Computationally Designed Peptides for Zika Virus Detection: An Incremental Construction Approach

 ,
,  , ,
, ,
Abstract
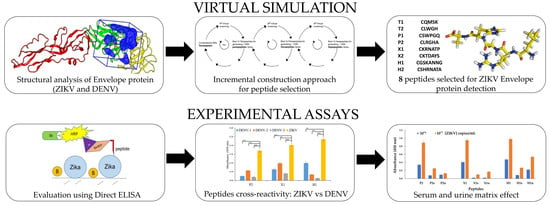
:
1. Introduction
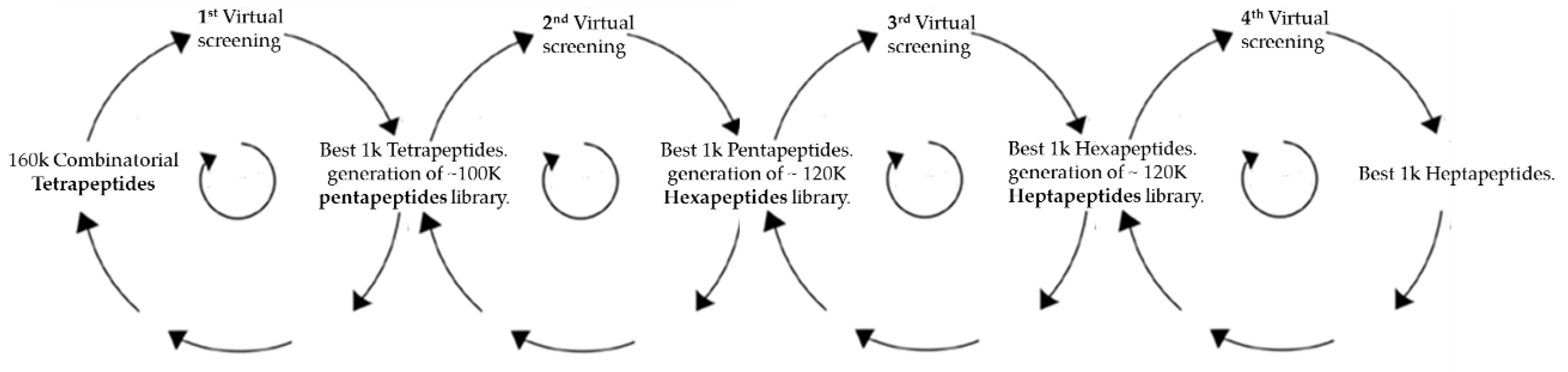
2. Materials and Methods
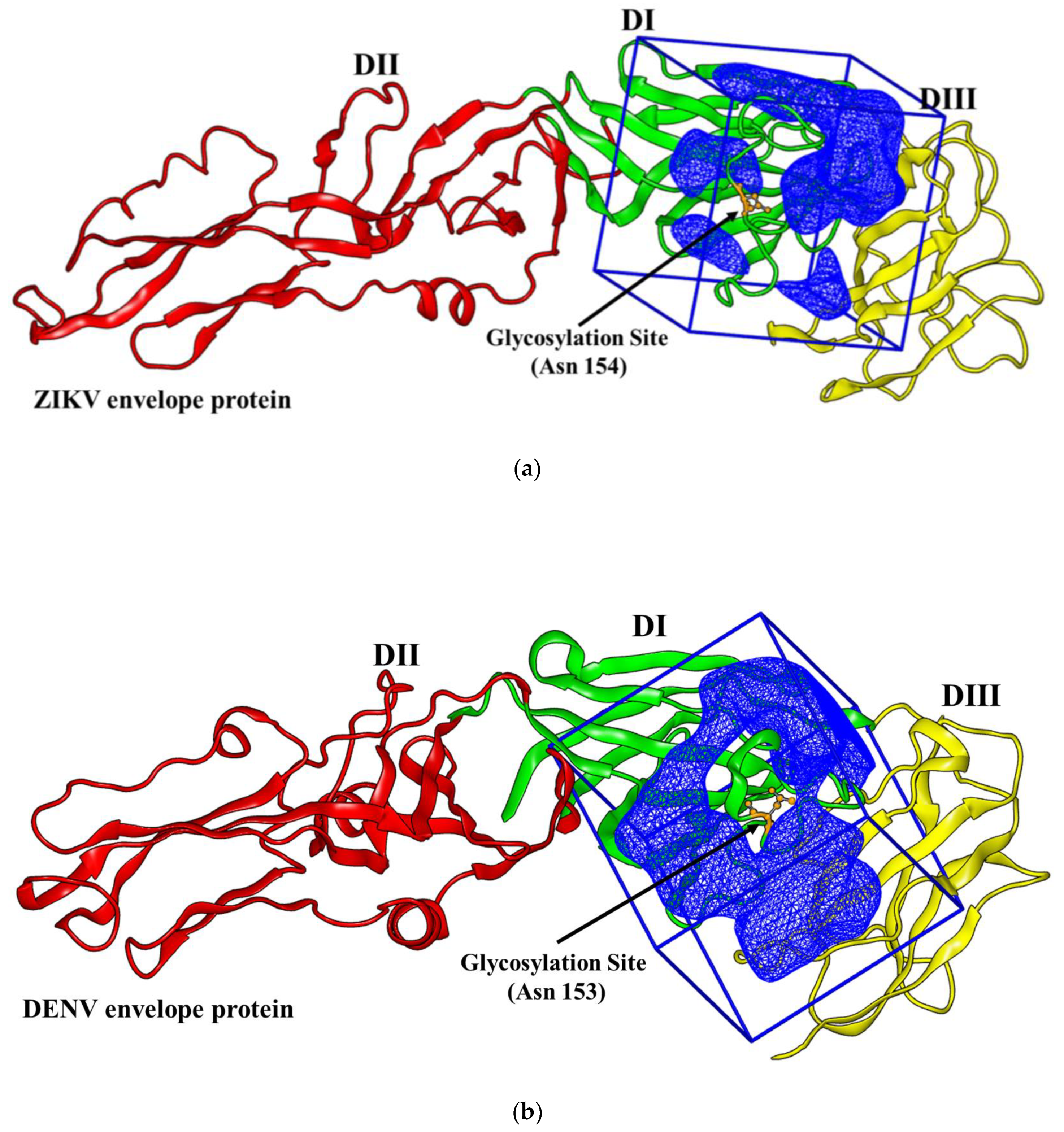
2.1. Virtual Docking
2.2. Experimental Setup
3. Results and Discussion
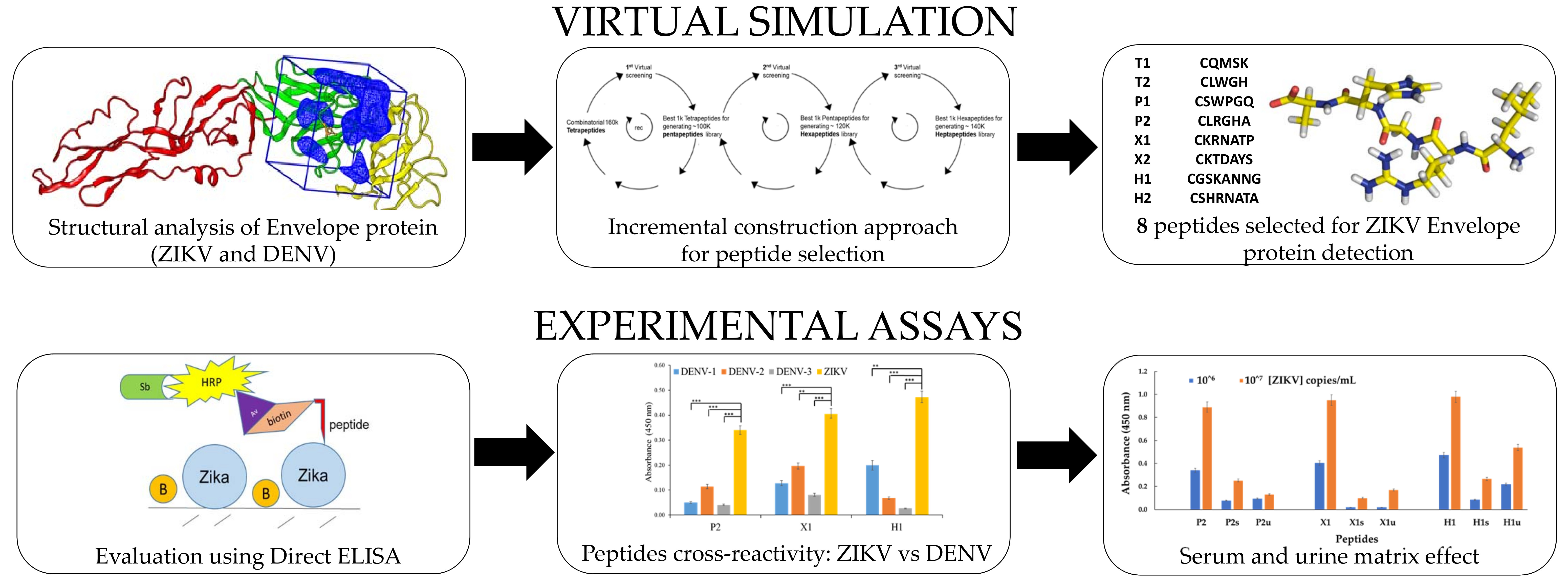
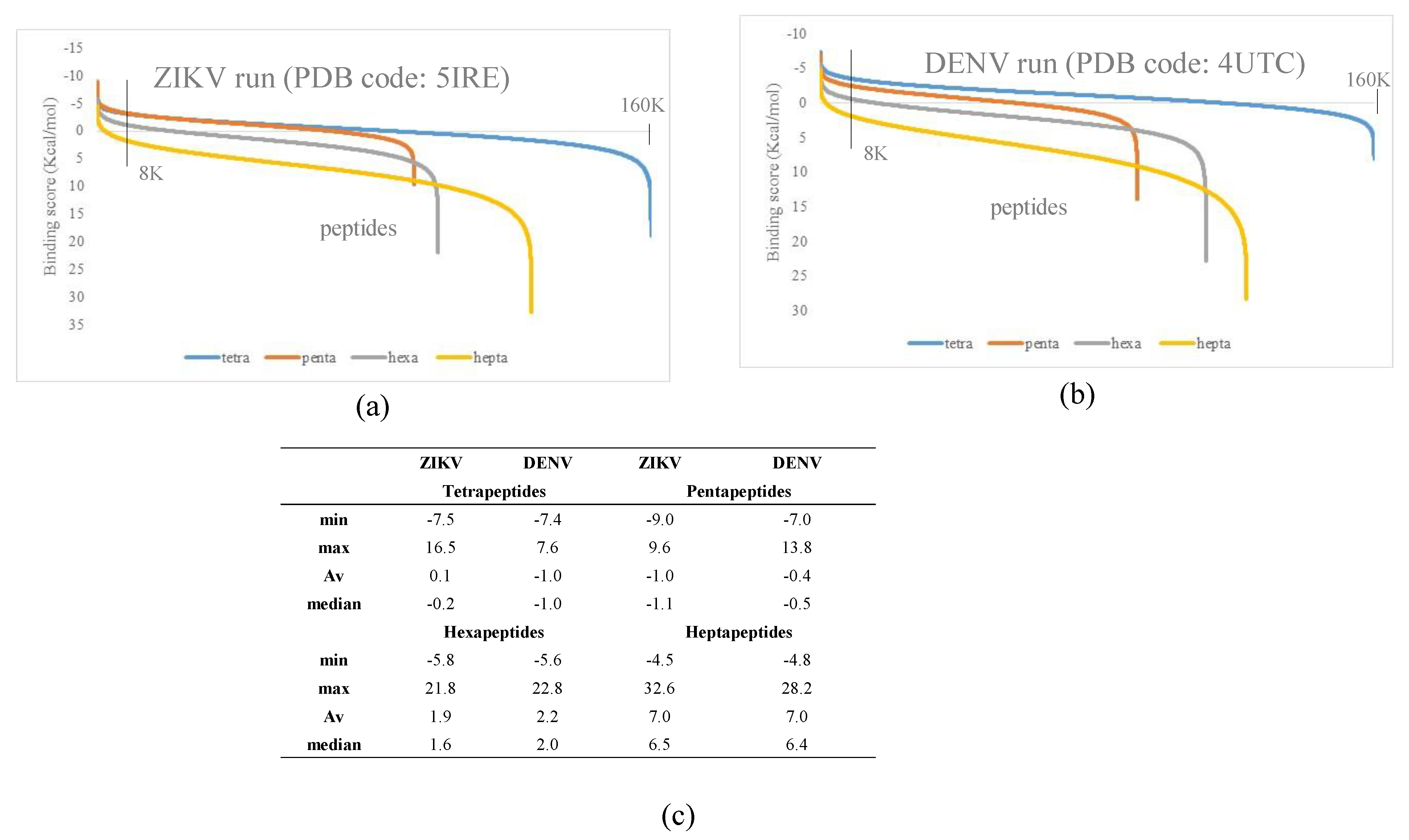
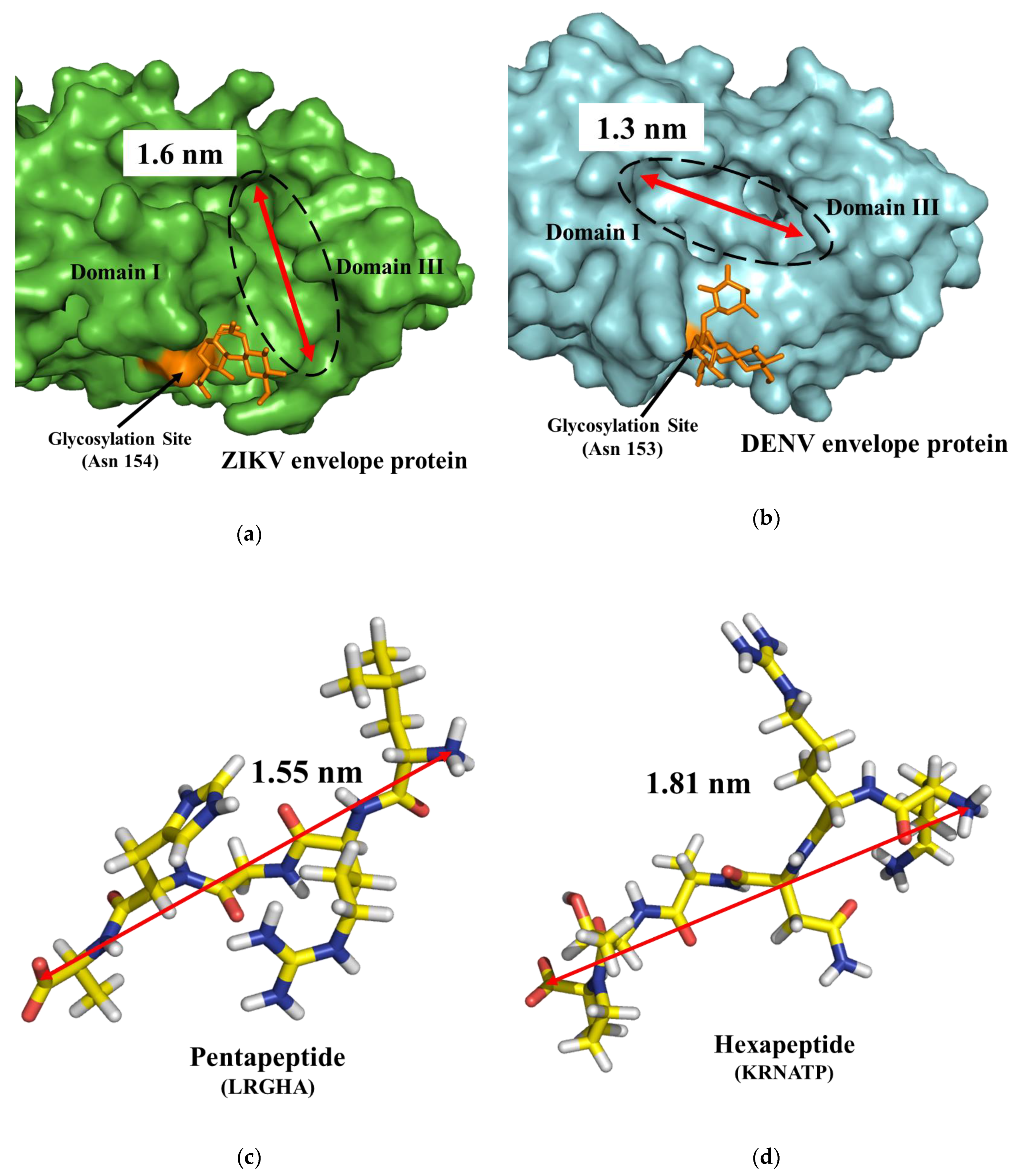
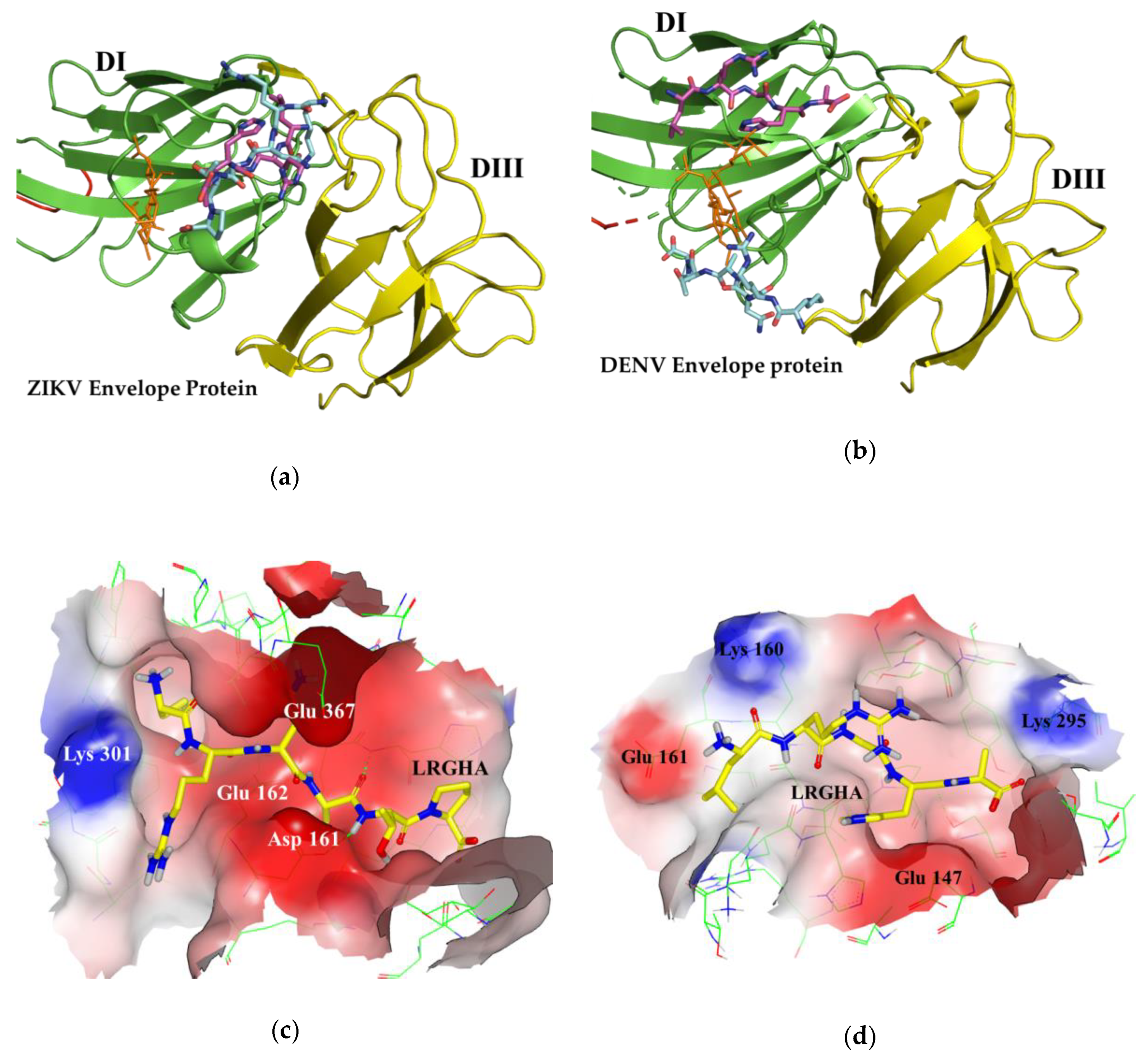
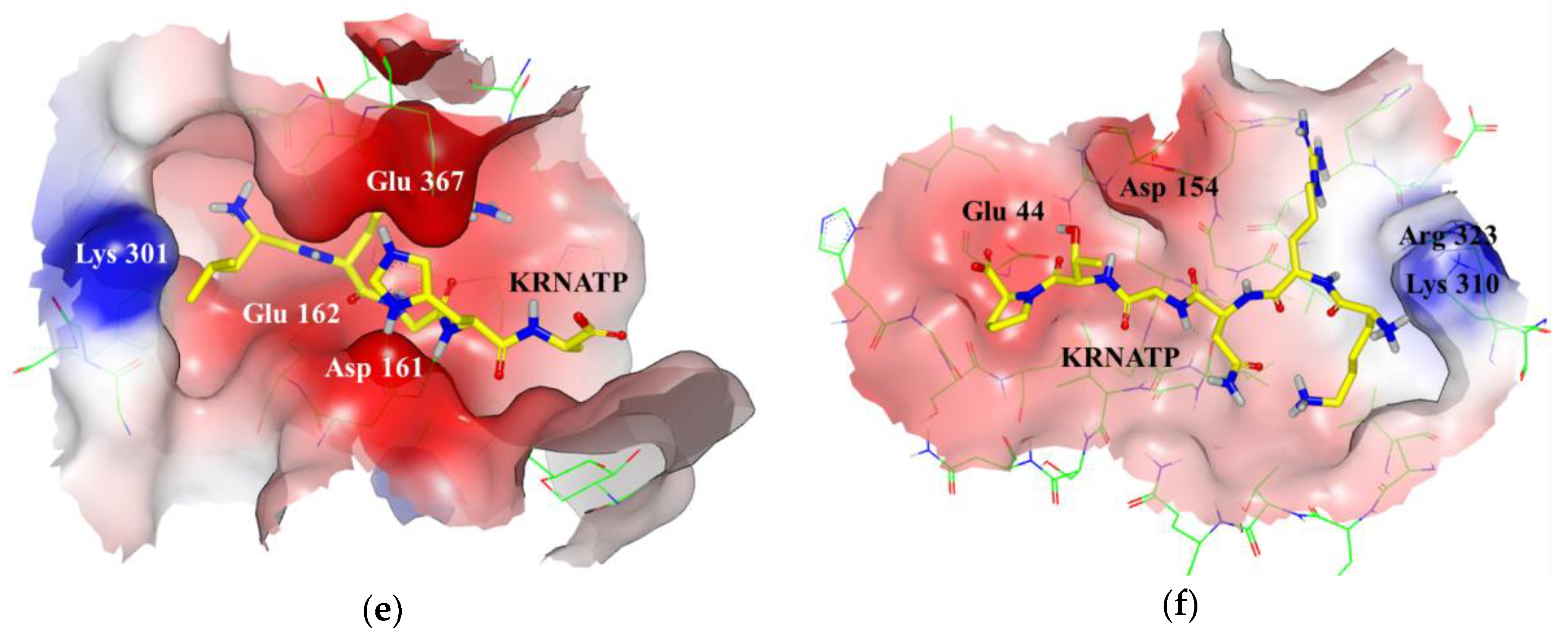
3.1. Docking Simulations
3.2. Experimental Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ioos, S.; Mallet, H.-P.; Goffart, I.L.; Gauthier, V.; Cardoso, T.; Herida, M. Current Zika virus epidemiology and recent epidemics. Med. Et Mal. Infect. 2014, 44, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.-Y.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Barba-Spaeth, G.; Dejnirattisai, W.; Rouvinski, A.; Vaney, M.-C.; Medits, I.; Sharma, A.; Simon-Lorière, E.; Sakuntabhai, A.; Cao-Lormeau, V.-M.; Haouz, A. Structural basis of potent Zika–dengue virus antibody cross-neutralization. Nature 2016, 536, 48. [Google Scholar] [CrossRef] [PubMed]
- Heffron, A.S.; Mohr, E.L.; Baker, D.; Haj, A.K.; Buechler, C.R.; Bailey, A.; Dudley, D.M.; Newman, C.M.; Mohns, M.S.; Koenig, M. Antibody responses to Zika virus proteins in pregnant and non-pregnant macaques. Plos Negl. Trop. Dis. 2018, 12, e0006903. [Google Scholar] [CrossRef] [PubMed]
