In Vitro and In Silico Mechanistic Insights into miR-21-5p-Mediated Topoisomerase Drug Resistance in Human Colorectal Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture and Transfection
2.3. Determination of Cell Proliferation and Cell Viability
2.4. Quantitative Real-time Polymerase Chain Reaction (qPCR)
2.5. Western Blot Analysis
2.6. Band-depletion Assay
2.7. Microarray, Connectivity Map (CMap), and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analyses
2.8. CellMinerCDB Analysis
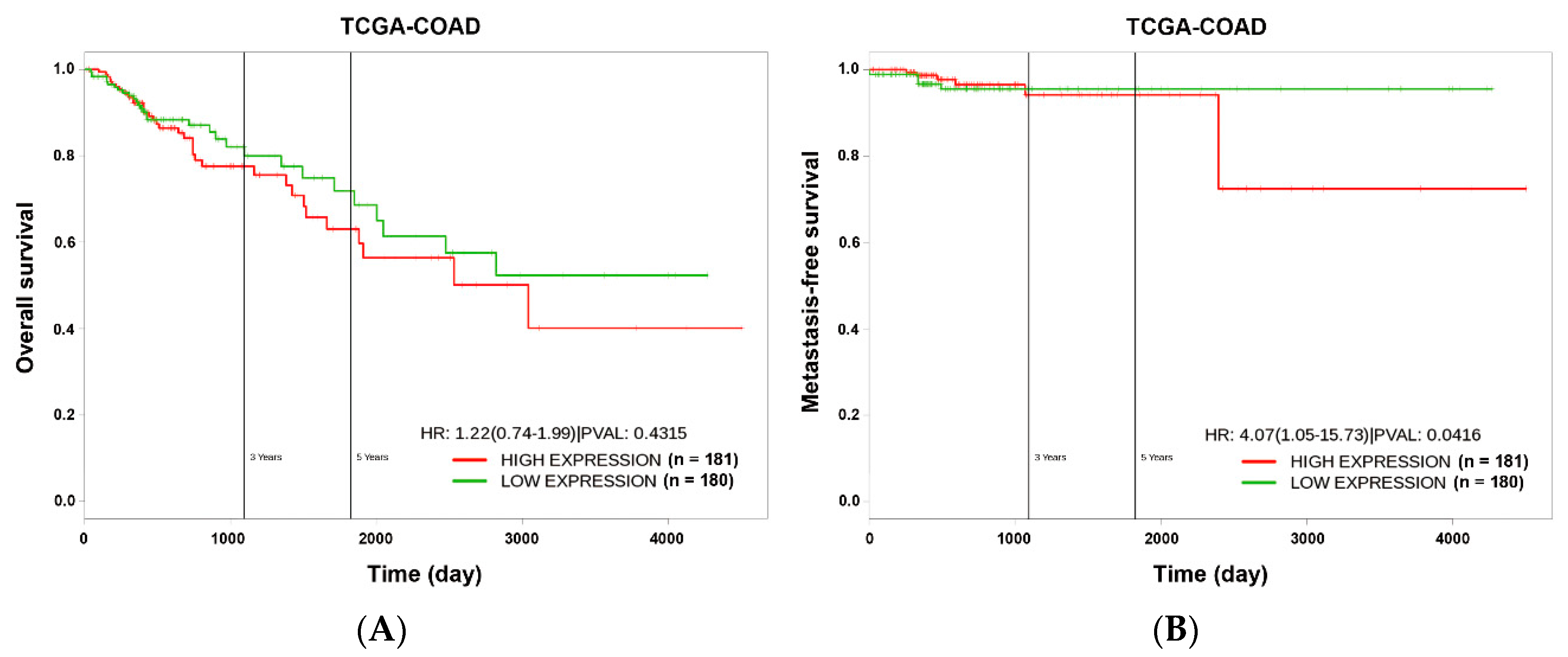
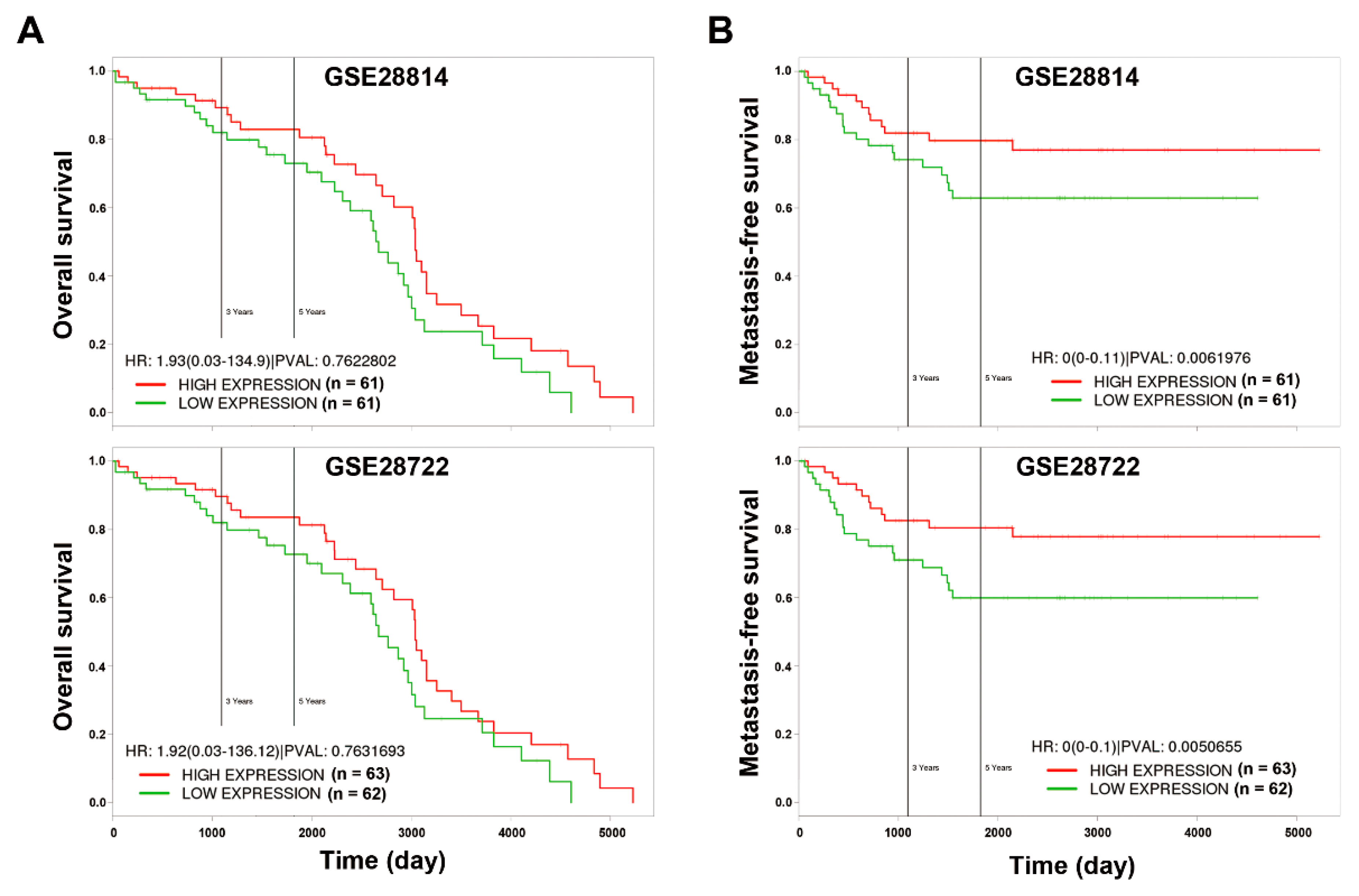
2.9. Kaplan–Meier Survival Analysis
3. Results
