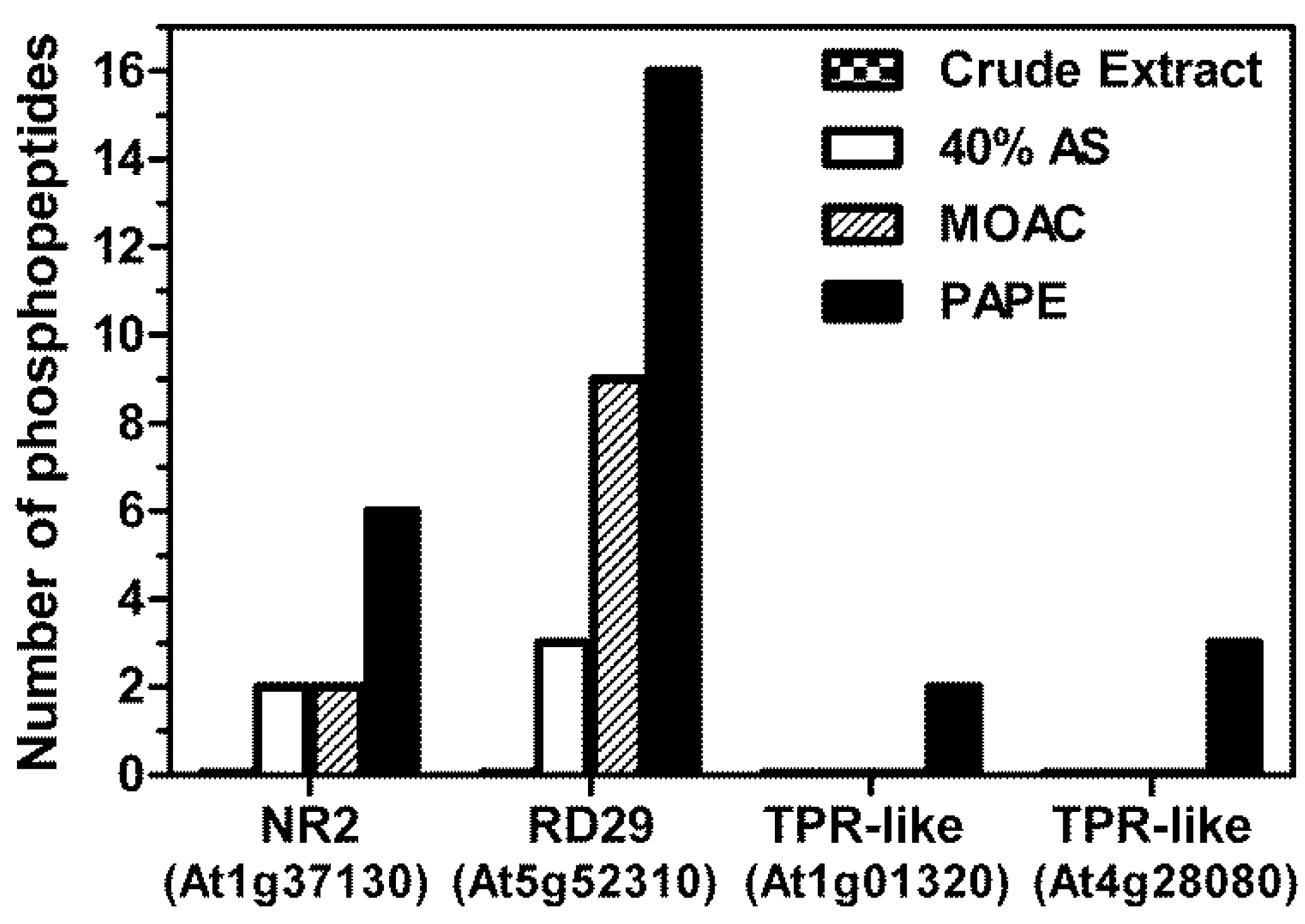
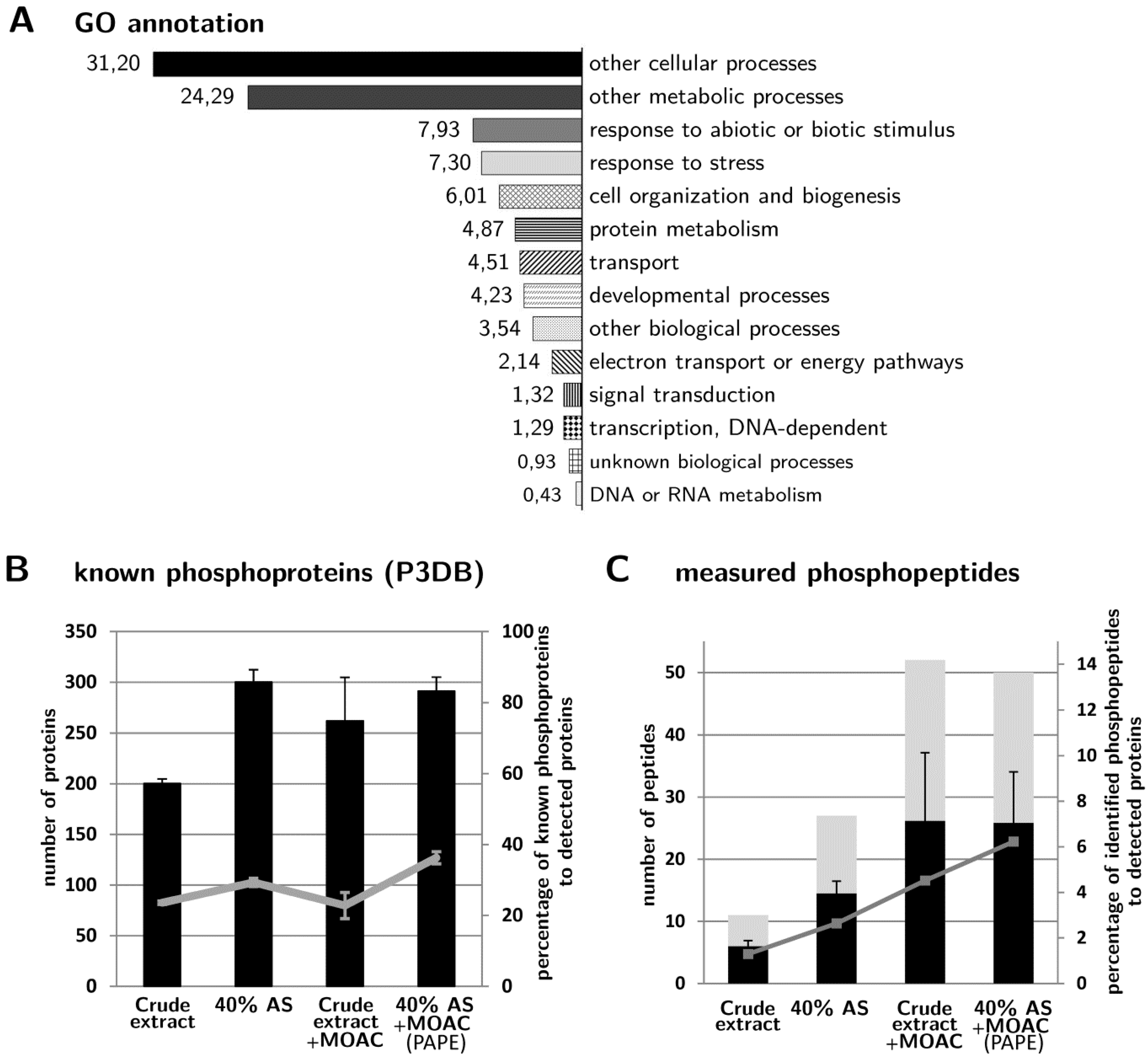
3.4. Validation of Phosphoprotein Enrichment by the PAPE Procedure

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Protein code | Description | Sequence | MH+[Da] | q-Value | PEP | pRS Score | # PSMs | pRS Site Probabilities | |
|---|---|---|---|---|---|---|---|---|---|---|
| Crude extract | 1 | AT1G14010.1 | emp24/gp25L/p24 family/GOLD family protein | SSIVLLILSILSPVTLSIR### | 2,184.20708 | 0.016281 | 0.3789499 | 58 | 11 | S(1): 15.3; S(2): 15.3; S(9): 84.3; S(12): 84.6; T(15): 0.5; S(17): 0.0 |
| 2 | AT2G40840.1 | disproportionating enzyme 2 | VEKPLGVFMNKSDQDDSVVVQFK | 2,689.27768 | 0.021343 | 0.6031149 | 33 | 1 | S(12): 0.4; S(17): 99.6 | |
| 3 | AT2G38280.1 | AMP deaminase, putative/myoadenylate deaminase, putative | SNGHVYVDEIPPGLPRLHTPSEGRASVHGASSIR | 3,832.73672 | 0.022388 | 0.6381906 | 32 | 2 | S(1): 33.1; Y(6): 33.1; T(19): 33.1; S(21): 4.1; S(26): 95.3; S(31): 50.7; S(32): 50.7 | |
| 4 | AT4G38740.1; AT2G21130.1 | rotamase CYP 1/ Cyclophilin-like peptidyl-prolyl cis-trans isomerase family protein | HTGPGILSMANAGANTNGSQFFICTVK | 2,873.30117 | 0.027543 | 0.4591594 | 37 | 3 | T(2): 0.7; S(8): 95.1; T(16): 2.0; S(19): 2.0; T(25): 0.1 | |
| 5 | AT4G23670.1 | polyketide cyclase/dehydrase and lipid transport superfamily protein | ATSGTYVTEVPLKGSAEK### | 1,917.91213 | 0.032722 | 0.4923883 | 48 | 4 | T(2): 24.5; S(3): 24.5; T(5): 24.5; Y(6): 24.5; T(8): 1.9; S(15): 0.0 | |
| 6 | AT1G70200.1 | RNA-binding (RRM/RBD/RNP motifs) family protein | QFTGQSLAFGKVIKQIK | 1,973.05167 | 0.047422 | 0.6522376 | 35 | 12 | T(3): 86.5; S(6): 13.5 | |
| 40% AS | 1 | AT5G56740.1 | histone acetyltransferase of the GNAT family 2 | LSQILVLPSFQGK | 1,509.80133 | 0.011279 | 0.2471017 | 30 | 4 | S(2): 0.4; S(9): 99.6 |
| 2 | AT1G23740.1 | oxidoreductase, zinc-binding dehydrogenase family protein | NAALATTTATTPVLRR | 1,736.90842 | 0.015443 | 0.4098554 | 40 | 1 | T(6): 12.5; T(7): 12.5; T(8): 59.5; T(10): 12.5; T(11): 3.0 | |
| 3 | AT5G52790.1 | CBS domain-containing protein with a domain of unknown function (DUF21) | LLDLLLGKRHSTLLGR### | 1,885.07854 | 0.023892 | 0.3993647 | 51 | 11 | S(11): 1.7; T(12): 98.3 | |
