The Biochemical Properties of Manganese in Plants


Abstract
1. Introduction—Manganese as an Essential Plant Nutrient
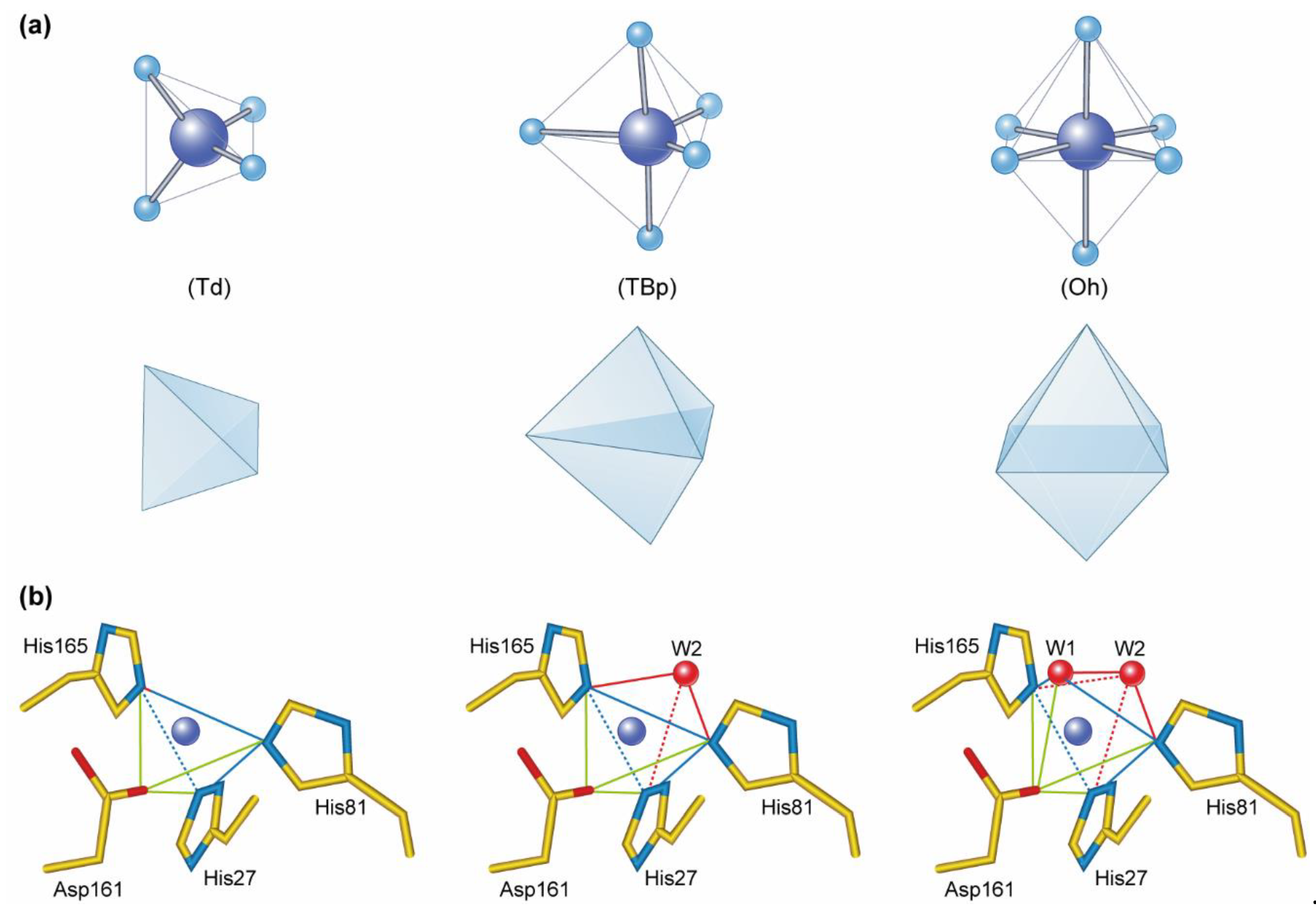
2. The Biological Chemistry of Manganese in Plants
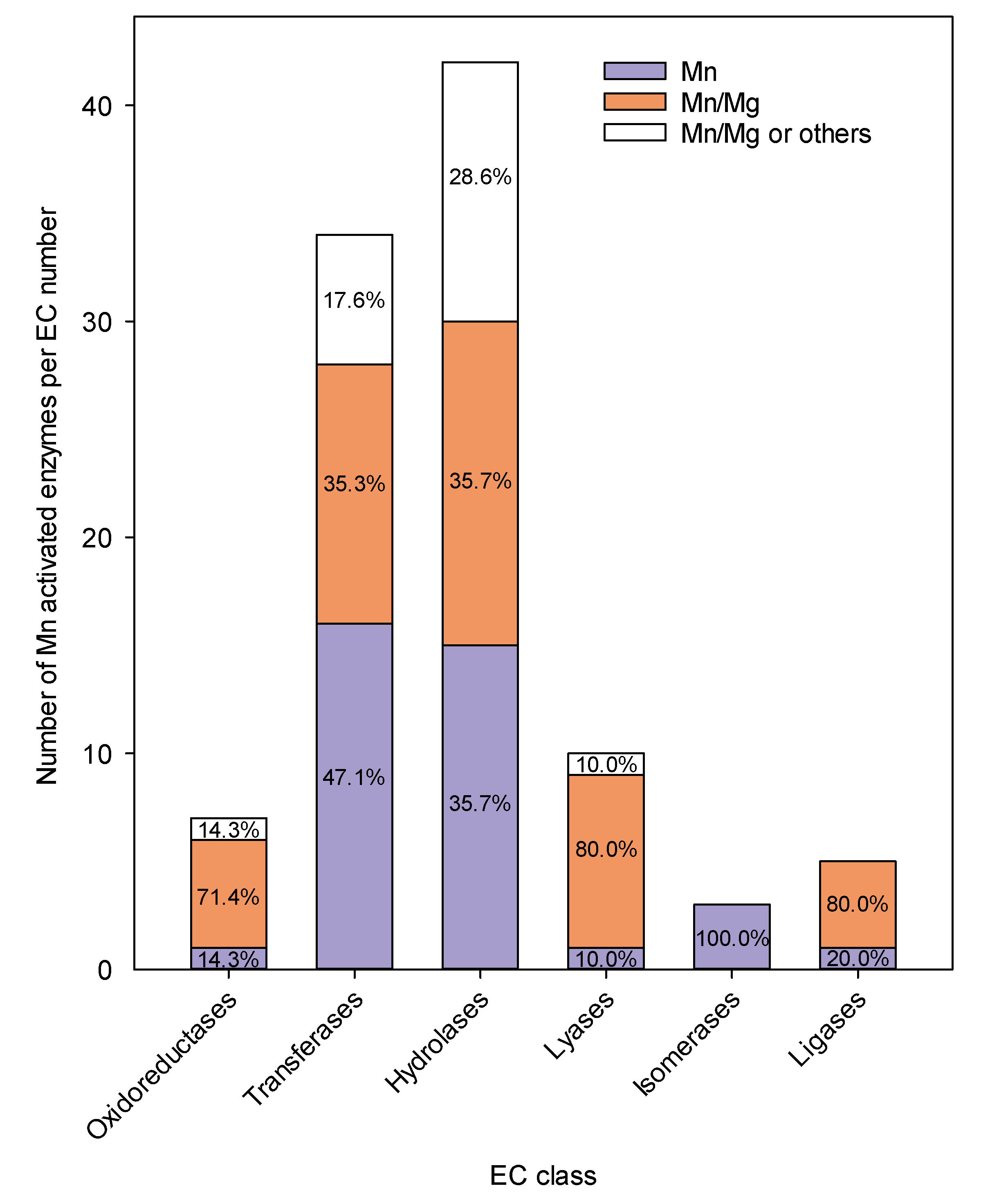
3. Manganese in Metalloenzymes
4. Metallation of Manganese Containing Metalloenzymes
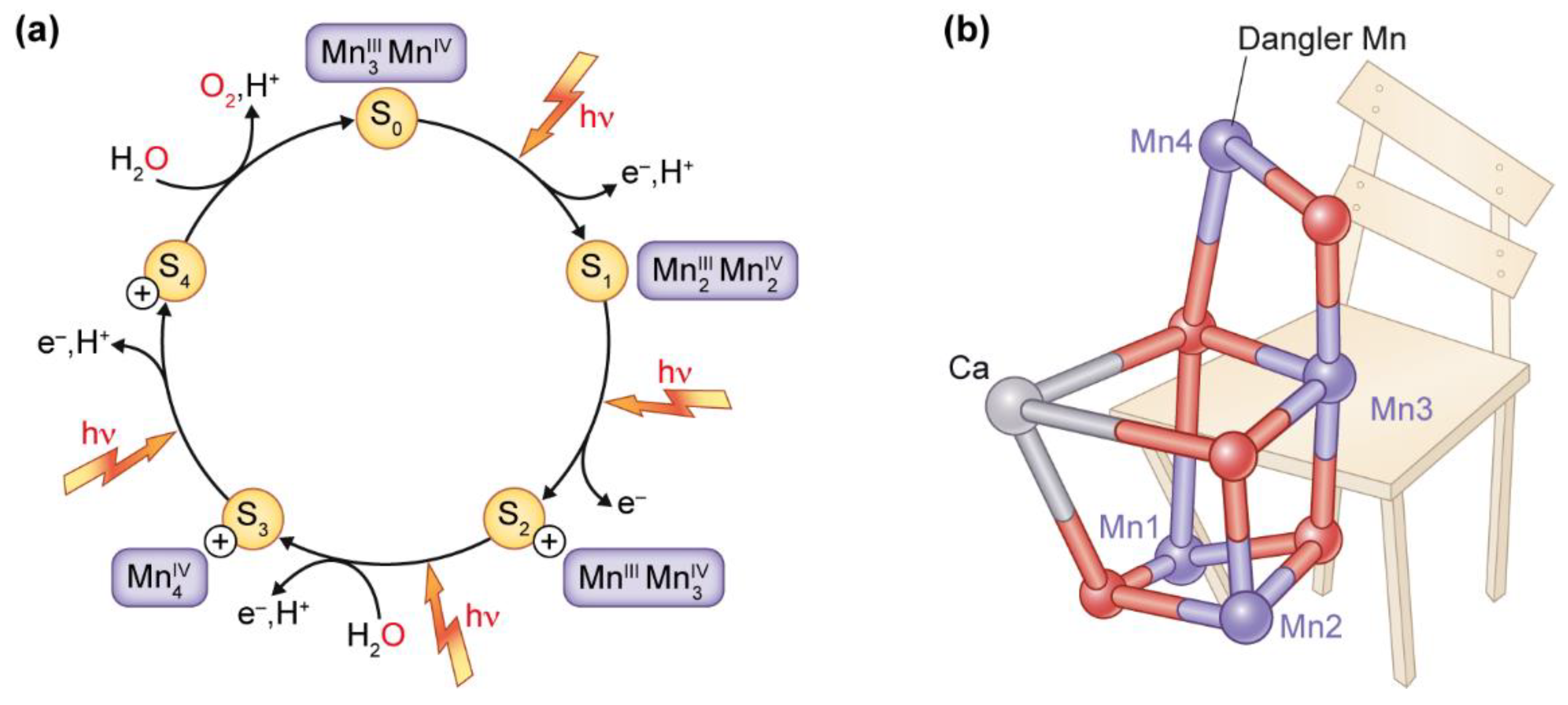
5. Manganese at the Active Site of Water Oxidation in Photosystem II
6. Manganese-Dependent Superoxide Dismutase
7. Manganese-Dependent Oxalate Oxidase
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saussure, T.D. Recherches Chimiques sur la Végétation; Nyon: Paris, France, 1804. [Google Scholar]
- McHargue, J.S. The effect of manganese on the growth of wheat: A source of manganese for agricultural purposes. J. Ind. Eng. Chem. 1919, 11, 332–335. [Google Scholar] [CrossRef]
- Loew, O.; Sawa, S. On the action of manganese compounds on plants. Bull. Coll. Agric. Tokyo 1902, 5, 161–172. [Google Scholar]
- Pirson, A. Ernährungs- und stoffwechselphysiologische Untersuchungen an Fontinalis und Chlorella. Zeitschrift für Botanik 1937, 31, 193–267. [Google Scholar]
- Pirson, A.; Tichy, C.; Wilhelmi, G. Stoffwechsel und mineralsalzernährung Einzelliger Grünalgen. Planta 1951, 40, 199–253. [Google Scholar] [CrossRef]
- Kok, B.; Forbush, B.; Mcgloin, M. Cooperation of charges in photosynthetic oxygen evolution-I. A linear four step mechanism. Photochem. Photobiol. 1970, 11, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Spector, M.; Winget, G.D. Purification of a manganese-containing protein involved in photosynthetic oxygen evolution and its use in reconstituting an active membrane. Proc. Natl. Acad. Sci. USA 1980, 77, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.B.; Jensen, P.E.; Husted, S. Manganese deficiency in plants: The impact on Photosystem II. Trends Plant Sci. 2016, 21, 622–632. [Google Scholar] [CrossRef]
- Hebbern, C.A.; Pedas, P.; Schjoerring, J.K.; Knudsen, L.; Husted, S. Genotypic differences in manganese efficiency: Field experiments with winter barley (Hordeum vulgare L.). Plant Soil 2005, 272, 233–244. [Google Scholar] [CrossRef]
- Schmidt, S.B.; Pedas, P.; Laursen, K.H.; Schjoerring, J.K.; Husted, S. Latent manganese deficiency in barley can be diagnosed and remediated on the basis of chlorophyll a fluorescence measurements. Plant Soil 2013, 372, 417–429. [Google Scholar] [CrossRef]
- Stoltz, E.; Wallenhammar, A.-C. Manganese application increases winter hardiness in barley. Field Crops Res. 2014, 164, 148–153. [Google Scholar] [CrossRef]
- Jiang, W.Z. Mn use efficiency in different wheat cultivars. Environ. Exp. Bot. 2006, 57, 41–50. [Google Scholar] [CrossRef]