- Priyamvada, L.; Quicke, K.M.; Hudson, W.H.; Onlamoon, N.; Sewatanon, J.; Edupuganti, S.; Pattanapanyasat, K.; Chokephaibulkit, K.; Mulligan, M.J.; Wilson, P.C. Human antibody responses after dengue virus infection are highly cross-reactive to Zika virus. Proc. Natl. Acad. Sci. 2016, 113, 7852–7857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stettler, K.; Beltramello, M.; Espinosa, D.A.; Graham, V.; Cassotta, A.; Bianchi, S.; Vanzetta, F.; Minola, A.; Jaconi, S.; Mele, F. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 2016, 353, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef]
- Zhao, H.; Fernandez, E.; Dowd, K.A.; Speer, S.D.; Platt, D.J.; Gorman, M.J.; Govero, J.; Nelson, C.A.; Pierson, T.C.; Diamond, M.S. Structural basis of Zika virus-specific antibody protection. Cell 2016, 166, 1016–1027. [Google Scholar] [CrossRef]
- Rather, I.A.; Lone, J.B.; Bajpai, V.K.; Park, Y.-H. Zika virus infection during pregnancy and congenital abnormalities. Front. Microbiol. 2017, 8, 581. [Google Scholar] [CrossRef]
- Martina, B.E.; Koraka, P.; Osterhaus, A.D. Dengue virus pathogenesis: An integrated view. Clin. Microbiol. Rev. 2009, 22, 564–581. [Google Scholar] [CrossRef]
- Goncalves, A.; Peeling, R.W.; Chu, M.C.; Gubler, D.J.; de Silva, A.M.; Harris, E.; Murtagh, M.; Chua, A.; Rodriguez, W.; Kelly, C. Innovative and new approaches to laboratory diagnosis of Zika and dengue: A meeting report. J. Infect. Dis. 2017, 217, 1060–1068. [Google Scholar] [CrossRef]
- Yu, X.; Yang, Y.-P.; Dikici, E.; Deo, S.K.; Daunert, S. Beyond antibodies as binding partners: The role of antibody mimetics in bioanalysis. Annu. Rev. Anal. Chem. 2017, 10, 293–320. [Google Scholar] [CrossRef]
- Gong, P.C.; Wang, L.; He, C.Y. Peptide aptamer: A powerful potential tool in plant functional genomics. Yi Chuan = Hered./Zhongguo Yi Chuan Xue Hui Bian Ji 2010, 32, 548–554. [Google Scholar] [CrossRef]