| 4 | AT4G28000.1 | P-loop containing nucleoside triphosphate hydrolases superfamily protein | HTRNLAPGSK | 1,160.55168 | 0.03559 | 0.754117 | 50 | 3 | T(2): 0.0; S(9): 100.0 | |
| 5 | AT1G72150.1 | PATELLIN 1 | SSFVFVSDFRNAPGLGKR | 2,064.01040 | 0.039425 | 0.6264254 | 39 | 1 | S(1): 1.0; S(2): 1.0; S(7): 98.0 | |
| 6 | AT2G04842.1 | threonyl-tRNA synthetase, putative/threonine-tRNA ligase, putative | SRFGGELGTIPVDDLINKINIAVETR### | 3,067.42545 | 0.041246 | 0.6307821 | 38 | 1 | S(1): 100.0; T(9): 100.0; T(25): 100.0 | |
| 7 | AT3G22760.1 | tesmin/TSO1-like CXC domain-containing protein | VIRNSDSIIEVGEDASK### | 1,911.89481 | 0.042639 | 0.8216446 | 52 | 1 | S(5): 0.0; S(7): 0.1; S(16): 99.9 | |
| 8 | AT3G16950.1; AT4G16155.1 | lipoamide dehydrogenase 1/ dihydrolipoyl dehydrogenases | DIIIATGSVPFVPK | 1,536.80143 | 0.043742 | 0.5158506 | 38 | 2 | T(6): 12.3; S(8): 87.7 | |
| Crude extract + MOAC | 1 | AT1G56220.4 | dormancy/auxin associated family protein | HHTFSFRPSSGNDQSEAGSAR### | 2,354.98525 | 0 | 7.2906E-05 | 36 | 16 | T(3): 13.7; S(5): 57.9; S(9): 13.7; S(10): 13.7; S(15): 1.1; S(19): 0.1 |
| 2 | AT2G17410.2 | ARID/BRIGHT DNA-binding domain-containing protein | HSEENQSPHHHANNVMEQDQAAEER | 3,004.19179 | 0 | 0.00011139 | 60 | 9 | S(2): 97.1; S(7): 2.9 | |
| 3 | AT5G52310.1 | low-temperature-responsive protein 78 (LTI78) | MDQTEEPPLNTHQQHPEEVEHHENGATK | 3,342.38857 | 0 | 2.1345E-05 | 36 | 16 | T(4): 96.1; T(11): 3.8; T(27): 0.0 | |
| 4 | AT5G55160.1 | small ubiquitin-like modifier 2 | SATPEEDKKPDQGAHINLK### | 2,237.97500 | 0.000487 | 0.00798287 | 31 | 2 | S(1): 100.0; T(3): 100.0 | |
| 5 | AT2G24270.2 | aldehyde dehydrogenase 11A3 | AGTGLFAEILDGEVYK### | 1,762.82077 | 0.000503 | 0.0115286 | 38 | 3 | T(3): 100.0; Y(15): 0.0 | |
| 6 | AT1G45207.2 | remorin family protein | GWSSERVPLR | 1,266.59882 | 0.00075 | 0.01489954 | 38 | 24 | S(3): 50.0; S(4): 50.0 | |
| 7 | AT1G01100.1; AT5G47700.1 | 60S acidic ribosomal protein family | STVGELACSYAVMILEDEGIAITADK | 2,836.31522 | 0.000976 | 0.01672018 | 48 | 4 | S(1): 25.0; T(2): 25.0; S(9): 25.0; Y(10): 25.0; T(23): 0.0 | |