- George, T.S.; French, A.S.; Brown, L.K.; Karley, A.J.; White, P.J.; Ramsay, L.; Daniell, T.J. Genotypic variation in the ability of landraces and commercial cereal varieties to avoid manganese deficiency in soils with limited manganese availability: Is there a role for root-exuded phytases? Physiol. Plant. 2014, 151, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, A.M.; Graham, D.R.; Langridge, P.; Sparrow, B.D.H.; Barker, J.S. RFLP mapping of manganese efficiency in barley. Theor. Appl. Genet. 2000, 101, 1100–1108. [Google Scholar] [CrossRef]
- Yang, X.E.; Chen, W.R.; Feng, Y. Improving human micronutrient nutrition through biofortification in the soil-plant system: China as a case study. Environ. Geochem. Health 2007, 29, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Waldron, K.J.; Rutherford, J.C.; Ford, D.; Robinson, N.J. Metalloproteins and metal sensing. Nature 2009, 460, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Richards, N.G.J. Biological functions controlled by manganese redox changes in mononuclear Mn-dependent enzymes. Essays Biochem. 2017, 61, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, O. Manganese complexes displaying superoxide dismutase activity: A balance between different factors. Bioorg. Chem. 2011, 39, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.A.; Sigman, D.M.; Morel, F.M.M. The bioinorganic chemistry of the ancient ocean: The co-evolution of cyanobacterial metal requirements and biogeochemical cycles at the Archean–Proterozoic boundary? Inorg. Chim. Acta 2003, 356, 308–318. [Google Scholar] [CrossRef]
- Irving, H.; Williams, R.J.P. Order of Stability of Metal Complexes. Nature 1948, 162, 746–747. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Retnoningrum, D.S.; Yoshida, H.; Arumsari, S.; Kamitori, S.; Ismaya, W.T. The first crystal structure of manganese superoxide dismutase from the genus Staphylococcus. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 135–142. [Google Scholar] [CrossRef]
- Irving, H.; Williams, R.J.P. 637. The stability of transition-metal complexes. J. Chem. Soc. 1953. [Google Scholar] [CrossRef]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G.L.; Thornton, J.M. Metal ions in biological catalysis: From enzyme databases to general principles. JBIC 2008, 13, 1205–1218. [Google Scholar] [CrossRef]
- Christianson, D.W. Structural chemistry and biology of manganese metalloenzymes. Prog. Biophys. Mol. Biol. 1997, 67. [Google Scholar] [CrossRef]
- Glusker, J.P. Structural aspects of metal liganding to functional groups in proteins. Adv. Protein Chem. 1991, 42, 1–76. [Google Scholar] [PubMed]
- Khrustalev, V.V.; Barkovsky, E.V.; Khrustaleva, T.A. Magnesium and manganese binding sites on proteins have the same predominant motif of secondary structure. J. Theor. Biol. 2016, 395, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, P.; Grover, N.; Westhof, E. Metal ion binding to RNA. Met. Ions Life Sci. 2011, 9, 1–35. [Google Scholar]
