Protein-Protein Interactions: Gene Acronym Redundancies and Current Limitations Precluding Automated Data Integration


Abstract
:1. Introduction

{kind=link}
{kind=link}
| Human interactome [reference] publication year | Description |
|---|---|
| 375,000 [5] 2005 | These authors used literature-mining algorithms and then estimated the number of protein interactions assuming 25,000 human genes. |
| 154,000–369,000 [6] 2006 | The authors quoted that their estimation includes protein complexes. |
| 650,000 [7] 2008 | This estimation relies on data retrieved from Y2H a experiments and database searches. |
| 130,000 [8] 2009 | This number of protein interactions exclusively considered binary interactions. |
| 13,217 b [9] 2012 | This estimation considered the longest protein isoform c of 20,846 human protein sequences. The size of the interactome was estimated using computational methods based on structural inference. The authors claimed that this estimation also includes self-interactions. |
2. Experimental
2.1. Human Gene Consensus Sequences and Acronyms
2.2. Human Protein Consensus Sequences and Acronyms
2.3. Database Searches and Protein Networking
2.4. Gene Redundancy
3. Results and Discussion
3.1. Evidence for Erroneous Protein-Protein Interactions after Database Searches
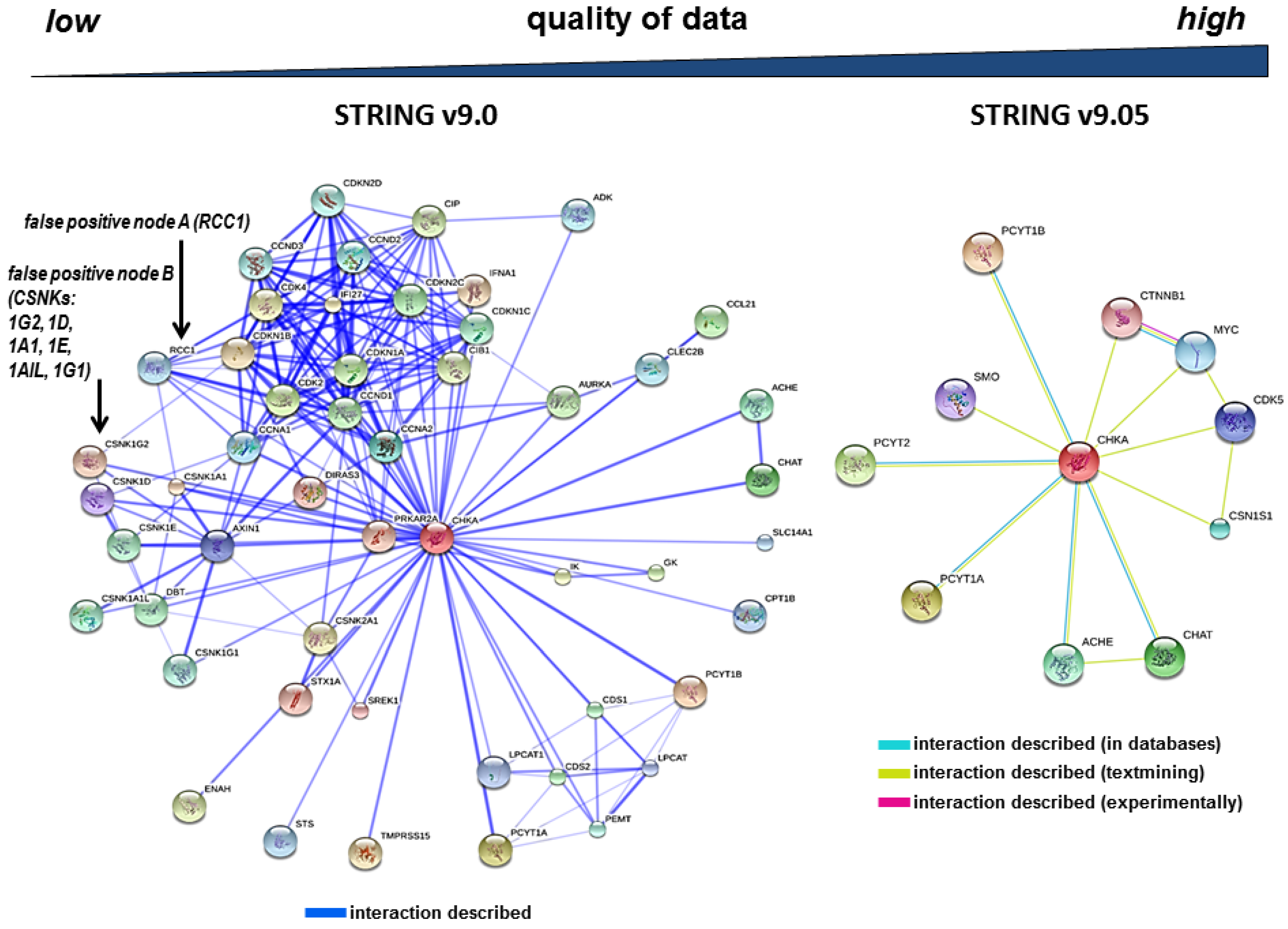
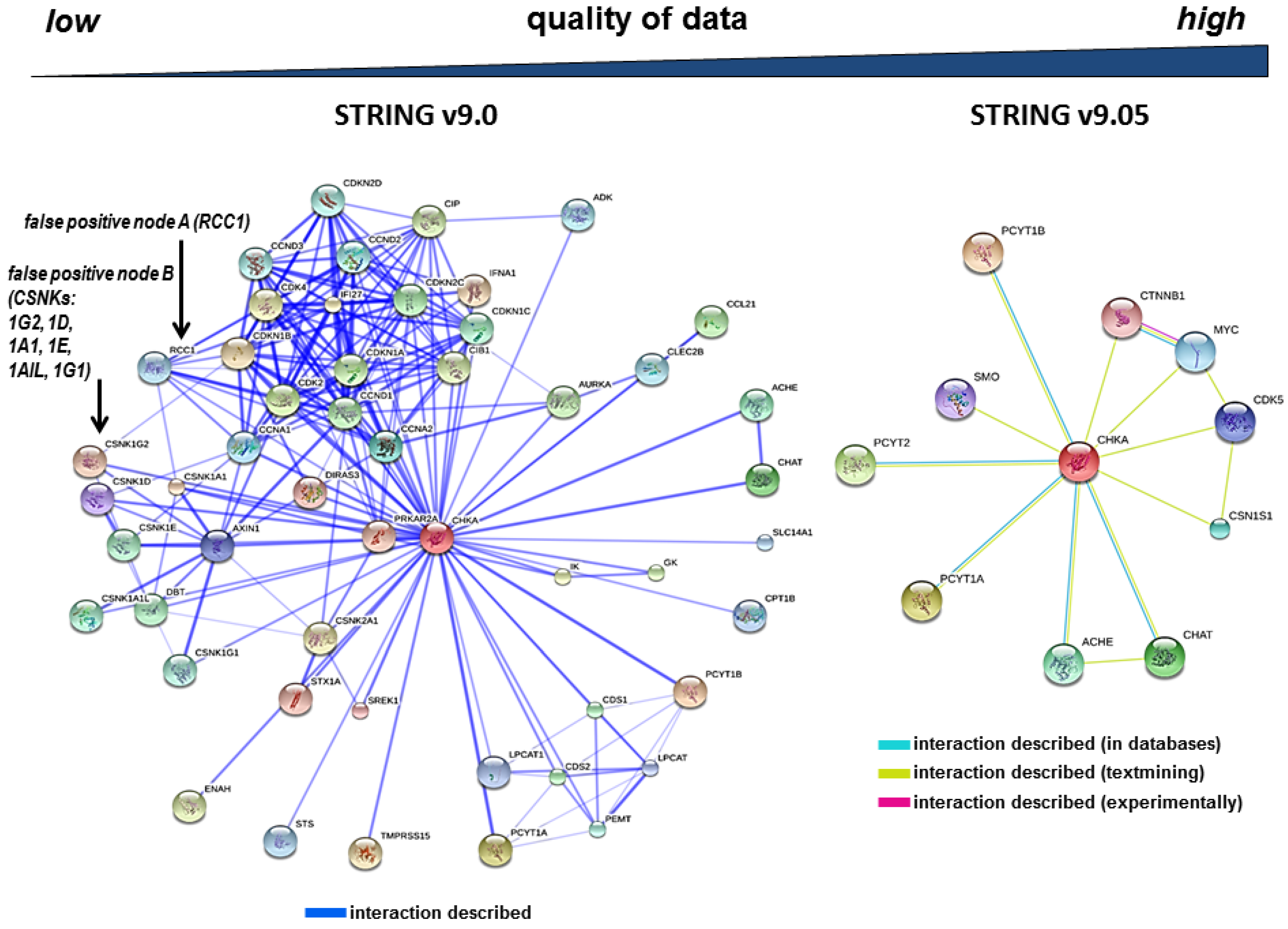
3.2. The Redundancy of Gene Acronyms
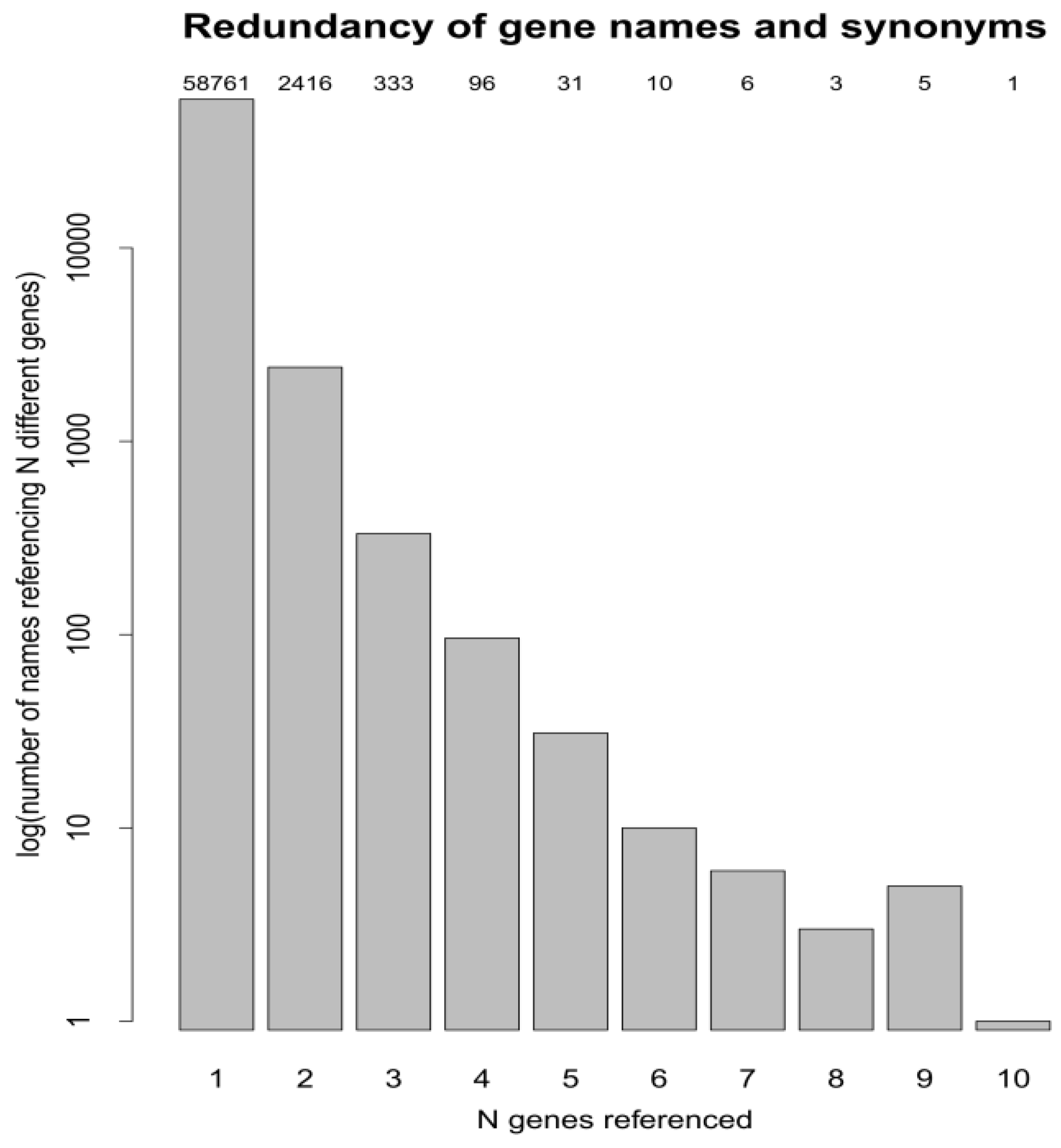
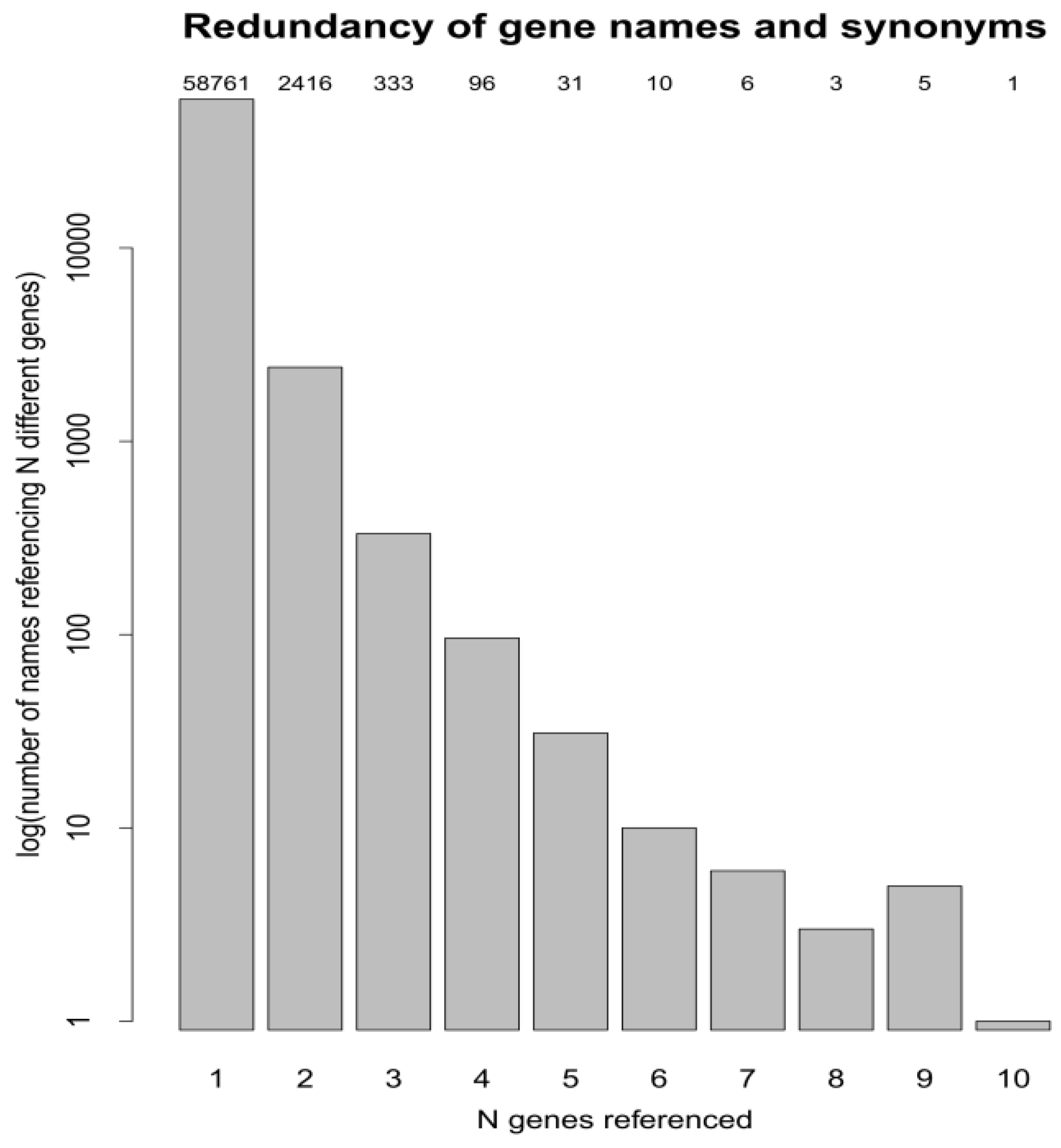
3.3. Protein Accessions, Protein Entries and Database Searches
3.4. Database Searches Fail to Include Dynamic Changes on Protein Networks
| Year Ref. | Experimental model used | HUMAN GENE ACRONYM HUGO acronym (bold), other synonyms | Human Swiss-Prot accession, entries | Protein description and synonyms | Detection method | Additional information |
|---|---|---|---|---|---|---|
| 1998 [46] | in vitro: Y2H, PSEN as bait and human brain cDNA. in vitro: COS-7 cells. | PSEN1,FAD, PS1, S182, PS-1, AD3 | P49768 PSN1_HUMAN | Presenilin-1, Protein S182. | Y2H and co-precipitation followed by Western blot. | Y2H experiments included in this report could not confirm PSEN2–DREAM interaction. |
| 1998 [46] 2008 [52] | in vitro: Y2H, PSEN as bait and human brain cDNA. in vitro: COS-7 cells. in vitro: SH-SY5Y cells. | PSEN2, AD3L, PS2, STM2, AD4, CMDV1 | P49810 PSN2_HUMAN | Presenilin-2, AD3LP, AD5, E5-1, STM-2. | Co-localization in COS-7 cells and co-precipitation followed by Western blot. Co-transfection, co-precipitation followed by Western blot | Y2H experiments were not able to confirm PSEN2-DREAM interaction. This interaction occurs in a Ca+2-independent manner. |