| 8 | AT4G12420.1 | cupredoxin superfamily protein | RPLTGPAKVATSIINGTYR | 2,175.06754 | 0.002939 | 0.08369295 | 36 | 3 | T(4): 99.1; T(11): 7.4; S(12): 91.8; T(17): 0.9; Y(18): 0.9 | |
| 9 | AT1G74920.1; AT3G48170.1 | aldehyde dehydrogenase 10A8/9 | SPLIVFDDVDLDK | 1,555.73259 | 0.009502 | 0.2584146 | 70 | 2 | S(1): 100.0 | |
| 10 | AT3G28710.1 | ATPase, V0/A0 complex, subunit C/D | AVNITINSIGTELTR### | 1,681.86216 | 0.015828 | 0.3155473 | 37 | 30 | T(5): 0.0; S(8): 77.3; T(11): 11.3; T(14): 11.3 | |
| 11 | AT1G73610.1 | GDSL-like Lipase/Acylhydrolase superfamily protein | SYETIAPQIIENIKAK### | 1,977.93410 | 0.017056 | 0.3870838 | 30 | 18 | S(1): 50.3; Y(2): 50.3; T(4): 99.3 | |
| 12 | AT2G41110.1 | calmodulin 2 | ADQLTDDQISEFK | 1,589.66288 | 0.019757 | 0.513722 | 50 | 5 | T(5): 100.0; S(10): 0.0 | |
| 13 | AT1G70200.1 | RNA-binding (RRM/RBD/RNP motifs) family protein | QFTGQSLAFGKVIKQIK | 1,973.05405 | 0.02041 | 0.4607051 | 40 | 7 | T(3): 50.0; S(6): 50.0 | |
| 14 | AT2G22400.1 | S-adenosyl-L-methionine-dependent methyltransferases superfamily protein | EIRKNQTLER | 1,366.68921 | 0.02499 | 0.5231273 | 38 | 1 | T(7): 100.0 | |
| 15 | AT4G30630.1 | unknown protein | LSESGGLEVPRKPSGERK### | 2,006.01230 | 0.031388 | 0.6183366 | 32 | 1 | S(2): 0.1; S(4): 0.2; S(14): 99.7 | |
| 16 | AT5G64090.1 | unknown protein | ASHDLNPQAILATR | 1,586.76555 | 0.035 | 0.6582299 | 57 | 1 | S(2): 0.0; T(13): 100.0 | |
| 17 | AT1G80380.3 | P-loop containing nucleoside triphosphate hydrolases superfamily protein | GNAGSHDLKLSVETLEALSKLTK### | 2,491.28416 | 0.043032 | 0.7887968 | 36 | 1 | S(5): 0.2; S(11): 0.1; T(14): 0.2; S(19): 9.4; T(22): 90.1 | |
| 40% AS + MOAC (PAPE) | 1 | AT5G52310.1 | low-temperature-responsive protein 78 (LTI78) | SHELDLKNESDIDKDVPTGFDGEPDFLAK | 3,311.49355 | 0 | 0.0027675 | 58 | 8 | S(1): 0.6; S(10): 98.8; T(18): 0.6 |
| 2 | AT1G01320.2 | tetratricopeptide repeat (TPR)-like superfamily protein | STQPSSGNAKTAGETSEEDGLKTDASSVEPPTLSSTVQSEAYHTK### | 4,690.11245 | 0.000811 | 0.04504684 | 41 | 10 | S(1): 2.7; T(2): 2.7; S(5): 2.7; S(6): 2.7; T(11): 2.7; T(15): 17.1; S(16): 17.1; T(23): 17.1; S(26): 17.1; S(27): 17.1; T(32): 0.5; S(34): 0.1; S(35): 0.1; T(36): 0.1; S(39): 0.0; Y(42): 0.0; T(44): 0.0 | |
