
Multi-Omics Analysis Revealed the AGR-FC.C3 Locus of Brassica napus as a Novel Candidate for Controlling Petal Color

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
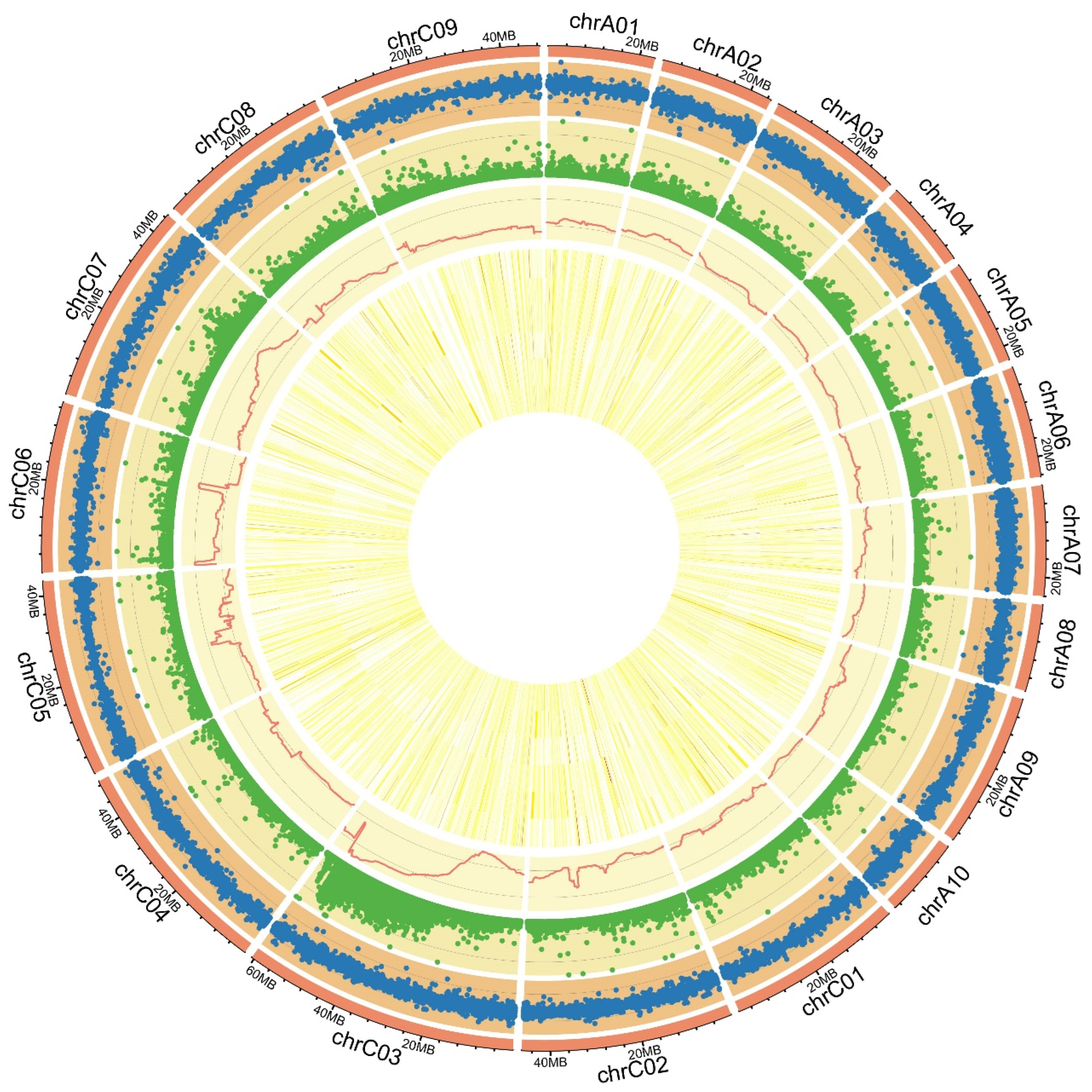
2.1. BSA for Petal Colors in B. napus
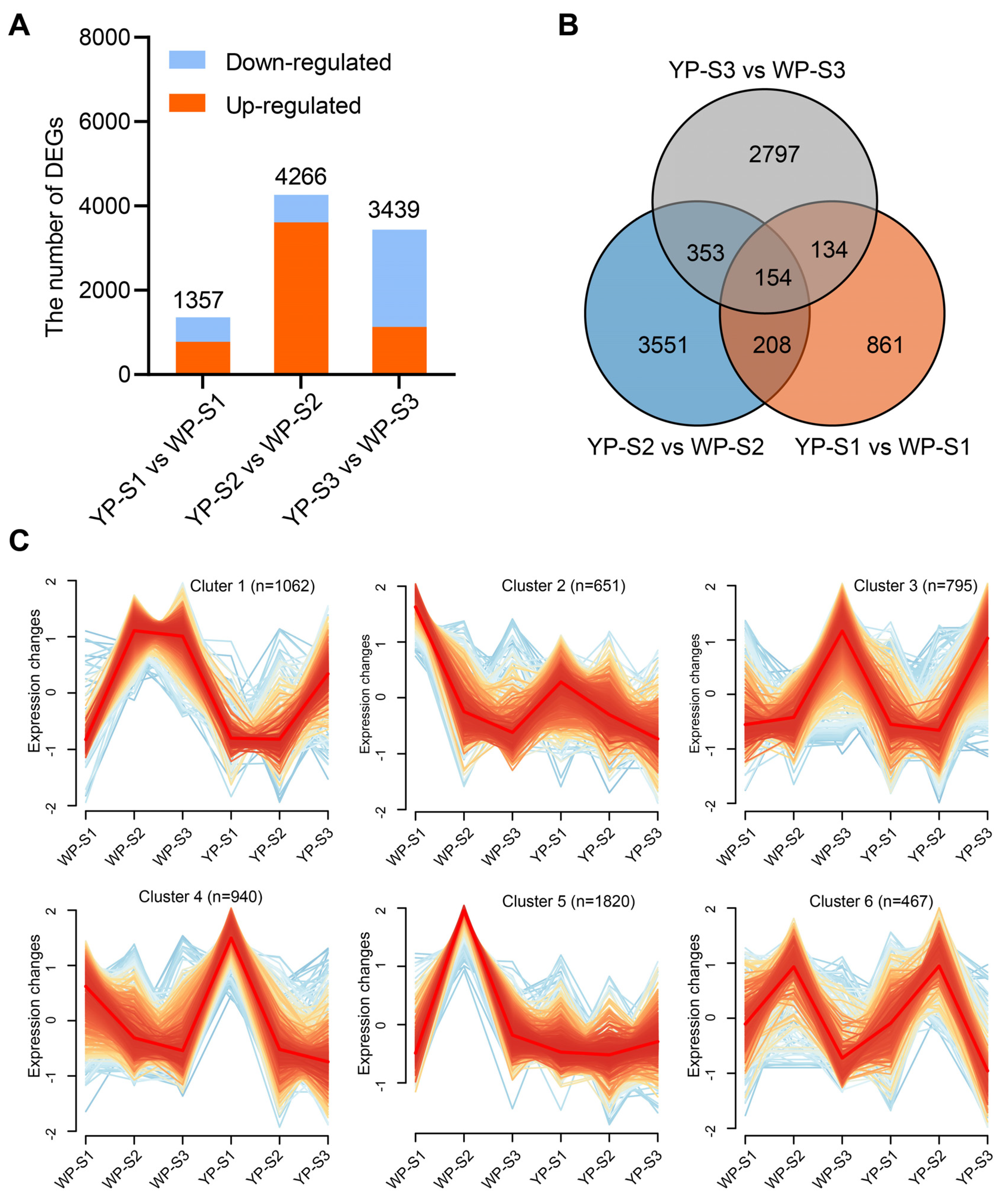
2.2. RNA-Seq and Differentially Expressed Genes (DEGs) Analysis between the YP and WP in B. napus
2.3. Overlapping Analysis of BSA and RNA-Seq Results
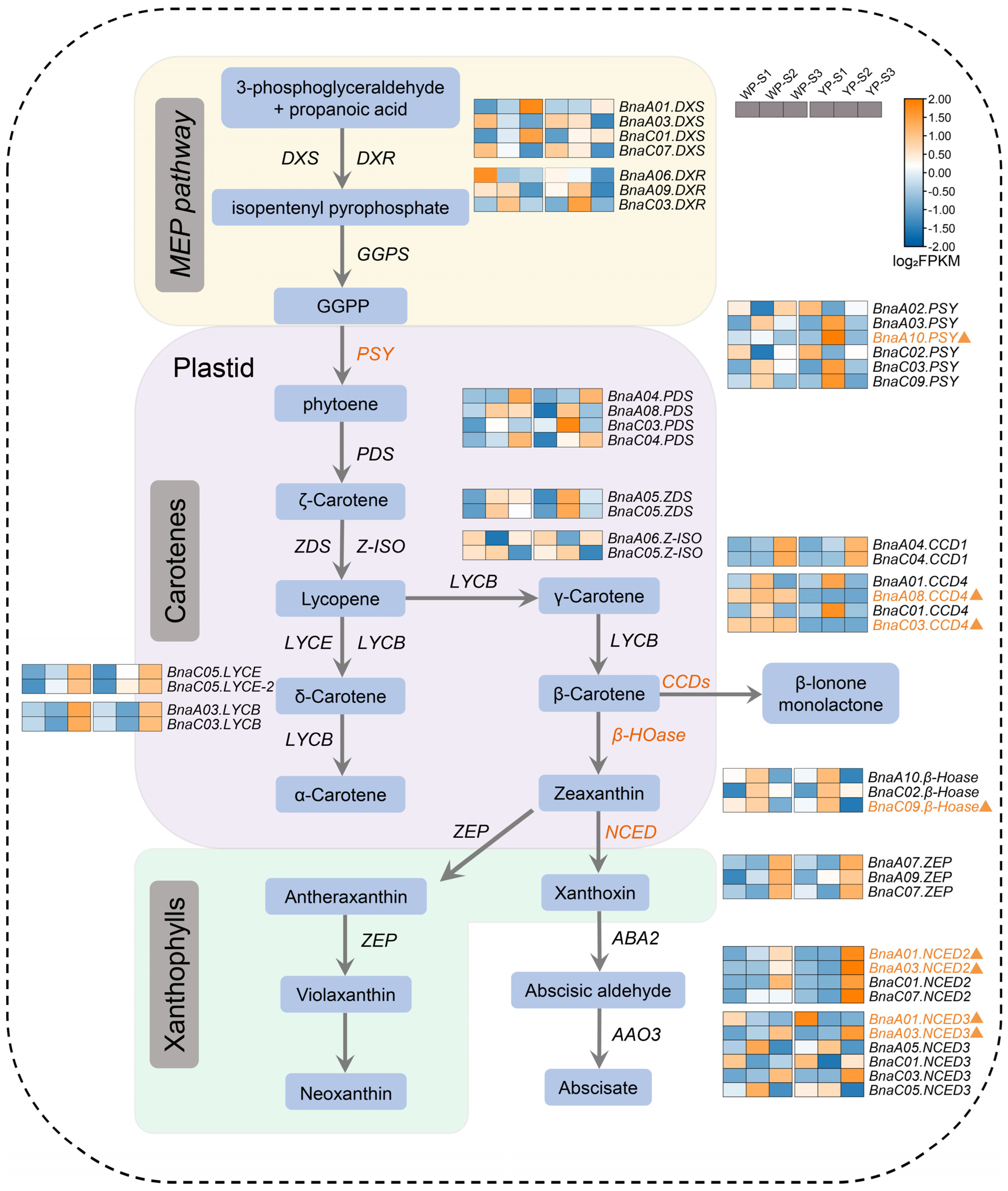
2.4. The Carotenoid Pathway Influences the Petal Colors in B. napus
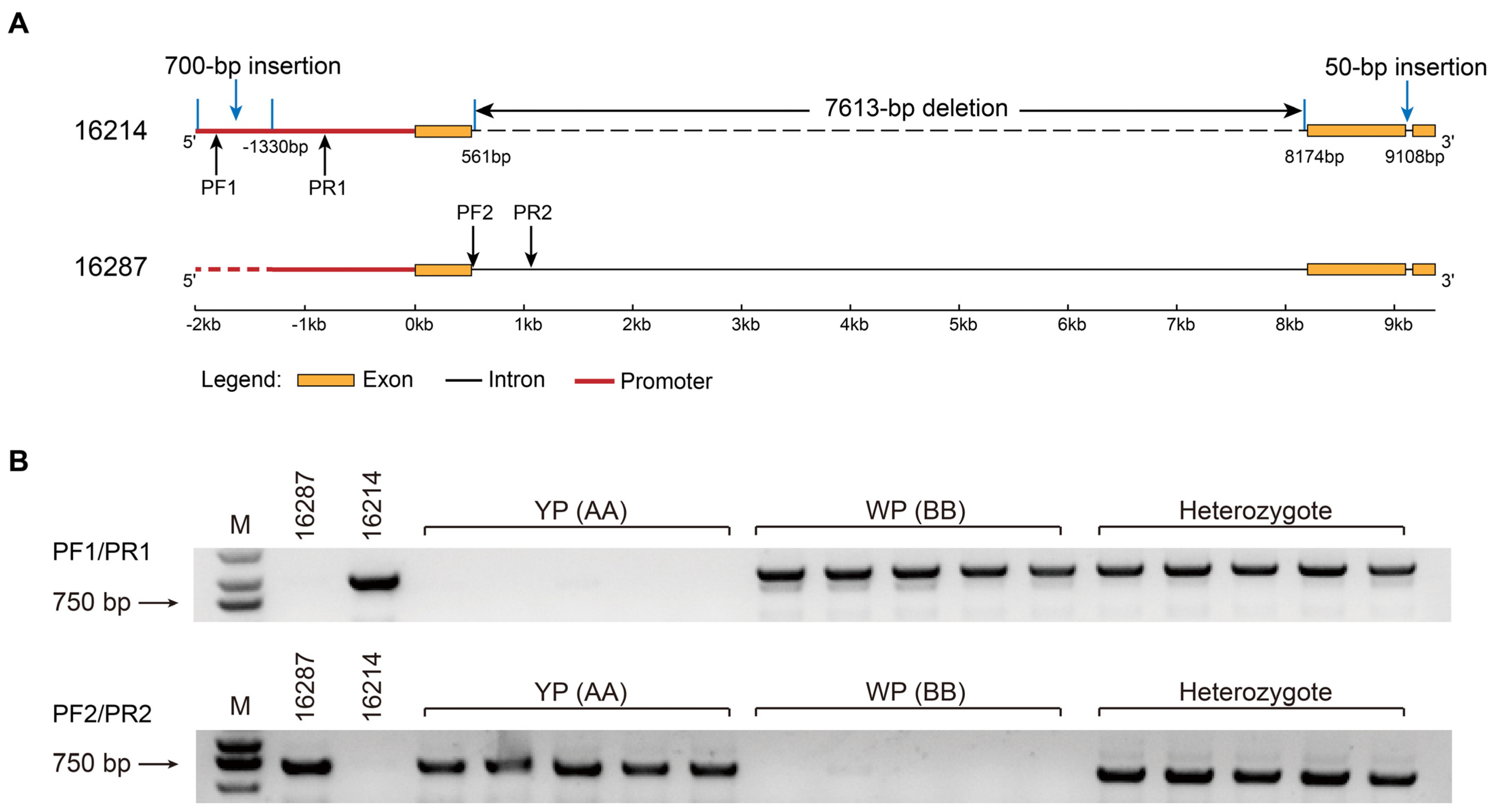
2.5. Application of Linkage Marker of BnaC03.CCD4 in B. napus
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. BSA Analysis of Two Pools of WP and YP in the F2 Population
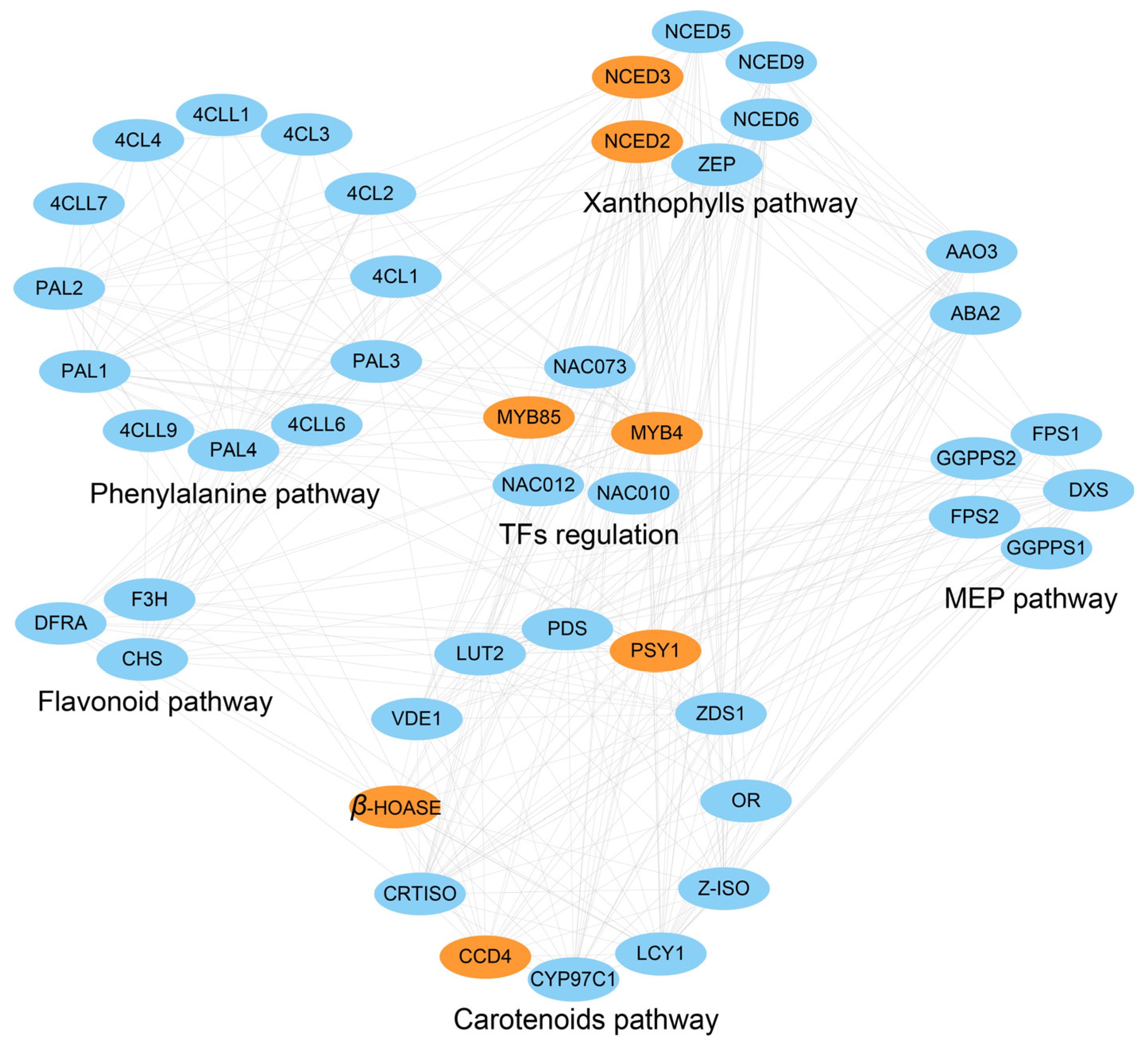
4.3. Identification of Petal Color-Related Candidate Genes and Potential Regulatory Network
4.4. RNA-Seq and DEGs Analysis of the WP and YP
4.5. The Expression Validation of DEGs via RT-qPCR
4.6. The Method for Carotenoid Measurement
4.7. The Molecular Marker Design and the Detection of B. napus with WP and YP
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xie, X.; Jiang, Y.; Xu, W.; Yang, W.; Lei, W.; Qian, D.; Gao, J.; Cai, F.; Yu, D.; Ke, L.; et al. Sef1, rapid-cycling Brassica napus for large-scale functional genome research in a controlled environment. Theor. Appl. Genet. 2023, 136, 163. [Google Scholar] [CrossRef]
- Aachary, A.A.; Thiyam, U. A Pursuit of the Functional Nutritional and Bioactive Properties of Canola Proteins and Peptides. Crit. Rev. Food Sci. Nutr. 2012, 52, 965–979. [Google Scholar] [CrossRef]
- Gacek, K.; Bartkowiak-Broda, I.; Batley, J. Genetic and Molecular Regulation of Seed Storage Proteins (SSPs) to Improve Protein Nutritional Value of Oilseed Rape (Brassica napus L.) Seeds. Front. Plant Sci. 2018, 9, 890. [Google Scholar] [CrossRef]
- Grotewold, E. The Genetics and Biochemistry of Floral Pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef]