- Pichon, V.; Brothier, F.; Combes, A. Aptamer-based-sorbents for sample treatment--a review. Anal. Bioanal. Chem. 2015, 407, 681–698. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yu, Z.; Han, X.; Lai, R.Y. Electrochemical aptamer-based sensors for food and water analysis: A review. Anal. Chim. Acta 2018. [Google Scholar] [CrossRef] [PubMed]
- Mascini, M.; Gaggiotti, S.; Della Pelle, F.; Wang, J.; Pingarrón, J.M.; Compagnone, D. Hairpin DNA-AuNPs as molecular binding elements for the detection of volatile organic compounds. Biosens. Bioelectron. 2019, 123, 124–130. [Google Scholar] [CrossRef]
- Mascini, M.; Montesano, C.; Perez, G.; Wang, J.; Compagnone, D.; Sergi, M. Selective solid phase extraction of JWH synthetic cannabinoids by using computationally designed peptides. Talanta 2017, 167, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, T.; Deng, J.; Zhang, M.; Shi, G.; Zhou, T. Quantum dot-DNA aptamer conjugates coupled with capillary electrophoresis: A universal strategy for ratiometric detection of organophosphorus pesticides. Talanta 2016, 146, 55–61. [Google Scholar] [CrossRef]
- Lin, S.; Gan, N.; Cao, Y.; Chen, Y.; Jiang, Q. Selective dispersive solid phase extraction-chromatography tandem mass spectrometry based on aptamer-functionalized UiO-66-NH2 for determination of polychlorinated biphenyls. J. Chromatogr. A 2016, 1446, 34–40. [Google Scholar] [CrossRef]
- Stobiecka, M.; Chałupa, A. Biosensors based on molecular beacons. Chem. Pap. 2015, 69, 62–76. [Google Scholar] [CrossRef]
- Palzkill, T.G.; Estes, M.K.; Atmar, R.L.; Rogers, J.D.; Ajami, N.J. Identification and characterization of a peptide affinity reagent for the detection of noroviruses in clinical samples. J. Clin. Microbiolo. 2013, 51, 1803–1808. [Google Scholar]