| 3 | AT3G11130.1; AT3G08530.1 | clathrin, heavy chain | EYSGKVDELIK### | 1,360.63437 | 0.000811 | 0.04561926 | 49 | 6 | Y(2): 99.7; S(3): 0.3 | |
| 4 | ATMG00285.1 | NADH dehydrogenase 2A | KSEFSTEAGSK### | 1,250.52198 | 0.001809 | 0.09635145 | 66 | 3 | S(2): 0.0; S(5): 9.0; T(6): 91.0; S(10): 0.0 | |
| 5 | AT1G20620.1 | catalase 3 | MDPYKYRPSSAYNAPFYTTNGGAPVSNNISSLTIGER | 4,118.89516 | 0.002241 | 0.04873965 | 47 | 14 | Y(4): 2.0; Y(6): 2.0; S(9): 15.6; S(10): 15.6; Y(12): 15.6; Y(17): 15.6; T(18): 15.6; T(19): 15.6; S(26): 2.0; S(30): 0.3; S(31): 0.1; T(33): 0.0 | |
| 6 | AT3G18780.2 | actin 2 | AEADDIQPIVCDNGTGMVKAGFAGDDAPR### | 3,070.31753 | 0.00444 | 0.09388046 | 45 | 2 | T(15): 100.0 | |
| 7 | AT5G09810.1; AT2G37620.1 | actin 1/7 | ADGEDIQPLVCDNGTGMVKAGFAGDDAPR### | 3,056.32534 | 0.005154 | 0.1148702 | 36 | 5 | T(15): 100.0 | |
| 8 | AT5G56180.1 | actin-related protein 8 | TVVLTGGSACLPGLSER### | 1,796.85564 | 0.005762 | 0.1187534 | 68 | 2 | T(1): 0.0; T(5): 0.0; S(8): 0.1; S(15): 99.8 | |
| 9 | AT1G49240.1 | actin 8 | ADADDIQPIVCDNGTGMVKAGFAGDDAPR### | 3,056.32534 | 0.008319 | 0.1784335 | 36 | 3 | T(15): 100.0 | |
| 10 | AT3G02830.1 | zinc finger protein 1 | NKAGIAGRVSLNMLGYPLR | 2,110.10227 | 0.016331 | 0.3097203 | 47 | 1 | S(10): 100.0; Y(16): 0.0 | |
| 11 | AT1G64790.1 | ILITYHIA | SPIVSAAAFENLVK | 1,525.75934 | 0.017784 | 0.3293382 | 48 | 5 | S(1): 10.3; S(5): 89.7 | |
| 12 | AT4G38770.1 | proline-rich protein 4 | KEVPPPVPVYKPPPK### | 1,751.95328 | 0.026158 | 0.4063287 | 34 | 1 | Y(10): 100.0 | |
| 13 | AT4G31120.2 | SHK1 binding protein 1 | DVHLGIEPTTATPNMFSW### | 2,095.92093 | 0.031227 | 0.430492 | 31 | 1 | T(9): 0.5; T(10): 0.5; T(12): 1.4; S(17): 97.6 | |
| 14 | AT5G16330.1 | NC domain-containing protein-related | RGTCTIAPSDPCDEVISR### | 2,193.86269 | 0.039343 | 0.684857 | 64 | 3 | T(3): 5.3; T(5): 0.4; S(9): 94.3; S(17): 100.0 |
4. Conclusions
Supplementary Materials
Supplementary File 1Supplementary File 2Supplementary File 3Author Contributions