- Walter, M.H.; Strack, D. Carotenoids and their cleavage products: Biosynthesis and functions. Nat. Prod. Rep. 2011, 28, 663–692. [Google Scholar] [CrossRef]
- Lu, C.-Y.; Nugent, G.; Wardley-Richardson, T.; Chandler, S.F.; Young, R.A.; Dalling, M.J. Agrobacterium-Mediated Transformation of Carnation (Dianthus caryophyllus L.). Bio/technology 1991, 9, 864–868. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tsuda, S.; Kusumi, T. Metabolic engineering to modify flower color. Plant Cell Physiol. 1998, 39, 1119–1126. [Google Scholar] [CrossRef]
- Mol, J.; Cornish, E.; Mason, J.; Koes, R. Novel coloured flowers. Curr. Opin. Biotechnol. 1999, 10, 198–201. [Google Scholar] [CrossRef]
- Fu, D.-H.; Jiang, L.-Y.; Mason, A.S.; Xiao, M.-L.; Zhu, L.-R.; Li, L.-Z.; Zhou, Q.-H.; Shen, C.-J.; Huang, C.-H. Research progress and strategies for multifunctional rapeseed: A case study of China. J. Integr. Agric. 2016, 15, 1673–1684. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Ohmiya, A.; Kishimoto, S.; Aida, R.; Yoshioka, S.; Sumitomo, K. Carotenoid Cleavage Dioxygenase (CmCCD4a) Contributes to White Color Formation in Chrysanthemum Petals. Plant Physiol. 2006, 142, 1193–1201. [Google Scholar] [CrossRef]
- Adami, M.; De Franceschi, P.; Brandi, F.; Liverani, A.; Giovannini, D.; Rosati, C.; Dondini, L.; Tartarini, S. Identifying a Carotenoid Cleavage Dioxygenase (ccd4) Gene Controlling Yellow/White Fruit Flesh Color of Peach. Plant Mol. Biol. Report. 2013, 31, 1166–1175. [Google Scholar] [CrossRef]
- Galpaz, N.; Ronen, G.; Khalfa, Z.; Zamir, D.; Hirschberg, J. A Chromoplast-Specific Carotenoid Biosynthesis Pathway Is Revealed by Cloning of the Tomato white-flower Locus. Plant Cell 2006, 18, 1947–1960. [Google Scholar] [CrossRef]
- Breitenbach, J.; Sandmann, G. ζ-Carotene cis isomers as products and substrates in the plant poly-cis carotenoid biosynthetic pathway to lycopene. Planta 2005, 220, 785–793. [Google Scholar] [CrossRef]
- Galpaz, N.; Burger, Y.; Lavee, T.; Tzuri, G.; Sherman, A.; Melamed, T.; Eshed, R.; Meir, A.; Portnoy, V.; Bar, E.; et al. Genetic and chemical characterization of an EMS induced mutation in Cucumis melo CRTISO gene. Arch. Biochem. Biophys. 2013, 539, 117–125. [Google Scholar] [CrossRef]