| 1996 [47] 1999 [53] 2011 [54] | in vitro: CHO cells. in vitro: HEK 293 cells. in vitro: CHO cells. | KCNIP3, CSEN, DREAM, KCHIP3 | Q9Y2W7, CSEN_HUMAN | Calsenilin, A-type potassium channel modulatory protein 3, DRE-antagonist modulator (DREAM), Kv channel-interacting protein 3 (KCHIP3) | Molecular mass from SDS-PAGE gels and Western blot. | Multimeric forms (monomers [47], dimers [47,54] and tetramers [47,53] described). |
| 2000 [53] | in vitro: HEK293, NB69, SK-NMC cells. | CREM, CREM-2, ICER, hCREM-2 | Q03060 CREM_HUMAN | cAMP-responsive element modulator, inducible cAMP early repressor (ICER). | Pull-down using CREM as bait. | DREAM–CREAM protein-protein interaction leads to loss of binding of the transcriptional repressor DREAM to target genes [53]. |
| 2000 [48] | in vitro: Y2H, KCNIP4 as baits. | KCNIP4, CALP, KCHIP4 | Q6PIL6 KCIP4_HUMAN | Kv channel-interacting protein 4 (KChIP4), A-type potassium channel modulatory protein 4, Calsenilin-like protein, Potassium channel-interacting protein 4. | Y2H and co-precipitation. | Y2H cDNA library was constructed from polyA+ RNA extracted from rat brain. |
| 2002 [55] | in vivo: rat brain nuclear extract. in vitro: HEK293, PC12 cells. | CREB1, CREB | P16220 CREB1_HUMAN | Cyclic AMP-responsive element binding protein 1. | Immunoprecipitation from brain nuclear extracts using anti-DREAM antibody. | DREAM–CREB1 protein-protein interaction prevents recruitment of CBP by phospho-CREB and affects CRE-dependent transcription. |
| 2004 [56] | in vivo: thyroid-derived FRTL-5 cells. in vitro: co-immunoprecipitation in CHO cells. | NKX2-1, BCH, BHC, NK-2, NKX2.1, NKX2A, TEBP,TITF1,TTF-1, TTF1 | P43699 NKX21_HUMAN | Homeobox protein Nkx-2.1, Homeobox protein NK-2 homolog A, Thyroid nuclear factor 1, Thyroid transcription factor 1. | Co-precipitation using GST-DREAM as bait in FRTL-5 thyroid-derived cells. | DREAM regulates the expression of the thyroglobulin gene. |
| 2005 [57] 2008 [58] | in vitro: co-incubation query protein (GST-DREAM) and bait protein (6His-VDR). | VDR, NR1/1 | P11473 VDR_HUMAN | Vitamin D3 receptor, 1,25-dihydroxyvitamin D3 receptor, Nuclear receptor subfamily 1 group I member 1. | Pull-down after incubation of Ni-Sepharose beads with a 1:1 protein mixture of GST-DREAM (Δ65-256) and 6His-VDR. | Ca2+ induces dimerization of DREAM and a binding interaction between DREAM and VDR. Chromatin immunoprecipitation showed that DREAM also binds to DNA, acting as a transcriptional regulator on vitamin D and retinoic acid response elements. |
| 2006 [59] | in vitro: Y2H, DREAM as bait. in vitro: co-immunoprecipitation in H4 cells. | CtBP1,CTBP | Q13363 CTBP1_HUMAN | C-terminal-binding protein 1. | Y2H using N-terminus of DREAM as bait and co-precipitation. | DREAM–CTBP may modulate transcriptional repression of c-fos. |