Acknowledgments
Conflicts of Interest
References and Notes
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis information resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2011, 40, D1202–D1210. [Google Scholar]
- Richards, C.L.; Rosas, U.; Banta, J.; Bhambhra, N.; Purugganan, M.D. Genome-wide patterns of Arabidopsis gene expression in nature. PLoS Genet. 2012, 8, e1002662. [Google Scholar] [CrossRef]
- Snoek, L.B.; Terpstra, I.R.; Dekter, R.; van den Ackerveken, G.; Peeters, A.J. Genetical genomics reveals large scale genotype-by-environment interactions in Arabidopsis thaliana. Front. Genet. 2013, 3, e317. [Google Scholar]
- Heazlewood, J.L. The green proteome: Challenges in plant proteomics. Front. Plant Sci. 2011, 2, e6. [Google Scholar] [CrossRef]
- Joshi, H.J.; Hirsch-Hoffmann, M.; Baerenfaller, K.; Gruissem, W.; Baginsky, S.; Schmidt, R.; Schulze, W.X.; Sun, Q.; van Wijk, K.J.; Egelhofer, V.; et al. MASCP Gator: An aggregation portal for the visualization of Arabidopsis proteomics data. Plant Physiol. 2011, 155, 259–270. [Google Scholar] [CrossRef]
- Petricka, J.J.; Schauer, M.A.; Megraw, M.; Breakfield, N.W.; Thompson, J.W.; Georgiev, S.; Soderblom, E.J.; Ohler, U.; Moseley, M.A.; Grossniklaus, U.; et al. The protein expression landscape of the Arabidopsis root. Proc. Natl. Acad. Sci. USA 2012, 109, 6811–6818. [Google Scholar] [CrossRef]
- Sakurai, T.; Yamada, Y.; Sawada, Y.; Matsuda, F.; Akiyama, K.; Shinozaki, K.; Hirai, M.Y.; Saito, K. PRIMe Update: Innovative content for plant metabolomics and integration of gene expression and metabolite accumulation. Plant Cell Physiol. 2013, 54, e5. [Google Scholar] [CrossRef]
- Nakagami, H.; Sugiyama, N.; Ishihama, Y.; Shirasu, K. Shotguns in the front line: Phosphoproteomics in plants. Plant Cell Physiol. 2012, 53, 118–124. [Google Scholar] [CrossRef]
- Guo, M.; Huang, B.X. Integration of phosphoproteomic, chemical, and biological strategies for the functional analysis of targeted protein phosphorylation. Proteomics 2013, 13, 424–437. [Google Scholar] [CrossRef]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002, 27, 514–520. [Google Scholar] [CrossRef]