- Lee, S.; Lee, S.-C.; Byun, D.H.; Lee, D.Y.; Park, J.Y.; Lee, J.H.; Lee, H.O.; Sung, S.H.; Yang, T.-J. Association of molecular markers derived from the BrCRISTO1 gene with prolycopene-enriched orange-colored leaves in Brassica rapa. Theor. Appl. Genet. 2014, 127, 179–191. [Google Scholar] [CrossRef]
- Li, H.; Yu, K.; Amoo, O.; Yu, Y.; Guo, M.; Deng, S.; Li, M.; Hu, L.; Wang, J.; Fan, C.; et al. Site-Directed Mutagenesis of the Carotenoid Isomerase Gene BnaCRTISO Alters the Color of Petals and Leaves in Brassica napus L. Front. Plant Sci. 2022, 13, 801456. [Google Scholar] [CrossRef]
- Li, S.; Li, X.; Wang, X.; Chang, T.; Peng, Z.; Guan, C.; Guan, M. Flavonoid Synthesis-Related Genes Determine the Color of Flower Petals in Brassica napus L. Int. J. Mol. Sci. 2023, 24, 6472. [Google Scholar] [CrossRef]
- Nishihara, M.; Nakatsuka, T.; Yamamura, S. Flavonoid components and flower color change in transgenic tobacco plants by suppression of chalcone isomerase gene. FEBS Lett. 2005, 579, 6074–6078. [Google Scholar] [CrossRef]
- Nakamura, N.; Fukuchi-Mizutani, M.; Miyazaki, K.; Suzuki, K.; Tanaka, Y.J. RNAi suppression of the anthocyanidin synthase gene in Torenia hybrida yields white flowers with higher frequency and better stability than antisense and sense suppression. Plant Biotechnol. 2006, 23, 13–17. [Google Scholar] [CrossRef]
- Tsuda, S.; Fukui, Y.; Nakamura, N.; Katsumoto, Y.; Yonekura-Sakakibara, K.; Fukuchi-Mizutani, M.; Ohira, K.; Ueyama, Y.; Ohkawa, H.; Holton, T.A.; et al. Flower color modification of Petunia hybrida commercial varieties by metabolic engineering. Plant Biotechnol. 2004, 21, 377–386. [Google Scholar] [CrossRef]
- Jez, J.M.; Bowman, M.E.; Dixon, R.A.; Noel, J.P. Structure and mechanism of the evolutionarily unique plant enzyme chalcone isomerase. Nat. Struct. Biol. 2000, 7, 786–791. [Google Scholar]