| 2006 [59] | in vitro: Y2H, DREAM as bait. in vitro: co-immunoprecipitation in H4 cells. | CtBP2, ribeye | P56545 CTBP2_HUMAN | C-terminal-binding protein 2. | Y2H using N-terminus of DREAM as bait and co-precipitation. | DREAM–CTBP may modulate transcriptional repression of c-fos. |
| 2007 [60] | in vitro: Y2H, DREAM as bait. in vitro: co-immunoprecipitation in PC12 cells. in vitro: DREAM Ser95 phosphorylation using HEK293. | GRK6, GPRK6 | P43250 GRK6_HUMAN | G protein-coupled receptor kinase 6, G protein-coupled receptor kinase GRK6. | Y2H and confirmed by co-precipitation of PC12 cell extracts and antibodies specific for GRK6. | A mutated DREAM insensitive to Ca+2 was used to preclude potential artifacts in Y2H screening. |
| 2007 [60] | in vitro: Y2H, DREAM as bait. in vitro: co-immunoprecipitation using PC 12 cells. in-vitro: DREAM Ser95 phosphorylation using HEK293. | ADRBK1, BARK1, BETA-AEK1,GRK2 | P25098 ARBK1_HUMAN | Beta-adrenergic receptor kinase 1, G-protein coupled receptor kinase 2. | Co-precipitation of PC12 cell extracts and antibodies specific for GRK6. | May regulate DREAM function through phosphorylation. |
| 2008 [61] | in vitro: Y2H, mouse G3GALT2 as bait and human brain cDNA. in vitro: co-immunoprecipitation in CHO-K1 cells. | B3GALT2, BETA3GALT2, GLCT2, beta3Gal-T2 | O43825 B3GT2_HUMAN | Beta-1,3-galactosyltransferase 2, Beta-1,3-GalTase 2, UDP-galactose:2-acetamido-2-deoxy-D-glucose 3beta-galactosyltransferase 2. | Y2H using N-terminus of GalT2 as bait. | DREAM is involved in the trafficking of glycosyl-transferases to Golgi and endoplasmic reticulum. |
| 2009 [62] | in vivo: thyroid glands from mice. in vitro: co-immunoprecipitation in CHO cells. | TSHR, CHNG1, LGR3, hTHSR-1 | P16473 TSHR_HUMAN | Thyrotropin receptor, Thyroid-stimulating hormone receptor. | Co-immunoprecipitation using mice thyroid protein extracts and cells transfected with hemagglutinin-tagged DREAM. | Activation of cAMP signaling pathway, thyroid enlargement and nodular development. |
| 2010 [63] | in vivo: co-immunoprecipitation using mouse hippocampal extracts. | DLG4, PSD-95, PSD95, SAP-90, SAP90 | P78352 DLG4_HUMAN | Disks large homolog 4, Postsynaptic density protein 95, Synapse-associated protein 90. | Co-immunoprecipitation. | DREAM modulates the function of postsynaptic NMDA receptor, synaptic plasticity, behavioral learning and memory. |
| 2010 [64] | in vivo: co-immunoprecipitation using rat brain extracts. | CACNA1H, Cav3.2 | O95180 CAC1H_HUMAN | Voltage-dependent T-type calcium channel subunit alpha-1H, Low-voltage-activated calcium channel alpha1 3.2 subunit, Voltage-gated calcium channel subunit alpha Cav3.2 | Co-immunoprecipitation. | Rat brain protein extracts. |