- Wang, D.; Harper, J.F.; Gribskov, M. Systematic trans-genomic comparison of protein kinases between Arabidopsis and Saccharomyces cerevisiae. Plant Physiol. 2003, 132, 2152–2165. [Google Scholar] [CrossRef]
- De la Fuente van Bentem, S.; Roitinger, E.; Anrather, D.; Csaszar, E.; Hirt, H. Phosphoproteomics as a tool to unravel plant regulatory mechanisms. Physiol. Plant. 2006, 126, 110–119. [Google Scholar] [CrossRef]
- Hunter, T. Signaling—2000 and beyond. Cell 2000, 100, 113–127. [Google Scholar] [CrossRef]
- Herskowitz, I. MAP kinase pathways in yeast: For mating and more. Cell 1995, 80, 187–197. [Google Scholar] [CrossRef]
- Hamel, L.P.; Nicole, M.C.; Sritubtim, S.; Morency, M.J.; Ellis, M.; Ehlting, J.; Beaudoin, N.; Barbazuk, B.; Klessig, D.; Lee, J.; et al. Ancient signals: Comparative genomics of plant MAPK and MAPKK gene families. Trends Plant Sci. 2006, 11, 192–198. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, S. MAPK cascades in plant disease resistance signaling. Annu. Rev. Phytopathol. 2013, 51, 245–266. [Google Scholar] [CrossRef]
- Di Palma, S.; Zoumaro-Djayoon, A.; Peng, M.; Post, H.; Preisinger, C.; Munoz, J.; Heck, A.J. Finding the same needles in the haystack? A comparison of phosphotyrosine peptides enriched by immuno-affinity precipitation and metal-based affinity chromatography. J. Proteomics 2013, 91, 331–337. [Google Scholar] [CrossRef]
- Yue, X.S.; Hummon, A.B. Combination of multistep IMAC enrichment with high-pH reverse phase separation for in-depth phosphoproteomic profiling. J. Proteome Res. 2013, 12, 4176–4186. [Google Scholar] [CrossRef]
- Wolschin, F.; Wienkoop, S.; Weckwerth, W. Enrichment of phosphorylated proteins and peptides from complex mixtures using metal oxide/hydroxide affinity chromatography (MOAC). Proteomics 2005, 5, 4389–4397. [Google Scholar] [CrossRef]
- Güzel, Y.; Rainer, M.; Mirza, M.R.; Messner, C.B.; Bonn, G.K. Highly selective recovery of phosphopeptides using trypsin-assisted digestion of precipitated lanthanide-phosphoprotein complexes. Analyst 2013, 138, 2897–2905. [Google Scholar] [CrossRef]