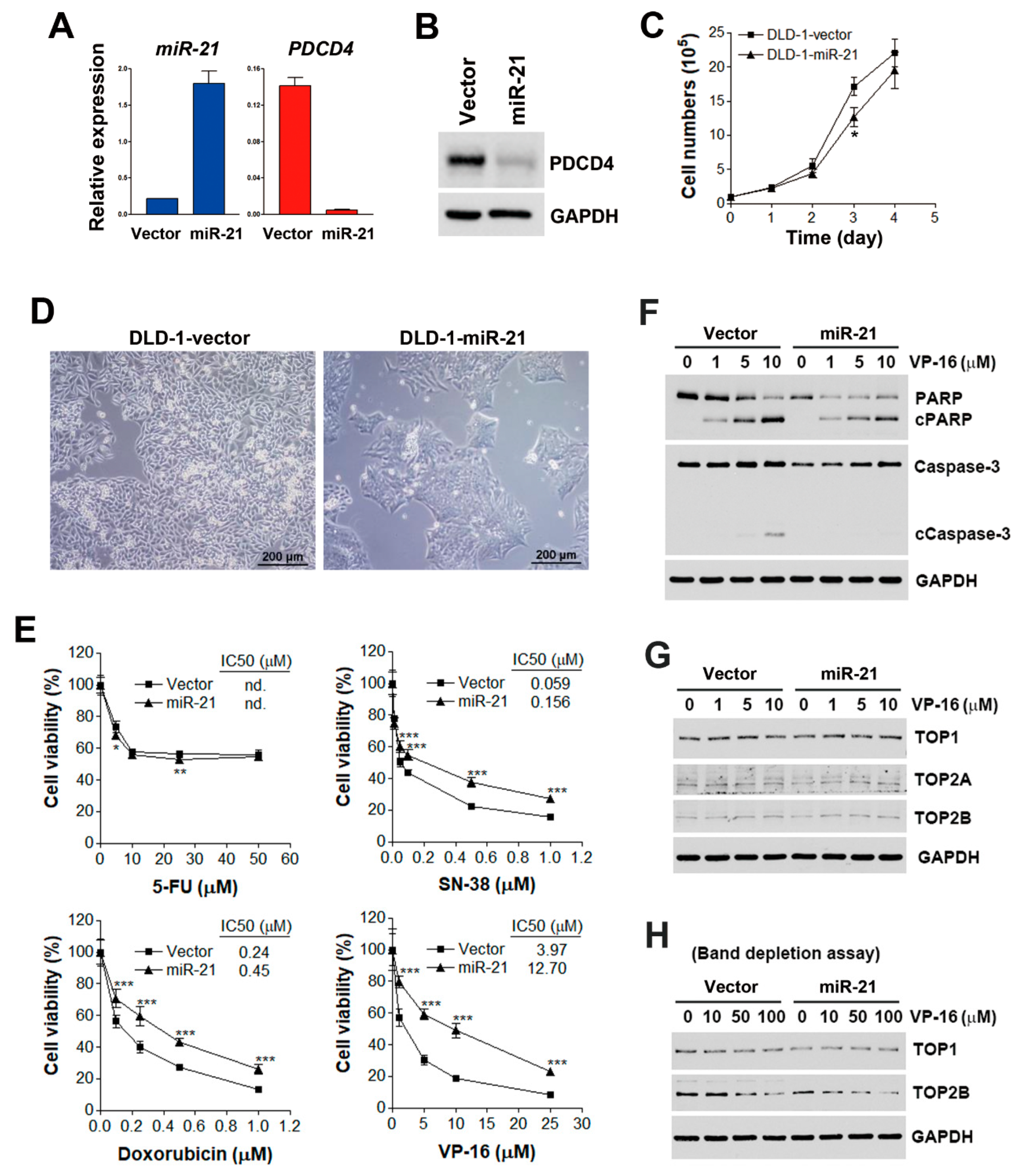
3.1. miR-21 Overexpression is Correlated with Drug Resistance to Topoisomerase Inhibitors
3.2. miR-21 Overexpression Attenuates VP-16-Induced Apoptosis without Affecting