| 2010 [64] | in vivo: co-immunoprecipitation using rat brain extracts. | CACNA1I, Cav 3.3, KIAA1120 | Q9P0X4 CAC1I_HUMAN | Voltage-dependent T-type calcium channel subunit alpha-1I, Voltage-gated calcium channel subunit alpha Cav3.3. | Co-immunoprecipitation. | Rat brain protein extracts. |
| 2010 [65] | in vitro: co-immunoprecipitation in HEK293 cells. in vivo: co-immunoprecipitation in rat hippocampus extracts. | GRIN1, GluN1, NR1, NMDAR1, NMDR1 * | Q05586 NMDZ1_HUMAN | Glutamate receptor ionotropic, Glutamate [NMDA] receptor subunit zeta-1, N-methyl-D-aspartate receptor subunit NR1. | Immunoprecipitation from rat hippocampus extracts. | This interaction supports the role of DREAM in learning and memory. |
| 2011 [66] | in vitro: Y2H, DREAM as bait and human brain cDNA. in vitro: co-immunoprecipitation in PC12 and HEK293 cells. | UBE2I, C358BE.1, P18, UBC9 | P63279 UBC9_HUMAN | SUMO-conjugating enzyme UBC9, SUMO-protein ligase, Ubiquitin carrier protein 9 Ubiquitin carrier protein I, Ubiquitin-conjugating enzyme E2, Ubiquitin-protein ligase I, p18. | Y2H and co-immunoprecipitation of PC12 cell protein extracts. | Sumoylation regulates nuclear localization of DREAM. A mutated DREAM insensitive to Ca+2 was used to preclude potential artifacts in Y2H screening. |
| 2011 [54] | in vitro: Y2H, Ca+2 insensitive DREAM as bait and human bone marrow cDNA. in vitro: co-immunoprecipitation in COS-7 cells. | Prdx3,AOP-1, AOP1, HBC189, MER5, PRO1748, SP-22, Prx-III | P30048 PRDX3_HUMAN | Thioredoxin-dependent peroxidereductase, mitochondrial, Antioxidant protein 1, HBC189, Peroxiredoxin III, Peroxiredoxin-3, Protein MER5 homolog. | Y2H and co-immunoprecipitation of CHO cell protein extracts. | Prdx3 is a mitochondrial protein. Unlikely to mediate the regulation of DREAM under basal conditions. The subcellular compartment where the redox regulation of DREAM in vivo takes place not yet characterized. |
| 2012 [67] | in vivo: co-immunoprecipitation using rat brain extracts. | CALM1,CAMI, CPVT4,DD132, PHKD, caM,CALML2 | P62158 CALM_HUMAN | Calmodulin. | Affinity capture followed by mass spectrometric identification of interacting proteins. | In the presence of Ca+2, DREAM binds to calmodulin. A list of proteins potentially binding to DREAM under Ca+2-dependent and independent conditions is included in [67]. |
| 2012 [67] | in vivo: co-immunoprecipitation using rat brain extracts. | PPP3R1, CALNB1, CNB, CNB1 | P63098 CANB1_HUMAN | Calcineurin subunit B type 1, Protein phosphatase 2B regulatory subunit 1, Protein phosphatase 3 regulatory subunit B alpha isoform 1. | Affinity capture and mass spectrometric identification of interacting proteins. | In the absence of Ca+2, DREAM binds to calcineurin subunit-B. A list of proteins potentially binding to DREAM under Ca+2-dependent and independent conditions is included in [67 ]. |
4. Conclusions
Electronic Supplementary Material