- Hwang, H.J.; Ryu, M.Y.; Park, C.Y.; Ahn, J.; Park, H.G.; Choi, C.; Ha, S.-D.; Park, T.J.; Park, J.P. High sensitive and selective electrochemical biosensor: Label-free detection of human norovirus using affinity peptide as molecular binder. Biosens. Bioelectron. 2017, 87, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Tambunan, U.S.F.; Chua, W.; Parikesit, A.A.; Kerami, D. Designing Disulfide Cyclic Peptide as Fusion Inhibitor That Targets DENV Envelope Protein. J. Teknol. 2016, 78. [Google Scholar] [CrossRef]
- Do Thi Hoang Kim, D.T.; Bao, H.P.; Nguyen Minh Ngoc, S.-J.Y. Development of a novel peptide aptamer-based immunoassay to detect Zika virus in serum and urine. Theranostics 2018, 8, 3629. [Google Scholar]
- Bunker, A.; Magarkar, A.; Viitala, T. Rational design of liposomal drug delivery systems, a review: Combined experimental and computational studies of lipid membranes, liposomes and their PEGylation. Biochim. Et Biophys. Acta (Bba)-Biomembr. 2016, 1858, 2334–2352. [Google Scholar] [CrossRef]
- Acebes, S.; Fernandez-Fueyo, E.; Monza, E.; Lucas, M.F.; Almendral, D.; Ruiz-Dueñas, F.J.; Lund, H.; Martinez, A.T.; Guallar, V. Rational enzyme engineering through biophysical and biochemical modeling. Acs Catal. 2016, 6, 1624–1629. [Google Scholar] [CrossRef]
- Xu, K.; Acharya, P.; Kong, R.; Cheng, C.; Chuang, G.-Y.; Liu, K.; Louder, M.K.; O’Dell, S.; Rawi, R.; Sastry, M. Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat. Med. 2018, 24, 857. [Google Scholar] [CrossRef]
- Michaeli, A.; Mezan, S.; Kühbacher, A.; Finkelmeier, D.; Elias, M.; Zatsepin, M.; Reed, S.G.; Duthie, M.S.; Rupp, S.; Lerner, I. Computationally Designed Bispecific MD2/CD14 Binding Peptides Show TLR4 Agonist Activity. J. Immunol. 2018, 201, 3383–3391. [Google Scholar] [CrossRef]