- Bock, C.W.; Katz, A.K.; Markham, G.D.; Glusker, J.P. Manganese as a Replacement for Magnesium and Zinc: Functional Comparison of the Divalent Ions. J. Am. Chem. Soc. 1999, 121, 7360–7372. [Google Scholar] [CrossRef]
- Williams, L.E.; Pittman, J.K. Dissecting pathways involved in manganese homeostasis and stress in higher plant cells. In Cell Biology of Metals and Nutrients; Hell, R., Mendel, R.-R., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 95–117. [Google Scholar]
- Hermans, C.; Conn, S.J.; Chen, J.; Xiao, Q.; Verbruggen, N. An update on magnesium homeostasis mechanisms in plants. Metallomics 2013, 5, 1170–1183. [Google Scholar] [CrossRef] [PubMed]
- Dudev, T.; Lim, C. Competition among Metal Ions for Protein Binding Sites: Determinants of Metal Ion Selectivity in Proteins. Chem. Rev. 2014, 114, 538–556. [Google Scholar] [CrossRef]
- Bloom, A.J. Metal regulation of metabolism. Curr. Opin. Chem. Biol. 2019, 49, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Lilley, R.M.; Wang, X.; Krausz, E.; Andrews, T.J. Complete Spectra of the Far-red Chemiluminescence of the Oxygenase Reaction of Mn2+-activated Ribulose-bisphosphate Carboxylase/Oxygenase Establish Excited Mn2+ as the Source. J. Biol. Chem. 2003, 278, 16488–16493. [Google Scholar] [CrossRef] [PubMed]
- Bloom, A.J.; Kameritsch, P. Relative association of Rubisco with manganese and magnesium as a regulatory mechanism in plants. Physiol. Plant. 2017, 161, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Socha, A.L.; Guerinot, M.L. Mn-euvering manganese: The role of transporter gene family members in manganese uptake and mobilization in plants. Front. Plant Sci. 2014, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Stengel, A.; Gugel, I.L.; Hilger, D.; Rengstl, B.; Jung, H.; Nickelsen, J. Initial steps of photosystem II de novo assembly and preloading with manganese take place in biogenesis centers in Synechocystis. Plant Cell 2012, 24, 660–675. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Whittaker, M.M.; Bachinger, H.P.; Whittaker, J.W. Calorimetric studies on the tight binding metal interactions of Escherichia coli manganese superoxide dismutase. J. Biol. Chem. 2004, 279, 27339–27344. [Google Scholar] [CrossRef]
- Bondarava, N.; Beyer, P.; Krieger-Liszkay, A. Function of the 23 kDa extrinsic protein of photosystem II as a manganese binding protein and its role in photoactivation. Biochim. Biophys. Acta 2005, 1708, 63–70. [Google Scholar] [CrossRef]
- Bondarava, N.; Un, S.; Krieger-Liszkay, A. Manganese binding to the 23 kDa extrinsic protein of photosystem II. Biochim. Biophys. Acta 2007, 1767, 583–588. [Google Scholar] [CrossRef]
- Cao, P.; Xie, Y.; Li, M.; Pan, X.; Zhang, H.; Zhao, X.; Su, X.; Cheng, T.; Chang, W. Crystal structure analysis of extrinsic PsbP protein of photosystem II reveals a manganese-induced conformational change. Mol. Plant 2015, 8, 664–666. [Google Scholar] [CrossRef]