Acknowledgments
Conflicts of Interest
References and Notes
- Schuler, G.D.; Boguski, M.S.; Stewart, E.A.; Stein, L.D.; Gyapay, G.; Rice, K.; White, R.E.; Rodriguez-Tome, P.; Aggarwal, A.; Bajorek, E.; et al. A gene map of the human genome. Science 1996, 274, 540–546. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Gray, K.A.; Daugherty, L.C.; Gordon, S.M.; Seal, R.L.; Wright, M.W.; Bruford, E.A. Genenames.org: the HGNC resources in 2013. Nucleic Acids Res. 2013, 41, D545–D552. [Google Scholar]
- Ramani, A.K.; Bunescu, R.C.; Mooney, R.J.; Marcotte, E.M. Consolidating the set of known human protein-protein interactions in preparation for large-scale mapping of the human interactome. Genome Biol. 2005, 6, R40. [Google Scholar] [CrossRef] [Green Version]
- Hart, G.T.; Ramani, A.K.; Marcotte, E.M. How complete are current yeast and human protein-interaction networks? Genome Biol. 2006, 7, e120. [Google Scholar]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar]
- Venkatesan, K.; Rual, J.F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.; Goh, K.I.; et al. An empirical framework for binary interactome mapping. Nat. Methods 2009, 6, 83–90. [Google Scholar] [CrossRef]
- Tyagi, M.; Hashimoto, K.; Shoemaker, B.A.; Wuchty, S.; Panchenko, A.R. Large-scale mapping of human protein interactome using structural complexes. EMBO Rep. 2012, 13, 266–271. [Google Scholar] [CrossRef]
- Casado-Vela, J.; Cebrian, A.; Gomez del Pulgar, M.T.; Sanchez-Lopez, E.; Vilaseca, M.; Menchen, L.; Diema, C.; Selles-Marchart, S.; Martinez-Esteso, M.J.; Yubero, N.; et al. Lights and shadows of proteomic technologies for the study of protein species including isoforms, splicing variants and protein post-translational modifications. Proteomics 2011, 11, 590–603. [Google Scholar] [CrossRef]
- Braun, P.; Tasan, M.; Dreze, M.; Barrios-Rodiles, M.; Lemmens, I.; Yu, H.; Sahalie, J.M.; Murray, R.R.; Roncari, L.; de Smet, A.S.; et al. An experimentally derived confidence score for binary protein-protein interactions. Nat. Methods 2009, 6, 91–97. [Google Scholar] [CrossRef]
- Casado-Vela, J.; Gonzalez-Gonzalez, M.; Matarraz, S.; Martínez-Esteso, M.J.; Vilella, M.; Sayagues, J.M.; Fuentes, M.; Lacal, J.C. Protein arrays: recent achievements and their application to study the human proteome. Curr. Proteomics 2013, in press. [Google Scholar]
- Mishra, S. Computational prediction of protein-protein complexes. BMC Res. Notes 2012, 5, e495. [Google Scholar] [CrossRef]
- Jessulat, M.; Pitre, S.; Gui, Y.; Hooshyar, M.; Omidi, K.; Samanfar, B.; Tan le, H.; Alamgir, M.; Green, J.; Dehne, F.; et al. Recent advances in protein-protein interaction prediction: Experimental and computational methods. Expert Opin. Drug Discov. 2011, 6, 921–935. [Google Scholar] [CrossRef]