- Singh, P.; Kaur, S.; Kaur, J.; Singh, G.; Bhatti, R. Rational design of small peptides for optimal inhibition of cyclooxygenase-2: Development of a highly effective anti-inflammatory agent. J. Med. Chem. 2016, 59, 3920–3934. [Google Scholar] [CrossRef]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- SZYBKI, version 1.5.7.; OpenEye Scientific Software: Santa Fe, NM, USA, 2012; Available online: http://www.eyesopen.com (accessed on 29 April 2019).
- OMEGA, version 2.4.6.; OpenEye Scientific Software: Santa Fe, NM, USA, 2012; Available online: http://www.eyesopen.com (accessed on 29 April 2019).
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Modeling 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Modeling 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Rouvinski, A.; Guardado-Calvo, P.; Barba-Spaeth, G.; Duquerroy, S.; Vaney, M.-C.; Kikuti, C.M.; Sanchez, M.E.N.; Dejnirattisai, W.; Wongwiwat, W.; Haouz, A. Recognition determinants of broadly neutralizing human antibodies against dengue viruses. Nature 2015, 520, 109. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible shape-guided docking for pose prediction. J. Chem. Inf. Modeling 2015, 55, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- OEDocking, version 3.0.0.; OpenEye Scientific Software: Santa Fe, NM, USA, 2012; Available online: http://www.eyesopen.com (accessed on 29 April 2019).
- VIDA, version 4.1.1.; OpenEye Scientific Software: Santa Fe, NM, USA, 2012; Available online: http://www.eyesopen.com (accessed on 29 April 2019).
- The PyMOL Molecular Graphics System; Version 2.1.; Schrodinger, LLC: New York, NY, USA, 2018; Available online: www.pymol.org (accessed on 29 April 2019).