- Ido, K.; Kakiuchi, S.; Uno, C.; Nishimura, T.; Fukao, Y.; Noguchi, T.; Sato, F.; Ifuku, K. The conserved His-144 in the PsbP protein is important for the interaction between the PsbP N-terminus and the Cyt b559 subunit of photosystem II. J. Biol. Chem. 2012, 287, 26377–26387. [Google Scholar] [CrossRef]
- Emerson, J.P.; Kovaleva, E.G.; Farquhar, E.R.; Lipscomb, J.D.; Que, L. Swapping metals in Fe- and Mn-dependent dioxygenases: Evidence for oxygen activation without a change in metal redox state. Proc. Natl. Acad. Sci. USA 2008, 105, 7347–7352. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Hu, R.-G.; Wang, B.-Z.; Chen, W.-F.; Liu, W.-Y.; Schröder, W.; Frank, P.; Ulbrich, N. Structural studies of an eukaryotic cambialistic superoxide dismutase purified from the mature seeds of camphor tree. Arch. Biochem. Biophys. 2002, 404, 218–226. [Google Scholar] [CrossRef]
- Tottey, S.; Waldron, K.J.; Firbank, S.J.; Reale, B.; Bessant, C.; Sato, K.; Cheek, T.R.; Gray, J.; Banfield, M.J.; Dennison, C.; et al. Protein-folding location can regulate manganese-binding versus copper- or zinc-binding. Nature 2008, 455, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Joliot, P. Kinetic studies of photosystem II in photosynthesis. Photochem. Photobiol. 1968, 8, 451–463. [Google Scholar] [CrossRef]
- Joliot, P.; Barbieri, G.; Chabaud, R. Un Nouveau modele des centres photochimiques du systeme II. Photochem. Photobiol. 1969, 10, 309–329. [Google Scholar] [CrossRef]
- Zouni, A.; Witt, H.-T.; Kern, J.; Fromme, P.; Krauss, N.; Saenger, W.; Orth, P. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature 2001, 409, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, K.N.; Iverson, T.M.; Maghlaoui, K.; Barber, J.; Iwata, S. Architecture of the Photosynthetic Oxygen-Evolving Center. Science 2004, 303, 1831–1838. [Google Scholar] [CrossRef]
- Kawakami, K.; Umena, Y.; Kamiya, N.; Shen, J.-R. Structure of the catalytic, inorganic core of oxygen-evolving photosystem II at 1.9 angstrom resolution. J. Photochem. Photobiol. B Biol. 2011, 104, 9–18. [Google Scholar] [CrossRef]
- Cao, P.; Su, X.; Pan, X.; Liu, Z.; Chang, W.; Li, M. Structure, assembly and energy transfer of plant photosystem II supercomplex. BBA Bioenergy 2018, 1859, 633–644. [Google Scholar] [CrossRef]
- Reiss, K.; Morzan, U.N.; Grigas, A.T.; Batista, V.S. Water Network Dynamics Next to the Oxygen-Evolving Complex of Photosystem II. Inorganics 2019, 7, 39. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, L. Why nature chose the Mn4CaO5 cluster as water-splitting catalyst in photosystem II: A new hypothesis for the mechanism of O-O bond formation. Dalton Trans. 2018, 47, 14381–14387. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 angstrom. Nature 2011, 473, U55–U65. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Chrysina, M.; Cox, N. Water oxidation in photosystem II. Photosynth. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vinyard, D.J.; Brudvig, G.W. Progress Toward a Molecular Mechanism of Water Oxidation in Photosystem II. Annu. Rev. Phys. Chem. 2017, 68, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, J.; Ananyev, G.M.; Dismukes, G.C. Photoassembly of the water-oxidizing complex in photosystem II. Coord. Chem. Rev. 2008, 252, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Burnap, R.L. D1 protein processing and Mn cluster assembly in light of the emerging Photosystem II structure. PCCP 2004, 6, 4803–4809. [Google Scholar] [CrossRef]
- Nixon, P.J.; Diner, B.A. Aspartate 170 of the photosystem II reaction center polypeptide D1 is involved in the assembly of the oxygen-evolving manganese cluster. Biochemistry 1992, 31, 942–948. [Google Scholar] [CrossRef]
- Schmidt, S.B.; Persson, D.P.; Powikrowska, M.; Frydenvang, J.; Schjoerring, J.K.; Jensen, P.E.; Husted, S. Metal binding in photosystem II super- and subcomplexes from barley thylakoids. Plant Physiol. 2015, 168, 1490–1502. [Google Scholar] [CrossRef]
- De Bang, T.C.; Petersen, J.; Pedas, P.R.; Rogowska-Wrzesinska, A.; Jensen, O.N.; Schjoerring, J.K.; Jensen, P.E.; Thelen, J.J.; Husted, S. A laser ablation ICP-MS based method for multiplexed immunoblot analysis: Applications to manganese-dependent protein dynamics of photosystem II in barley (Hordeum vulgare L.). Plant J. 2015, 83, 555–565. [Google Scholar] [CrossRef]
- Schmidt, S.B.; Powikrowska, M.; Krogholm, K.S.; Naumann-Busch, B.; Schjoerring, J.K.; Husted, S.; Jensen, P.E.; Pedas, P.R. Photosystem II functionality in barley responds dynamically to changes in leaf manganese status. Front. Plant Sci. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Husted, S.; Laursen, K.H.; Hebbern, C.A.; Schmidt, S.B.; Pedas, P.; Haldrup, A.; Jensen, P.E. Manganese deficiency leads to genotype-specific changes in fluorescence induction kinetics and state transitions. Plant Physiol. 2009, 150, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, F.; López-Gorgé, J.; Gómez, M.; del Río, L.A. Manganese superoxide dismutase from a higher plant. Planta 1980, 150, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, F.; López-Gorgé, J.; del Río, L.A. Characterization of a Manganese Superoxide Dismutase from the Higher Plant Pisum sativum. Plant Physiol. 1982, 70, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, V.M.; Sevilla, F.; López-Gorgé, J.; del Río, L.A. Evidence for manganese(III) binding to the mangano superoxide dismutase from a higher plant (Pisum sativum L.). J. Inorg. Biochem. 1982, 16, 79–84. [Google Scholar] [CrossRef]