- Xia, J.F.; Wang, S.L.; Lei, Y.K. Computational methods for the prediction of protein-protein interactions. Protein Pept. Lett. 2010, 17, 1069–1078. [Google Scholar] [CrossRef]
- Skrabanek, L.; Saini, H.K.; Bader, G.D.; Enright, A.J. Computational prediction of protein-protein interactions. Mol. Biotechnol. 2008, 38, 1–17. [Google Scholar] [CrossRef]
- Pitre, S.; Alamgir, M.; Green, J.R.; Dumontier, M.; Dehne, F.; Golshani, A. Computational methods for predicting protein-protein interactions. Adv. Biochem. Eng. Biotechnol. 2008, 110, 247–267. [Google Scholar]
- Keskin, O.; Tuncbag, N.; Gursoy, A. Characterization and prediction of protein interfaces to infer protein-protein interaction networks. Curr. Pharm. Biotechnol. 2008, 9, 67–76. [Google Scholar] [CrossRef]
- Gomez, S.M.; Choi, K.; Wu, Y. Prediction of protein-protein interaction networks. Curr. Protoc. Bioinformatics 2008. [Google Scholar] [CrossRef]
- Fernandez-Suarez, X.M.; Galperin, M.Y. The 2013 Nucleic Acids Research Database Issue and the online molecular biology database collection. Nucleic Acids Res. 2013, 41, D1–D7. [Google Scholar] [CrossRef]
- Elefsinioti, A.; Sarac, O.S.; Hegele, A.; Plake, C.; Hubner, N.C.; Poser, I.; Sarov, M.; Hyman, A.; Mann, M.; Schroeder, M.; et al. Large-scale de novo prediction of physical protein-protein association. Mol. Cell. Proteomics 2011, 10. [Google Scholar] [CrossRef]
- Klingstrom, T.; Plewczynski, D. Protein-protein interaction and pathway databases, a graphical review. Brief. Bioinform. 2011, 12, 702–713. [Google Scholar] [CrossRef]
- Aranda, B.; Blankenburg, H.; Kerrien, S.; Brinkman, F.S.; Ceol, A.; Chautard, E.; Dana, J.M.; De Las Rivas, J.; Dumousseau, M.; Galeota, E.; et al. PSICQUIC and PSISCORE: Accessing and scoring molecular interactions. Nat. Methods 2011, 8, 528–529. [Google Scholar] [CrossRef] [Green Version]
- Blankenburg, H.; Finn, R.D.; Prlic, A.; Jenkinson, A.M.; Ramirez, F.; Emig, D.; Schelhorn, S.E.; Buch, J.; Lengauer, T.; Albrecht, M. DASMI: Exchanging, annotating and assessing molecular interaction data. Bioinformatics 2009, 25, 1321–1328. [Google Scholar]
- Garcia-Garcia, J.; Schleker, S.; Klein-Seetharaman, J.; Oliva, B. BIPS: BIANA Interolog Prediction Server. A tool for protein-protein interaction inference. Nucleic Acids Res. 2012, 40, W147–W151. [Google Scholar]
- Schleker, S.; Sun, J.; Raghavan, B.; Srnec, M.; Muller, N.; Koepfinger, M.; Murthy, L.; Zhao, Z.; Klein-Seetharaman, J. The current Salmonella-host interactome. Proteomics Clin. Appl. 2012, 6, 117–133. [Google Scholar] [CrossRef]
- Orchard, S. Molecular interaction databases. Proteomics 2012, 12, 1656–1662. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Available online: www.ncbi.nlm.nih.gov/gene (accessed on 23 May 2013).