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Magnani, D.M.; Rogers, T.F.; Beutler, N.; Ricciardi, M.J.; Bailey, V.K.; Gonzalez-Nieto, L.; Briney, B.; Sok, D.; Le, K.; Strubel, A. Neutralizing human monoclonal antibodies prevent Zika virus infection in macaques. Sci. Transl. Med. 2017, 9, eaan8184. [Google Scholar] [CrossRef]
- Perez, G.; Mascini, M.; Lanzone, V.; Sergi, M.; Del Carlo, M.; Esposito, M.; Compagnone, D. Peptides trapping dioxins: A docking-based inverse screening approach. J. Chem. 2013. [Google Scholar] [CrossRef]
- Pawley, D.C.; Ricciardi, M.J.; Dikici, E.; Deo, S.K.; Daunert, S. Highly Sensitive and Selective Direct Detection of Zika Virus Particles in Human Bodily Fluids for Accurate Early Diagnosis of Infection. ACS Omega 2019, 4, 6808–6818. [Google Scholar] [CrossRef] [Green Version]
- Campos, R.d.M.; Cirne-Santos, C.; Meira, G.L.; Santos, L.L.; de Meneses, M.D.; Friedrich, J.; Jansen, S.; Ribeiro, M.S.; da Cruz, I.C.; Schmidt-Chanasit, J. Prolonged detection of Zika virus RNA in urine samples during the ongoing Zika virus epidemic in Brazil. J. Clin. Virol. 2016, 77, 69–70. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | Occurrence (%) | |||
| ZIKV | DENV | |||
| Side chain type | Receptor Active Site | |||
| Aliphatic | 45 | 49 | ||
| Polar | 26 | 19 | ||
| Aromatic | 4 | 4 | ||
| Negative | 12 | 13 | ||
| Positive | 13 | 15 | ||
| (b) | Occurrence (%) | Occurrence (%) | ||
| ZIKV | DENV | ZIKV | DENV | |
| Side chain type | Tetrapeptide | Pentapeptide | ||
| Aliphatic | 44 | 61 | 51 | 60 |
| Polar | 31 | 19 | 32 | 32 |
| Aromatic | 8 | 5 | 7 | 2 |
| Negative | 2 | 1 | 1 | 0 |
| Positive | 14 | 14 | 9 | 6 |
| Hexapeptide | Heptapeptide | |||
| Aliphatic | 50 | 46 | 49 | 60 |
| Polar | 33 | 42 | 36 | 32 |
| Aromatic | 5 | 8 | 4 | 4 |
| Negative | 3 | 0 | 1 | 1 |
| Positive | 10 | 3 | 9 | 3 |
| (a) | Docking Score Rank | (b) | ||||||
|---|---|---|---|---|---|---|---|---|
| Peptide in Simulation | ZIKV | DENV | Peptide in Experimental | Label | Iso-Point (pH) | Net Charge at pH 7 | Water Sol. | MW |
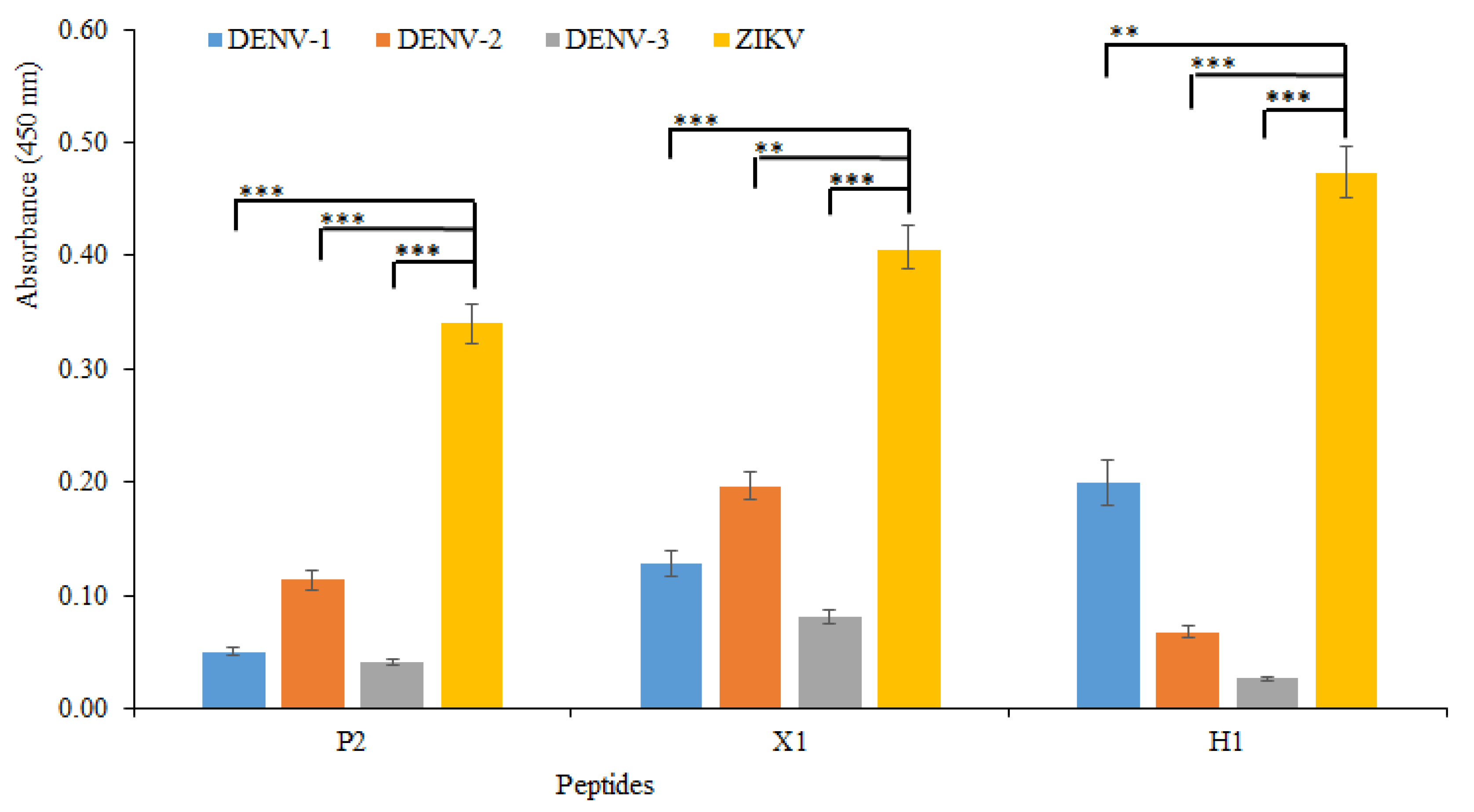
| QMSK | 15 | 32607 | C-QMSK | T1 | 9.13 | 0.9 | good | 595 |
| LWGH | 48 | 101726 | C-LWGH | T2 | 7.09 | 0.0 | poor | 614 |