- Shimizu, K.; Ohnishi, N.; Morikawa, N.; Ishigami, A.; Otake, S.; Rabah, I.O.; Sakata, Y.; Hashimoto, F. A 94-bp Deletion of Anthocyanidin Synthase Gene in Acyanic Flower Lines of Lisianthus [Eustoma grandiflorum (Raf.) Shinn.]. J. Jpn. Soc. Hortic. Sci. 2011, 80, 434–442. [Google Scholar] [CrossRef]
- Hao, P.; Liu, H.; Lin, B.; Ren, Y.; Huang, L.; Jiang, L.; Hua, S. BnaA03.ANS Identified by Metabolomics and RNA-seq Partly Played Irreplaceable Role in Pigmentation of Red Rapeseed (Brassica napus) Petal. Front. Plant Sci. 2022, 13, 940765. [Google Scholar] [CrossRef]
- Li, R.; Zeng, Q.; Zhang, X.; Jing, J.; Ge, X.; Zhao, L.; Yi, B.; Tu, J.; Fu, T.; Wen, J.; et al. Xanthophyll esterases in association with fibrillins control the stable storage of carotenoids in yellow flowers of rapeseed (Brassica juncea). New Phytol. 2023, 240, 285–301. [Google Scholar] [CrossRef]
- Ding, L.; Zhao, Z.G.; Ge, X.H.; Li, Z.Y. Different timing and spatial separation of parental chromosomes in intergeneric somatic hybrids between Brassica napus and Orychophragmus violaceus. Genet. Mol. Res. 2014, 13, 2611–2618. [Google Scholar] [CrossRef]
- Fu, W.; Chen, D.; Pan, Q.; Li, F.; Zhao, Z.; Ge, X.; Li, Z. Production of red-flowered oilseed rape via the ectopic expression of Orychophragmus violaceus OvPAP2. Plant Biotechnol. J. 2018, 16, 367–380. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, C.; Wang, Y.; Yao, X.; Wang, F.; Wu, J.; King, G.J.; Liu, K. Disruption of a CAROTENOID CLEAVAGE DIOXYGENASE 4 gene converts flower colour from white to yellow in Brassica species. New Phytol. 2015, 206, 1513–1526. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, S.; Yuan, G.; Ma, X.; Heng, S.; Yi, B.; Ma, C.; Shen, J.; Tu, J.; Fu, T.; et al. Gene silencing of BnaA09.ZEP and BnaC09.ZEP confers orange color in Brassica napus flowers. Plant J. 2020, 104, 932–949. [Google Scholar] [CrossRef]
- Zhao, C.; Safdar, L.B.; Xie, M.; Shi, M.; Dong, Z.; Yang, L.; Cheng, X.; Liu, Y.; Bai, Z.; Xiang, Y.; et al. Mutation of the PHYTOENE DESATURASE 3 gene causes yellowish-white petals in Brassica napus. Crop J. 2021, 9, 1124–1134. [Google Scholar] [CrossRef]