- Alzheimer and neuronal disease laboratory_data repository. Available online: www.cnb.csic.es/~naranjo/ (accessed on 23 May 2013).
- Uniprot protein knowledgebase/Swiss-Prot. Available online: http://www.uniprot.org/uniprot/ (accessed on 16 May 2013).
- UniProt Frequently Asked Questions_question_48. Available online: http://www.uniprot.org/faq/48 (accessed on 23 May 2013).
- Uniprot link to CHKA_HUMAN, P35790, Choline kinase. Available online: http://www.uniprot.org/uniprot/P35790 (accessed on 23 May 2013).
- Dubois, T.; Howell, S.; Zemlickova, E.; Aitken, A. Identification of casein kinase Ialpha interacting protein partners. FEBS Lett. 2002, 517, 167–171. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef]
- String 9.05_web interface. Available online: http://string-db.org/ (accessed on 23 May 2013).
- Psicquic View_web interface. Available online: http://www.ebi.ac.uk/Tools/webservices/psicquic/view/main.xhtml (accessed on 23 May 2013).
- Casado-Vela, J.; Cebrian, A.; Gomez del Pulgar, M.T.; Lacal, J.C. Approaches for the study of cancer: Towards the integration of genomics, proteomics and metabolomics. Clin. Transl. Oncol. 2011, 13, 617–628. [Google Scholar]
- UniProt Frequently Asked Questions_question_6. Available online: www.uniprot.org/faq/6 (accessed on 23 May 2013).
- Gene Symbol Redundancy Checker. Available online: https://dl.dropboxusercontent.com/u/ 77276631/SymbolRedundancy.zip (accessed on 23 May 2013).
- Swiss-Prot incorporated the International Protein Index Database. Available online: http://www.uniprot.org/news/2011/05/03/release (accessed on 23 May 2013).
- Navarro-Munoz, M.; Ibernon, M.; Bonet, J.; Perez, V.; Pastor, M.C.; Bayes, B.; Casado-Vela, J.; Navarro, M.; Ara, J.; Espinal, A.; et al. Uromodulin and alpha(1)-antitrypsin urinary peptide analysis to differentiate glomerular kidney diseases. Kidney Blood Press. Res. 2012, 35, 314–325. [Google Scholar] [CrossRef]
- Tamm, I.; Horsfall, F.L., Jr. A mucoprotein derived from human urine which reacts with influenza, mumps, and Newcastle disease viruses. J. Exp. Med. 1952, 95, 71–97. [Google Scholar] [CrossRef]
- Klein, J.; Jupp, S.; Moulos, P.; Fernandez, M.; Buffin-Meyer, B.; Casemayou, A.; Chaaya, R.; Charonis, A.; Bascands, J.L.; Stevens, R.; et al. The KUPKB: A novel Web application to access multiomics data on kidney disease. FASEB J. 2012, 26, 2145–2153. [Google Scholar] [CrossRef]
- Casado-Vela, J.; del Pulgar, T.G.; Cebrian, A.; Alvarez-Ayerza, N.; Lacal, J.C. Human urine proteomics: Building a list of human urine cancer biomarkers. Expert Rev. Proteomics 2011, 8, 347–360. [Google Scholar] [CrossRef]
- Hegde, S.R.; Manimaran, P.; Mande, S.C. Dynamic changes in protein functional linkage networks revealed by integration with gene expression data. PLoS Comput. Biol. 2008, 4, e1000237. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Choi, E.K.; Luo, Y.; Lilliehook, C.; Crowley, A.C.; Merriam, D.E.; Wasco, W. Calsenilin: A calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med. 1998, 4, 1177–1181. [Google Scholar] [CrossRef]
- Carrion, A.M.; Link, W.A.; Ledo, F.; Mellstrom, B.; Naranjo, J.R. DREAM is a Ca2+-regulated transcriptional repressor. Nature 1999, 398, 80–84. [Google Scholar] [CrossRef]
- An, W.F.; Bowlby, M.R.; Betty, M.; Cao, J.; Ling, H.P.; Mendoza, G.; Hinson, J.W.; Mattsson, K.I.; Strassle, B.W.; Trimmer, J.S.; Rhodes, K.J. Modulation of A-type potassium channels by a family of calcium sensors. Nature 2000, 403, 553–556. [Google Scholar] [CrossRef]
- Mellstrom, B.; Naranjo, J.R. Ca2+-dependent transcriptional repression and derepression: DREAM, a direct effector. Semin. Cell Dev. Biol. 2001, 12, 59–63. [Google Scholar] [CrossRef]
- Pathguide.org. Available online: http://www.pathguide.org/ (accessed on 23 May 2013).