| SWPGQ | 4 | 55575 | C-SWPGQ | P1 | 2.98 | −0.1 | poor | 676 |
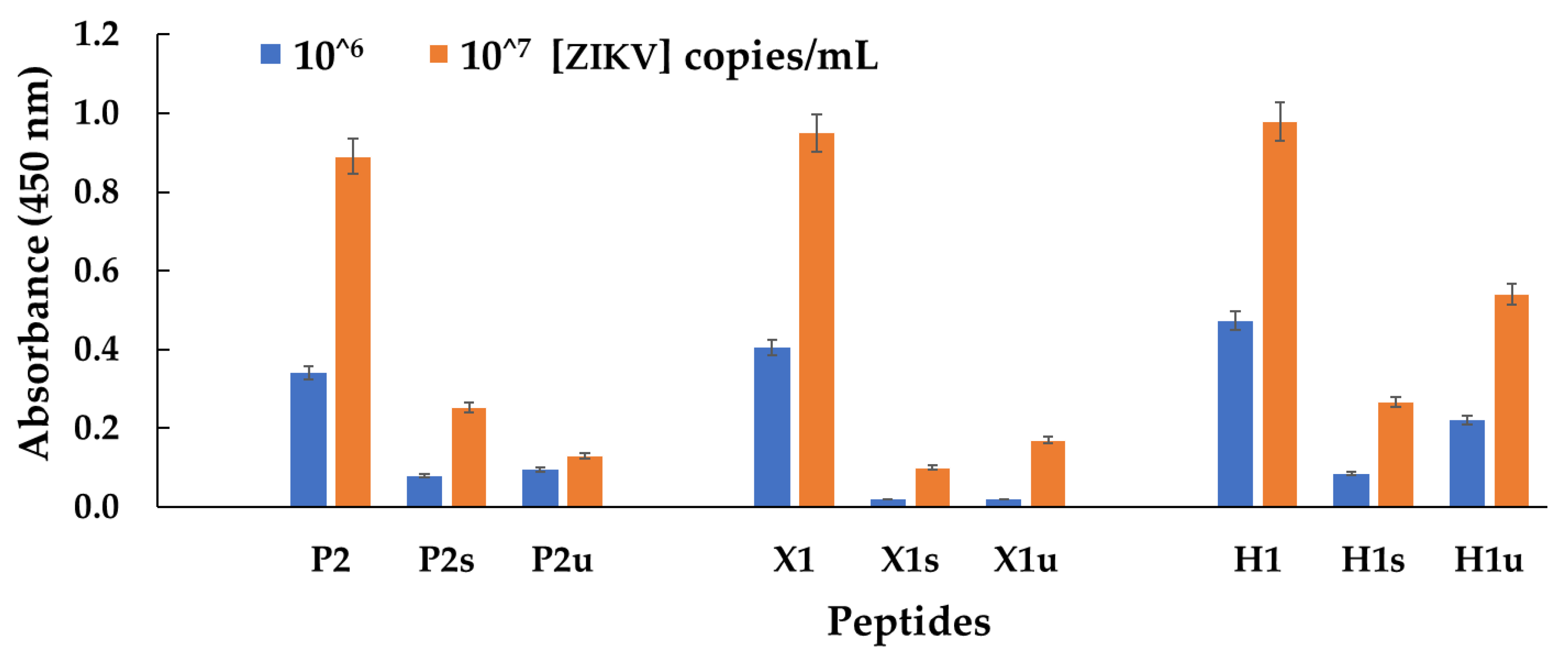
| LRGHA | 53 | 74900 | C-LRGHA | P2 | 9.21 | 1.0 | good | 655 |
| KRNATP | 16 | 85123 | C-KRNATP | X1 | 10.46 | 1.9 | good | 788 |
| KTDAYS | 120 | 95558 | C-KTDAYS | X2 | 5.92 | −0.1 | good | 786 |
| GSKANNG | 1 | 63937 | C-GSKANNG | H1 | 9.13 | 0.9 | good | 749 |
| SHRNATA | 5 | 94782 | C-SHRNATA | H2 | 9.21 | 1.0 | good | 858 |
| ZIKV | 1P | 2P | 3P | 4P | Av | ||||
| QMSK | 21 | 10 | 8 | 11 | 13 | ||||
| LWGH | 17 | 11 | 39 | 16 | 21 | ||||
| best occurring AA: QSGH | 21 | 18 | 39 | 16 | 23 | ||||
| DENV | 1P | 2P | 3P | 4P | Av | ||||
| QMSK | 0 | 3 | 4 | 0 | 2 | ||||
| LWGH | 1 | 0 | 36 | 1 | 10 | ||||
| best occurring AA: GPGP | 26 | 21 | 36 | 21 | 26 | ||||
| ZIKV | 1P | 2P | 3P | 4P | 5P | Av | |||
| SWPGQ | 29 | 18 | 31 | 39 | 2 | 24 | |||
| LRGHA | 11 | 6 | 21 | 21 | 8 | 13 | |||
| best occurring AA: SWPGG | 29 | 18 | 31 | 39 | 19 | 27 | |||
| DENV | 1P | 2P | 3P | 4P | 5P | Av | |||
| SWPGQ | 8 | 1 | 10 | 20 | 0 | 8 | |||
| LRGHA | 21 | 0 | 13 | 7 | 12 | 11 | |||
| best occurring AA: LGASG | 21 | 38 | 35 | 43 | 38 | 35 | |||
| ZIKV | 1P | 2P | 3P | 4P | 5P | 6P | Av | ||
| KRNATP | 10 | 6 | 32 | 57 | 37 | 43 | 31 | ||
| KTDAYS | 10 | 10 | 3 | 57 | 3 | 3 | 14 | ||
| best occurring AA: FPNATP | 13 | 14 | 32 | 57 | 37 | 43 | 33 | ||
| DENV | 1P | 2P | 3P | 4P | 5P | 6P | Av | ||
| KRNATP | 2 | 0 | 2 | 34 | 18 | 2 | 9 | ||
| KTDAYS | 2 | 2 | 0 | 34 | 0 | 12 | 8 | ||
| best occurring AA: GSSASC | 18 | 27 | 33 | 34 | 21 | 19 | 25 | ||
| ZIKV | 1P | 2P | 3P | 4P | 5P | 6P | 7P | Av | |
| GSKANNG | 27 | 3 | 6 | 14 | 7 | 6 | 11 | 11 | |
| SHRNATA | 9 | 13 | 12 | 36 | 66 | 50 | 14 | 28 | |
| best occurring AA: GFPNATP | 27 | 19 | 16 | 36 | 66 | 50 | 40 | 36 | |
| DENV | 1P | 2P | 3P | 4P | 5P | 6P | 7P | Av | |
| GSKANNG | 40 | 8 | 0 | 9 | 6 | 1 | 14 | 11 | |
| SHRNATA | 13 | 1 | 0 | 11 | 19 | 24 | 5 | 10 | |
| best occurring AA: GGPTPGP | 40 | 36 | 25 | 19 | 22 | 27 | 36 | 29 |
| T1 | T2 | P1 | P2 | X1 | X2 | H1 | H2 | 4G2 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Blocking | PF | BT | PF | PF | PF | PF | PF | PF | PF | |