- Yu, Q.; Rengel, Z. Micronutrient deficiency influences plant growth and activities of superoxide dismutases in narrow-leafed lupins. Ann. Bot. 1999, 83, 175–182. [Google Scholar] [CrossRef]
- Tewari, R.K.; Kumar, P.; Sharma, P.N. Oxidative stress and antioxidant responses of mulberry (Morus alba) plants subjected to deficiency and excess of manganese. Acta Physiol. Plant. 2013, 35, 3345–3356. [Google Scholar] [CrossRef]
- Del Río, L.A.; Sevilla, F.; Gómez, M.; Yañez, J.; López, J. Superoxide dismutase: An enzyme system for the study of micronutrient interactions in plants. Planta 1978, 140, 221–225. [Google Scholar] [CrossRef]
- Del Río, L.A.; Lyon, D.S.; Olah, I.; Glick, B.; Salin, M.L. Immunocytochemical evidence for a peroxisomal localization of manganese superoxide dismutase in leaf protoplasts from a higher plant. Planta 1983, 158, 216–224. [Google Scholar] [CrossRef]
- Bowler, C.; Vancamp, W.; Vanmontagu, M.; Inze, D. Superoxide-dismutase in plants. Crit. Rev. Plant Sci. 1994, 13, 199–218. [Google Scholar] [CrossRef]
- Miller, A.-F. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012, 586, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Abreu, I.A.; Cabelli, D.E. Superoxide dismutases-a review of the metal-associated mechanistic variations. Biochim. Biophys. Acta 2010, 1804, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.A.; Gutman, C.T.; Maliekal, J.; Miller, A.F.; Brunold, T.C. Geometric and electronic structures of manganese-substituted iron superoxide dismutase. Inorg. Chem. 2013, 52, 3356–3367. [Google Scholar] [CrossRef] [PubMed]
- Beyer, W.F., Jr.; Fridovich, I. In vivo competition between iron and manganese for occupancy of the active site region of the manganese-superoxide dismutase of Escherichia coli. J. Biol. Chem. 1991, 266, 303–308. [Google Scholar] [PubMed]
- Krauss, I.R.; Merlino, A.; Pica, A.; Rullo, R.; Bertoni, A.; Capasso, A.; Amato, M.; Riccitiello, F.; De Vendittis, E.; Sica, F. Fine tuning of metal-specific activity in the Mn-like group of cambialistic superoxide dismutases. RSC Adv. 2015, 5, 87876–87887. [Google Scholar] [CrossRef]
- Mishra, P.; Sharma, P. Superoxide Dismutases (SODs) and Their Role in Regulating Abiotic Stress induced Oxidative Stress in Plants. In Reactive Oxygen, Nitrogen and Sulfur Species in Plants; Hasanuzzaman, V.F.M., Nahar, K., Fujita, M., Eds.; Wiley: Hoboken, NJ, USA, 2019; pp. 53–88. [Google Scholar]
- Su, Z.; Chai, M.-F.; Lu, P.-L.; An, R.; Chen, J.; Wang, X.-C. AtMTM1, a novel mitochondrial protein, may be involved in activation of the manganese-containing superoxide dismutase in Arabidopsis. Planta 2007, 226, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Noctor, G. Redox Regulation in Photosynthetic Organisms: Signaling, Acclimation, and Practical Implications. Antioxid. Redox Signal. 2009, 11, 861–905. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Kropat, J.; Tottey, S.; Del Campo, J.A.; Merchant, S.S. Manganese deficiency in Chlamydomonas results in loss of photosystem II and MnSOD function, sensitivity to peroxides, and secondary phosphorus and iron deficiency. Plant Physiol. 2007, 143, 263–277. [Google Scholar] [CrossRef]