- Rivas, M.; Villar, D.; Gonzalez, P.; Dopazo, X.M.; Mellstrom, B.; Naranjo, J.R. Building the DREAM interactome. Sci. China Life Sci. 2011, 54, 786–792. [Google Scholar] [CrossRef]
- Fedrizzi, L.; Lim, D.; Carafoli, E.; Brini, M. Interplay of the Ca2+-binding protein DREAM with presenilin in neuronal Ca2+ signaling. J. Biol. Chem. 2008, 283, 27494–27503. [Google Scholar] [CrossRef]
- Ledo, F.; Carrion, A.M.; Link, W.A.; Mellstrom, B.; Naranjo, J.R. DREAM-alphaCREM interaction via leucine-charged domains derepresses downstream regulatory element-dependent transcription. Mol. Cell. Biol. 2000, 20, 9120–9126. [Google Scholar] [CrossRef]
- Rivas, M.; Aurrekoetxea, K.; Mellstrom, B.; Naranjo, J.R. Redox signaling regulates transcriptional activity of the Ca2+-dependent repressor DREAM. Antioxid. Redox Signal. 2011, 14, 1237–1243. [Google Scholar] [CrossRef]
- Ledo, F.; Kremer, L.; Mellstrom, B.; Naranjo, J.R. Ca2+-dependent block of CREB-CBP transcription by repressor DREAM. EMBO J. 2002, 21, 4583–4592. [Google Scholar] [CrossRef]
- Rivas, M.; Mellstrom, B.; Naranjo, J.R.; Santisteban, P. Transcriptional repressor DREAM interacts with thyroid transcription factor-1 and regulates thyroglobulin gene expression. J. Biol. Chem. 2004, 279, 33114–33122. [Google Scholar]
- Scsucova, S.; Palacios, D.; Savignac, M.; Mellstrom, B.; Naranjo, J.R.; Aranda, A. The repressor DREAM acts as a transcriptional activator on Vitamin D and retinoic acid response elements. Nucleic Acids Res. 2005, 33, 2269–2279. [Google Scholar] [CrossRef]
- Lusin, J.D.; Vanarotti, M.; Li, C.; Valiveti, A.; Ames, J.B. NMR structure of DREAM: Implications for Ca(2+)-dependent DNA binding and protein dimerization. Biochemistry 2008, 47, 2252–2264. [Google Scholar] [CrossRef]
- Zaidi, N.F.; Kuplast, K.G.; Washicosky, K.J.; Kajiwara, Y.; Buxbaum, J.D.; Wasco, W. Calsenilin interacts with transcriptional co-repressor C-terminal binding protein(s). J. Neurochem. 2006, 98, 1290–1301. [Google Scholar] [CrossRef]
- Ruiz-Gomez, A.; Mellstrom, B.; Tornero, D.; Morato, E.; Savignac, M.; Holguin, H.; Aurrekoetxea, K.; Gonzalez, P.; Gonzalez-Garcia, C.; Cena, V.; et al. G protein-coupled receptor kinase 2-mediated phosphorylation of downstream regulatory element antagonist modulator regulates membrane trafficking of Kv4.2 potassium channel. J. Biol. Chem. 2007, 282, 1205–1215. [Google Scholar]
- Quintero, C.A.; Valdez-Taubas, J.; Ferrari, M.L.; Haedo, S.D.; Maccioni, H.J. Calsenilin and CALP interact with the cytoplasmic tail of UDP-Gal:GA2/GM2/GD2 beta-1,3-galactosyltransferase. Biochem. J. 2008, 412, 19–26. [Google Scholar] [CrossRef]
- Rivas, M.; Mellstrom, B.; Torres, B.; Cali, G.; Ferrara, A.M.; Terracciano, D.; Zannini, M.; Morreale de Escobar, G.; Naranjo, J.R. The DREAM protein is associated with thyroid enlargement and nodular development. Mol. Endocrinol. 2009, 23, 862–870. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.J.; Mellstrom, B.; Wang, H.; Ren, M.; Domingo, S.; Kim, S.S.; Li, X.Y.; Chen, T.; Naranjo, J.R.; Zhuo, M. DREAM (downstream regulatory element antagonist modulator) contributes to synaptic depression and contextual fear memory. Mol. Brain 2010, 3, e3. [Google Scholar] [CrossRef]
- Anderson, D.; Mehaffey, W.H.; Iftinca, M.; Rehak, R.; Engbers, J.D.; Hameed, S.; Zamponi, G.W.; Turner, R.W. Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat. Neurosci. 2010, 13, 333–337. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, P.; Liang, P.; Liu, T.; Liu, X.; Liu, X.Y.; Zhang, B.; Han, T.; Zhu, Y.B.; Yin, D.M.; et al. The DREAM protein negatively regulates the NMDA receptor through interaction with the NR1 subunit. J. Neurosci. 2010, 30, 7575–7586. [Google Scholar] [CrossRef]
- Palczewska, M.; Casafont, I.; Ghimire, K.; Rojas, A.M.; Valencia, A.; Lafarga, M.; Mellstrom, B.; Naranjo, J.R. Sumoylation regulates nuclear localization of repressor DREAM. Biochim. Biophys. Acta 2010, 1813, 1050–1058. [Google Scholar]
- Ramachandran, P.L.; Craig, T.A.; Atanasova, E.A.; Cui, G.; Owen, B.A.; Bergen, H.R., 3rd; Mer, G.; Kumar, R. The potassium channel interacting protein 3 (DREAM/KChIP3) heterodimerizes with and regulates calmodulin function. J. Biol. Chem. 2012, 287, 39439–39448. [Google Scholar]
- Full list of human genes, acronyms and descriptions (NCBI). Available online: ftp://ftp.ncbi.nih.gov/gene/DATA/GENE_INFO/Mammalia/ (accessed on 23 May 2013).
- DASMI_web interface. Available online: http://dasmi.de/dasmiweb.php (accessed on 23 May 2013).
- BIPS_web interface. Available online: http://sbi.imim.es/web/index.php/research/servers/bips. (accessed on 23 May 2013).
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Casado-Vela, J.; Matthiesen, R.; Sellés, S.; Naranjo, J.R. Protein-Protein Interactions: Gene Acronym Redundancies and Current Limitations Precluding Automated Data Integration. Proteomes 2013, 1, 3-24. https://doi.org/10.3390/proteomes1010003
Casado-Vela J, Matthiesen R, Sellés S, Naranjo JR. Protein-Protein Interactions: Gene Acronym Redundancies and Current Limitations Precluding Automated Data Integration. Proteomes. 2013; 1(1):3-24. https://doi.org/10.3390/proteomes1010003
Chicago/Turabian StyleCasado-Vela, Juan, Rune Matthiesen, Susana Sellés, and José Ramón Naranjo. 2013. "Protein-Protein Interactions: Gene Acronym Redundancies and Current Limitations Precluding Automated Data Integration" Proteomes 1, no. 1: 3-24. https://doi.org/10.3390/proteomes1010003
APA StyleCasado-Vela, J., Matthiesen, R., Sellés, S., & Naranjo, J. R. (2013). Protein-Protein Interactions: Gene Acronym Redundancies and Current Limitations Precluding Automated Data Integration. Proteomes, 1(1), 3-24. https://doi.org/10.3390/proteomes1010003