- Fila, J.; Honys, D. Enrichment techniques employed in phosphoproteomics. Amino Acids 2011, 43, 1025–1047. [Google Scholar] [CrossRef]
- Cellar, N.A.; Kuppannan, K.; Langhorst, M.L.; Ni, W.; Xu, P.; Young, S.A. Cross species applicability of abundant protein depletion columns for ribulose-1,5-bisphosphate carboxylase/ oxygenase. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 861, 29–39. [Google Scholar] [CrossRef]
- Xi, J.; Wang, X.; Li, S.; Zhou, X.; Yue, L.; Fan, J.; Hao, D. Polyethylene glycol fractionation improved detection of low-abundant proteins by two-dimensional electrophoresis analysis of plant proteome. Phytochemistry 2006, 67, 2341–2348. [Google Scholar] [CrossRef]
- Aryal, U.K.; Krochko, J.E.; Ross, A.R. Identification of phosphoproteins in Arabidopsis thaliana leaves using polyethylene glycol fractionation, immobilized metal-ion affinity chromatography, two-dimensional gel electrophoresis and mass spectrometry. J. Proteome Res. 2011, 11, 425–437. [Google Scholar] [CrossRef]
- Widjaja, I.; Naumann, K.; Roth, U.; Wolf, N.; Mackey, D.; Dangl, J.L.; Scheel, D.; Lee, J. Combining subproteome enrichment and Rubisco depletion enables identification of low abundance proteins differentially regulated during plant defense. Proteomics 2009, 9, 138–147. [Google Scholar] [CrossRef]
- Lassowskat, I. Optimisation, Extension and application of metal-oxide affinity chromatography for Proteome analysis of Arabidopsis thaliana. Diplom (Master) Thesis, Martin-Luther-Universität Halle-Wittenberg, Halle/Saale, Germany, 2008. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Agrawal, G.K.; Thelen, J.J. Development of a simplified, economical polyacrylamide gel staining protocol for phosphoproteins. Proteomics 2005, 5, 4684–4688. [Google Scholar] [CrossRef]
- Taverner, T.; Karpievitch, Y.V.; Polpitiya, A.D.; Brown, J.N.; Dabney, A.R.; Anderson, G.A.; Smith, R.D. DanteR: An extensible R-based tool for quantitative analysis of -omics data. Bioinformatics 2012, 28, 2404–2406. [Google Scholar] [CrossRef]
- The Arabidopsis information resource. Available online: http://arabidopsis.org/tools/bulk/go/index.jsp (accessed on 1 February 2013).