- Chen, D.; Jin, Q.; Pan, J.; Liu, Y.; Tang, Y.; E, Y.; Xu, L.; Yang, T.; Qiu, J.; Chen, X.; et al. Fine mapping of genes controlling pigment accumulation in oilseed rape (Brassica napus L.). Mol. Breed. 2023, 43, 19. [Google Scholar] [CrossRef]
- Triques, K.; Sturbois, B.; Gallais, S.; Dalmais, M.; Chauvin, S.; Clepet, C.; Aubourg, S.; Rameau, C.; Caboche, M.; Bendahmane, A. Characterization of Arabidopsis thaliana mismatch specific endonucleases: Application to mutation discovery by TILLING in pea. Plant J. 2007, 51, 1116–1125. [Google Scholar] [CrossRef]
- Duchêne, A.M.; Giritch, A.; Hoffmann, B.; Cognat, V.; Lancelin, D.; Peeters, N.M.; Zaepfel, M.; Maréchal-Drouard, L.; Small, I.D. Dual targeting is the rule for organellar aminoacyl-tRNA synthetases in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2005, 102, 16484–16489. [Google Scholar] [CrossRef] [PubMed]
- Rehrauer, H.; Aquino, C.; Gruissem, W.; Henz, S.R.; Hilson, P.; Laubinger, S.; Naouar, N.; Patrignani, A.; Rombauts, S.; Shu, H.; et al. AGRONOMICS1: A New Resource for Arabidopsis Transcriptome Profiling. Plant Physiol. 2010, 152, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Giglione, C.; Serero, A.; Pierre, M.; Boisson, B.; Meinnel, T. Identification of eukaryotic peptide deformylases reveals universality of N-terminal protein processing mechanisms. EMBO J. 2000, 19, 5916–5929. [Google Scholar] [CrossRef] [PubMed]
- Bernacki, M.J.; Rusaczonek, A.; Czarnocka, W.; Karpiński, S. Salicylic Acid Accumulation Controlled by LSD1 Is Essential in Triggering Cell Death in Response to Abiotic Stress. Cells 2021, 10, 962. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Gao, X.; Gao, X.; Zhang, S.; Zheng, Y.; Zhang, N.; Hong, B.; Zhao, X.; Gu, Z. Flower color mutation, pink to orange, through CmGATA4—CCD4a-5 module regulates carotenoids degradation in chrysanthemum. Plant Sci. 2022, 322, 111290. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wang, J.; Wang, R.; Duan, M.; Qiao, C.; Chen, X.; Ma, G.; Zhou, X.; Zhu, M.; Jing, F.; et al. Comparative transcriptomic and metabolomic analyses of carotenoid biosynthesis reveal the basis of white petal color in Brassica napus. Planta 2021, 253, 8. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Liu, S.; Hu, L.; Shi, J.; Chen, J. Transcriptome analysis and metabolic profiling reveal the key role of carotenoids in the petal coloration of Liriodendron tulipifera. Hortic. Res. 2020, 7, 70. [Google Scholar] [CrossRef]
- Zhou, N.; Yan, Y.; Wen, Y.; Zhang, M.; Huang, Y. Integrated transcriptome and metabolome analysis unveils the mechanism of color-transition in Edgeworthia chrysantha tepals. BMC Plant Biol. 2023, 23, 567. [Google Scholar] [CrossRef]
- Zeng, H.; Zheng, T.; Li, Y.; Chen, Q.; Xue, Y.; Tang, Q.; Xu, H.; Chen, M. Characterization Variation of the Differential Coloring Substances in Rapeseed Petals with Different Colors Using UPLC-HESI-MS/MS. Molecules 2023, 28, 5670. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Hua, S.; Ma, T.; Ma, X.; Chen, Y.; Wu, L.; Zhao, L.; Yi, B.; Ma, C.; Tu, J.; et al. Genetic and multi-omics analyses reveal BnaA07.PAP2In−184−317 as the key gene conferring anthocyanin-based color in Brassica napus flowers. J. Exp. Bot. 2022, 73, 6630–6645. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, Z.; Chen, C.; Wu, W.; Ren, N.; Jiang, C.; Yu, J.; Zhao, Y.; Zheng, X.; Yang, Q.; et al. The C–S–A gene system regulates hull pigmentation and reveals evolution of anthocyanin biosynthesis pathway in rice. J. Exp. Bot. 2018, 69, 1485–1498. [Google Scholar] [CrossRef] [PubMed]
- Petroni, K.; Tonelli, C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 2011, 181, 219–229. [Google Scholar] [CrossRef]
- Agarwal, P.; Mitra, M.; Banerjee, S.; Roy, S. MYB4 transcription factor, a member of R2R3-subfamily of MYB domain protein, regulates cadmium tolerance via enhanced protection against oxidative damage and increases expression of PCS1 and MT1C in Arabidopsis. Plant Sci. 2020, 297, 110501. [Google Scholar] [CrossRef]