- Leonowicz, G.; Trzebuniak, K.F.; Zimak-Piekarczyk, P.; Ślesak, I.; Mysliwa-Kurdziel, B. The activity of superoxide dismutases (SODs) at the early stages of wheat deetiolation. PLoS ONE 2018, 13, e0194678. [Google Scholar] [CrossRef]
- Bowler, C.; Slooten, L.; Vandenbranden, S.; De Rycke, R.; Botterman, J.; Sybesma, C.; Van Montagu, M.; Inze, D. Manganese superoxide dismutase can reduce cellular damage mediated by oxygen radicals in transgenic plants. EMBO J. 1991, 10, 1723–1732. [Google Scholar] [CrossRef]
- Allen, R.D. Dissection of Oxidative Stress Tolerance Using Transgenic Plants. Plant Physiol. 1995, 107, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, M.M.; Whittaker, J.W. Characterization of recombinant barley oxalate oxidase expressed by Pichia pastoris. J. Biol. Inorg. Chem. 2002, 7, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Requena, L.; Bornemann, S. Barley (Hordeum vulgare) oxalate oxidase is a manganese-containing enzyme. Biochem. J. 1999, 343, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Caliskan, M.; Cuming, A.C. Spatial specificity of H2O2-generating oxalate oxidase gene expression during wheat embryo germination. Plant J. 1998, 15, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Kotsira, V.P.; Clonis, Y.D. Oxalate oxidase from barley roots: Purification to homogeneity and study of some molecular, catalytic, and binding properties. Arch. Biochem. Biophys. 1997, 340, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Liao, Y.Y.; Leung, D.W.M.; Wang, H.Y.; Chen, B.L.; Peng, X.X.; Liu, E.E. Divergent biochemical and enzymatic properties of oxalate oxidase isoforms encoded by four similar genes in rice. Phytochemistry 2015, 118, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Vuletic, M.; Sukalovic, V.H. Characterization of cell wall oxalate oxidase from maize roots. Plant Sci. 2000, 157, 257–263. [Google Scholar] [CrossRef]
- Darvill, J.E.; McNeil, M.; Darvill, A.G.; Albersheim, P. Structure of Plant Cell Walls: XI. Glucuronoarabinoxylan, a second hemicellulose in the primary cell walls of suspension-cultured sycamore cells. Plant Physiol. 1980, 66, 1135–1139. [Google Scholar] [CrossRef]
- Woo, E.J.; Dunwell, J.M.; Goodenough, P.W.; Marvier, A.C.; Pickersgill, R.W. Germin is a manganese containing homohexamer with oxalate oxidase and superoxide dismutase activities. Nat. Struct. Biol. 2000, 7, 1036–1040. [Google Scholar] [CrossRef]
- Woo, E.J.; Dunwell, J.M.; Goodenough, P.W.; Pickersgill, R.W. Barley oxalate oxidase is a hexameric protein related to seed storage proteins: Evidence from X-ray crystallography. FEBS Lett. 1998, 437, 87–90. [Google Scholar] [CrossRef]