- Vizcaino, J.A.; Cote, R.G.; Csordas, A.; Dianes, J.A.; Fabregat, A.; Foster, J.M.; Griss, J.; Alpi, E.; Birim, M.; Contell, J.; et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: Status in 2013. Nucleic Acids Res. 2013, 41, D1063–D1069. [Google Scholar] [CrossRef]
- Görg, A.; Weiss, W.; Dunn, M.J. Current two-dimensional electrophoresis technology for proteomics. Proteomics 2004, 4, 3665–3685. [Google Scholar] [CrossRef]
- Pace, C.N.; Trevino, S.; Prabhakaran, E.; Scholtz, J.M. Protein structure, stability and solubility in water and other solvents. Philos. Trans. R Soc. Lond. B Biol. Sci. 2004, 359, 1225–1235. [Google Scholar] [CrossRef]
- Stensballe, A.; Andersen, S.; Jensen, O.N. Characterization of phosphoproteins from electrophoretic gels by nanoscale Fe(III) affinity chromatography with off-line mass spectrometry analysis. Proteomics 2001, 1, 207–222. [Google Scholar] [CrossRef]
- Gao, J.; Agrawal, G.K.; Thelen, J.J.; Xu, D. P3DB: A plant protein phosphorylation database. Nucleic Acids Res. 2009, 37, D960–D962. [Google Scholar] [CrossRef]
- Yao, Q.; Bollinger, C.; Gao, J.; Xu, D.; Thelen, J.J. P(3)DB: An integrated database for plant protein phosphorylation. Front. Plant Sci. 2012, 3, e206. [Google Scholar]
- Durek, P.; Schmidt, R.; Heazlewood, J.L.; Jones, A.; MacLean, D.; Nagel, A.; Kersten, B.; Schulze, W.X. PhosPhAt: The Arabidopsis thaliana phosphorylation site database. An update. Nucleic Acids Res. 2009, 38, D828–D834. [Google Scholar]
- Heazlewood, J.L.; Durek, P.; Hummel, J.; Selbig, J.; Weckwerth, W.; Walther, D.; Schulze, W.X. PhosPhAt: A database of phosphorylation sites in Arabidopsis thaliana and a plant-specific phosphorylation site predictor. Nucleic Acids Res. 2008, 36, D1015–D1021. [Google Scholar]
- Boersema, P.J.; Mohammed, S.; Heck, A.J. Phosphopeptide fragmentation and analysis by mass spectrometry. J. Mass Spectrom. 2009, 44, 861–878. [Google Scholar] [CrossRef]
- Thingholm, T.E.; Jensen, O.N.; Larsen, M.R. Enrichment and separation of mono- and multiply phosphorylated peptides using sequential elution from IMAC prior to mass spectrometric analysis. Methods Mol. Biol. 2009, 527, 67–78. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lassowskat, I.; Naumann, K.; Lee, J.; Scheel, D. PAPE (Prefractionation-Assisted Phosphoprotein Enrichment): A Novel Approach for Phosphoproteomic Analysis of Green Tissues from Plants. Proteomes 2013, 1, 254-274. https://doi.org/10.3390/proteomes1030254
Lassowskat I, Naumann K, Lee J, Scheel D. PAPE (Prefractionation-Assisted Phosphoprotein Enrichment): A Novel Approach for Phosphoproteomic Analysis of Green Tissues from Plants. Proteomes. 2013; 1(3):254-274. https://doi.org/10.3390/proteomes1030254
Chicago/Turabian StyleLassowskat, Ines, Kai Naumann, Justin Lee, and Dierk Scheel. 2013. "PAPE (Prefractionation-Assisted Phosphoprotein Enrichment): A Novel Approach for Phosphoproteomic Analysis of Green Tissues from Plants" Proteomes 1, no. 3: 254-274. https://doi.org/10.3390/proteomes1030254