- Panda, C.; Li, X.; Wager, A.; Chen, H.Y.; Li, X. An importin-beta-like protein mediates lignin-modification-induced dwarfism in Arabidopsis. Plant J. 2020, 102, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Geng, P.; Zhang, S.; Liu, J.; Zhao, C.; Wu, J.; Cao, Y.; Fu, C.; Han, X.; He, H.; Zhao, Q. MYB20, MYB42, MYB43, and MYB85 Regulate Phenylalanine and Lignin Biosynthesis during Secondary Cell Wall Formation. Plant Physiol. 2020, 182, 1272–1283. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAS and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef]
- Gismondi, A.; Di Marco, G.; Camoni, L.; Montesano, C.; Braglia, R.; Marra, M.; Canini, A. MicroRNA Expression Profiles in Moringa oleifera Lam. Seedlings at Different Growth Conditions. J. Plant Growth Regul. 2022, 42, 2115–2123. [Google Scholar] [CrossRef]
- Camargo-Ramírez, R.; Val-Torregrosa, B.; San Segundo, B. MiR858-Mediated Regulation of Flavonoid-Specific MYB Transcription Factor Genes Controls Resistance to Pathogen Infection in Arabidopsis. Plant Cell Physiol. 2018, 59, 190–204. [Google Scholar] [CrossRef]
- ValdÉS-LÓPez, O.; Arenas-Huertero, C.; RamÍRez, M.; Girard, L.; SÁNchez, F.; Vance, C.P.; Luis Reyes, J.; HernÁNdez, G. Essential role of MYB transcription factor: PvPHR1 and microRNA: PvmiR399 in phosphorus-deficiency signalling in common bean roots. Plant Cell Environ. 2008, 31, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Liu, Z.; Zhao, C.; Zhao, J.; Zheng, Y.; Zhang, H.; Tan, C.; Zhang, Z.; Xue, L.; Lei, J. Comparative Transcriptome Analysis Uncovers the Regulatory Roles of MicroRNAs Involved in Petal Color Change of Pink-Flowered Strawberry. Front. Plant Sci. 2022, 13, 854508. [Google Scholar] [CrossRef]
- Rong, H.; Han, X.; Xin, Y.; Ni, Z.; Zhang, W.; Xu, L.A. Small RNA and Degradome Sequencing Reveal Roles of miRNAs in the Petal Color Fading of Malus Crabapple. Int. J. Mol. Sci. 2023, 24, 11384. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Long, Y.; Li, D.; Yin, Y.; Tian, J.; Chen, L.; Liu, L.; Zhao, W.; Zhao, Y.; et al. Identification of QTLs associated with oil content in a high-oil Brassica napus cultivar and construction of a high-density consensus map for QTLs comparison in B. napus. PLoS ONE 2013, 8, e80569. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tong, C.; Zhang, X.; Song, A.; Hu, M.; Dong, W.; Chen, F.; Wang, Y.; Tu, J.; Liu, S.; et al. A high-quality Brassica napus genome reveals expansion of transposable elements, subgenome evolution and disease resistance. Plant Biotechnol. J. 2020, 19, 615–630. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Chao, H.; Wang, H.; Wang, X.; Guo, L.; Gu, J.; Zhao, W.; Li, B.; Chen, D.; Raboanatahiry, N.; Li, M. Genetic dissection of seed oil and protein content and identification of networks associated with oil content in Brassica napus. Sci. Rep. 2017, 7, 46295. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Podnar, J.; Deiderick, H.; Huerta, G.; Hunicke-Smith, S. Next-Generation Sequencing RNA-Seq Library Construction. Curr. Protoc. Mol. Biol. 2014, 106, 4.21.1–4.21.19. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Cheng, X.; Liu, X.; He, J.; Tang, M.; Li, H.; Li, M. The genome wide analysis of Tryptophan Aminotransferase Related gene family, and their relationship with related agronomic traits in Brassica napus. Front. Plant Sci. 2022, 13, 1098820. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Y.; Li, H.; Liu, X.; Cheng, X.; Chen, W.; Wu, M.; Chen, L.; He, J.; Chao, H.; Jia, H.; et al. Multi-Omics Analysis Revealed the AGR-FC.C3 Locus of Brassica napus as a Novel Candidate for Controlling Petal Color. Plants 2024, 13, 507. https://doi.org/10.3390/plants13040507
Ding Y, Li H, Liu X, Cheng X, Chen W, Wu M, Chen L, He J, Chao H, Jia H, et al. Multi-Omics Analysis Revealed the AGR-FC.C3 Locus of Brassica napus as a Novel Candidate for Controlling Petal Color. Plants. 2024; 13(4):507. https://doi.org/10.3390/plants13040507
Chicago/Turabian StyleDing, Yiran, Huaixin Li, Xinmin Liu, Xin Cheng, Wang Chen, Mingli Wu, Liurong Chen, Jianjie He, Hongbo Chao, Haibo Jia, and et al. 2024. "Multi-Omics Analysis Revealed the AGR-FC.C3 Locus of Brassica napus as a Novel Candidate for Controlling Petal Color" Plants 13, no. 4: 507. https://doi.org/10.3390/plants13040507