- Dumas, B.; Freyssinet, G.; Pallett, K.E. Tissue-Specific Expression of Germin-Like Oxalate Oxidase during Development and Fungal Infection of Barley Seedlings. Plant Physiol. 1995, 107, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Collinge, D.B.; Thordal-Christensen, H. Germin-like oxalate oxidase, a H2O2-producing enzyme, accumulates in barley attacked by the powdery mildew fungus. Plant J. 1995, 8, 139–145. [Google Scholar] [CrossRef]
- McCay-Buis, T.S.; Huber, D.M.; Graham, R.D.; Phillips, J.D.; Miskin, K.E. Manganese seed content and take-all of cereals. J. Plant Nutr. 1995, 18, 1711–1721. [Google Scholar] [CrossRef]
- Leplat, F.; Pedas, P.R.; Rasmussen, S.K.; Husted, S. Identification of manganese efficiency candidate genes in winter barley (Hordeum vulgare) using genome wide association mapping. BMC Genom. 2016, 17, 775. [Google Scholar] [CrossRef]
- Thompson, C.; Dunwell, J.M.; Johnstone, C.E.; Lay, V.; Ray, J.; Schmitt, M.; Watson, H.; Nisbet, G. Degradation of oxalic acid by transgenic oilseed rape plants expressing oxalate oxidase. Euphytica 1995, 85, 169–172. [Google Scholar] [CrossRef]
- Dong, X.; Ji, R.; Guo, X.; Foster, S.J.; Chen, H.; Dong, C.; Liu, Y.; Hu, Q.; Liu, S. Expressing a gene encoding wheat oxalate oxidase enhances resistance to Sclerotinia sclerotiorum in oilseed rape (Brassica napus). Planta 2008, 228, 331. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Ca | Mg | Mn | Fe | Co | Ni | Cu | Zn | |
|---|---|---|---|---|---|---|---|---|
| Ionic radius (Å) of the free metal ion | 0.99 | 0.66 | 0.78 | 0.76 | 0.74 | 0.73 | 0.72 | 0.72 |
| Second ionization potential (eV): | 11.87 | 15.04 | 15.64 | 16.18 | 17.06 | 18.15 | 20.29 | 17.96 |
| HSAB classification: | H | H | HB | B | B | B | B | B |
| Ligands: | O | O | N, O, S | N, S, O | N, O, S | S, N | S, N | N, S, O |
| Coordination numbers: | 6,7,8 | 6 | 4, 5, 6 | 4,5,6 | 4,5,6 | 4,5,6 | 4,5,6 | 4,5,6 |
| Dominating geometries: | Oh, IR, IR | Oh | Td, D3h, Oh | Td/D4h, C4v/D3H, Oh | Td, C4v/D3h, Oh | Oh, Td, D4h | Td/D4h, C4v/D3h, Oh | Td, C4v/D3h, Oh |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, S.B.; Husted, S. The Biochemical Properties of Manganese in Plants. Plants 2019, 8, 381. https://doi.org/10.3390/plants8100381
Schmidt SB, Husted S. The Biochemical Properties of Manganese in Plants. Plants. 2019; 8(10):381. https://doi.org/10.3390/plants8100381
Chicago/Turabian StyleSchmidt, Sidsel Birkelund, and Søren Husted. 2019. "The Biochemical Properties of Manganese in Plants" Plants 8, no. 10: 381. https://doi.org/10.3390/plants8100381
APA StyleSchmidt, S. B., & Husted, S. (2019). The Biochemical Properties of Manganese in Plants. Plants, 8(10), 381. https://doi.org/10.3390/plants8100381