| Incubation buffer | PBS | PBS | PBS | PBS | PBS | PBS | PBS | PBS | PBST | |
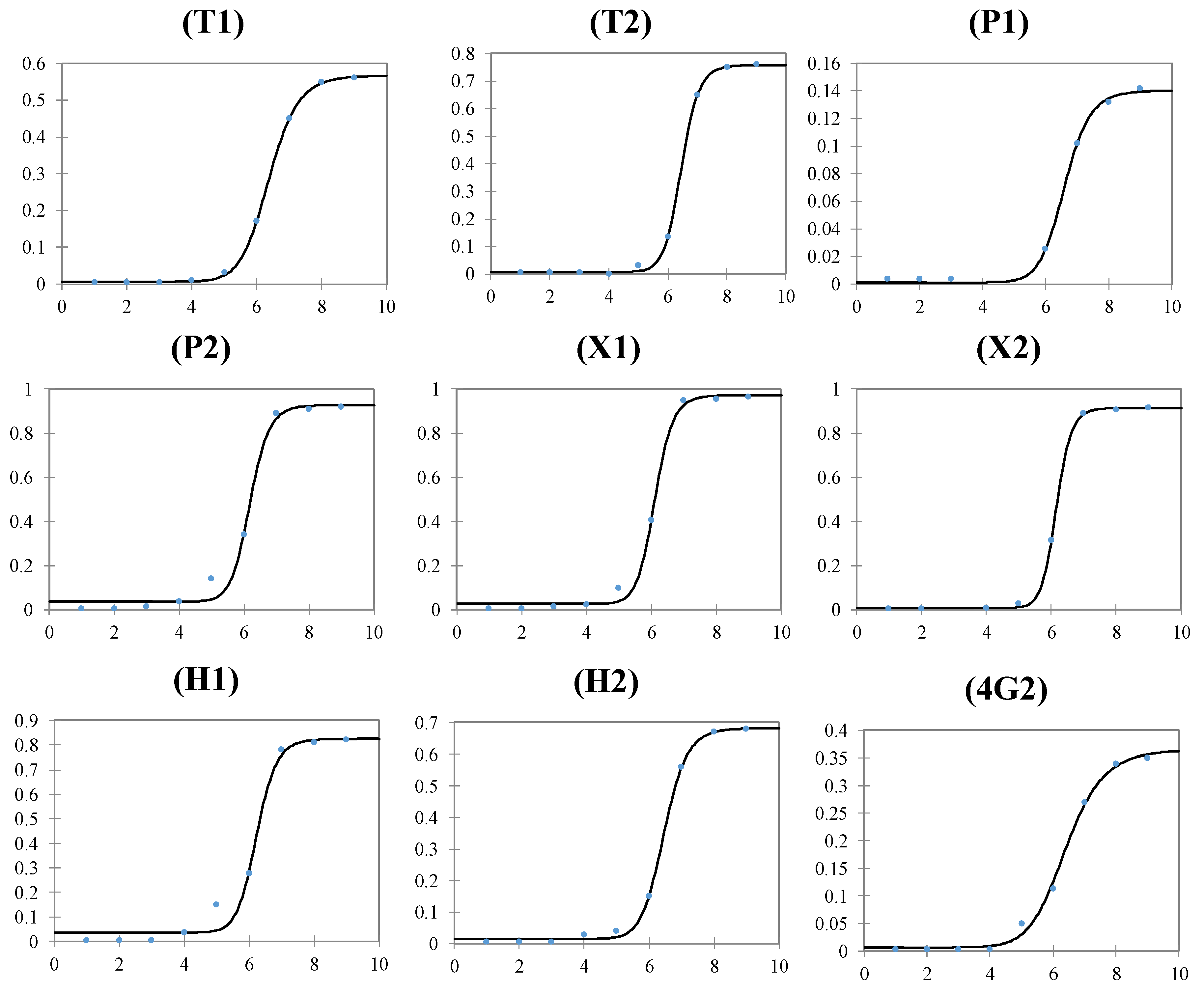
| FPLR—Dynamic Range | (log[ZIKV], copies/mL) | 6-8 | 6-8 | 7-8 | 5-7 | 5-7 | 6-8 | 5-7 | 6-8 | 6-8 |
| LOD | (log[ZIKV], copies/mL) | 5.8 | 5.8 | 6.8 | 4.7 | 4.8 | 5.8 | 4.5 | 5.7 | 5.8 |
| FPLR—C50 | (log[ZIKV], copies/mL) | 6.4 | 6.5 | 6.6 | 6.1 | 6.1 | 6.2 | 6.2 | 6.5 | 6.4 |
| FPLR—slope | Absorbance/(log[ZIKV], copies/mL) | 14.2 | 21.7 | 16.3 | 20.9 | 21.9 | 26.7 | 20.4 | 18.3 | 10.7 |
| FPLR—maximum | Absorbance (450 nm) | 0.568 | 0.759 | 0.140 | 0.928 | 0.973 | 0.915 | 0.826 | 0.683 | 0.365 |
| FPLR—minimum | Absorbance (450 nm) | 0.007 | 0.008 | 0.001 | 0.036 | 0.026 | 0.007 | 0.036 | 0.014 | 0.007 |
| FPLR- R2 | 1.000 | 0.999 | 0.997 | 0.991 | 0.996 | 1.000 | 0.987 | 0.999 | 0.996 | |
| Peptide Concentration | (μM) | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 1 μg/mL |
| Intra-day reproducibility | CV(%) | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <10 |
| Inter-day and batch-to-batch reproducibility | CV(%) | <10 | <10 | <10 | <10 | <10 | <10 | <10 | <10 | nd |
| Long-term stability | (Month) | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | nd |
| Assay time after Plate Coating | (h) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mascini, M.; Dikici, E.; Robles Mañueco, M.; Perez-Erviti, J.A.; Deo, S.K.; Compagnone, D.; Wang, J.; Pingarrón, J.M.; Daunert, S. Computationally Designed Peptides for Zika Virus Detection: An Incremental Construction Approach. Biomolecules 2019, 9, 498. https://doi.org/10.3390/biom9090498
Mascini M, Dikici E, Robles Mañueco M, Perez-Erviti JA, Deo SK, Compagnone D, Wang J, Pingarrón JM, Daunert S. Computationally Designed Peptides for Zika Virus Detection: An Incremental Construction Approach. Biomolecules. 2019; 9(9):498. https://doi.org/10.3390/biom9090498
Chicago/Turabian StyleMascini, Marcello, Emre Dikici, Marta Robles Mañueco, Julio A. Perez-Erviti, Sapna K. Deo, Dario Compagnone, Joseph Wang, José M. Pingarrón, and Sylvia Daunert. 2019. "Computationally Designed Peptides for Zika Virus Detection: An Incremental Construction Approach" Biomolecules 9, no. 9: 498. https://doi.org/10.3390/biom9090498