Expressions or Activities of Topoisomerases
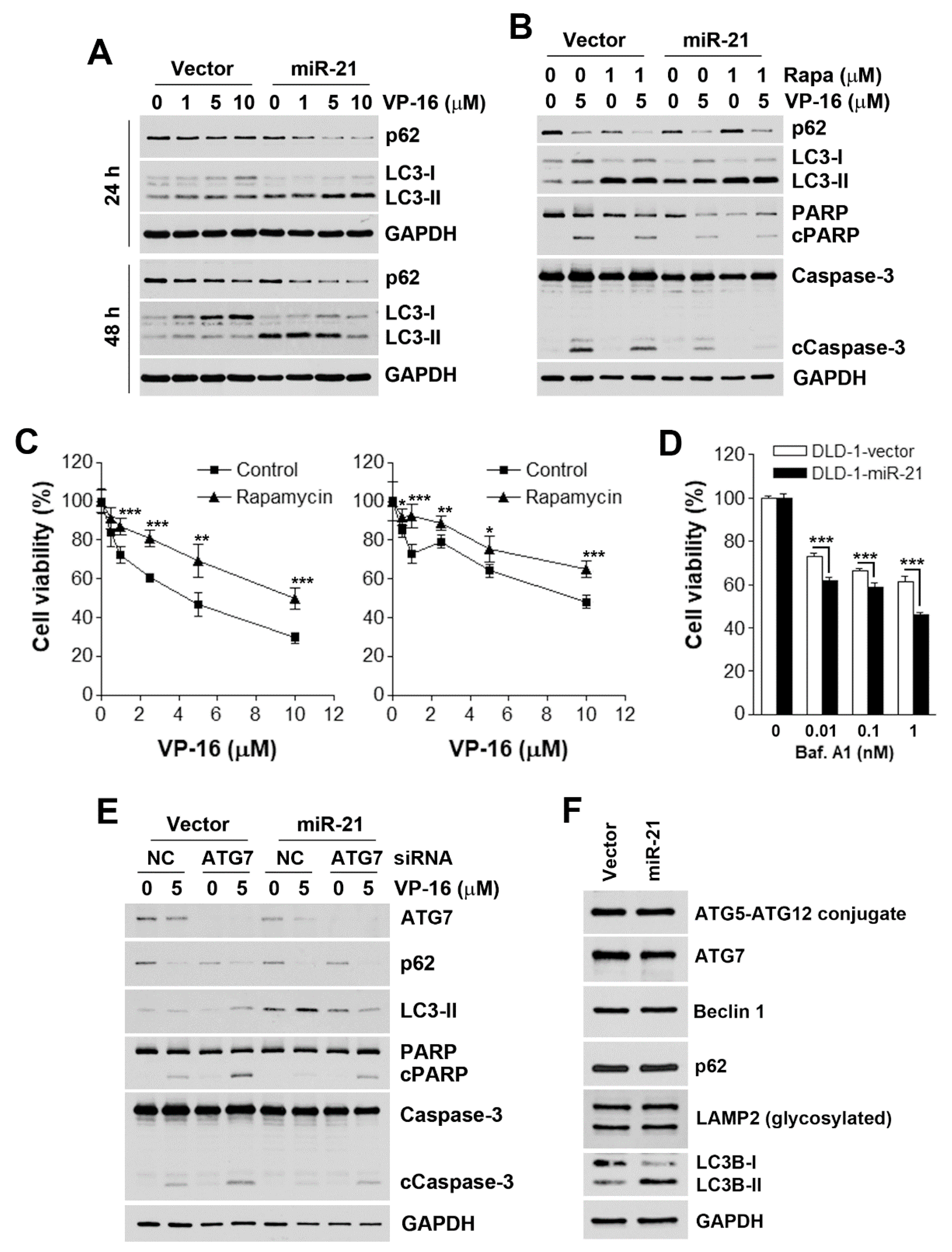
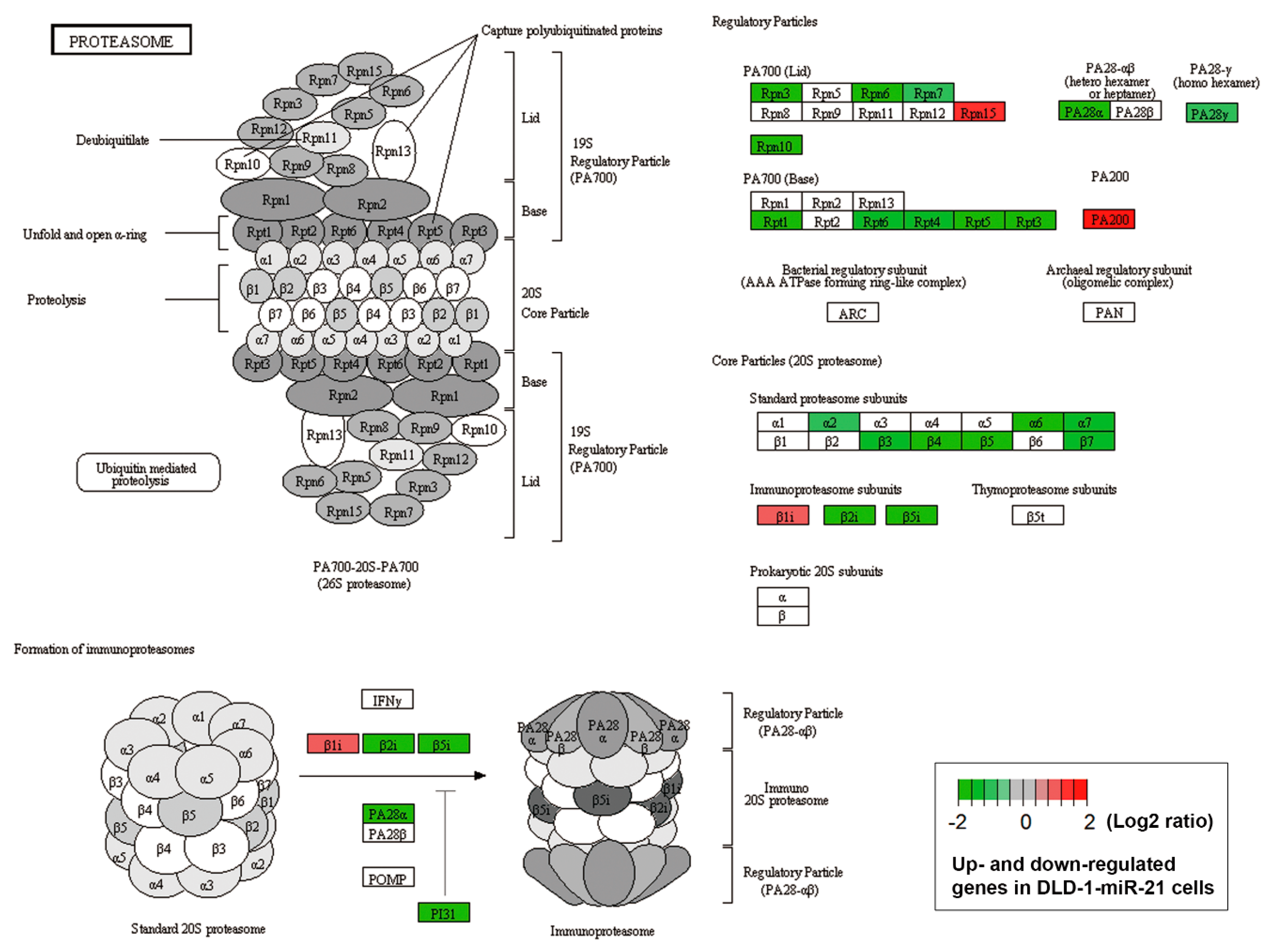
3.3. Accelerated Autophagic flux is Associated with miR-21-Induced Drug Resistance
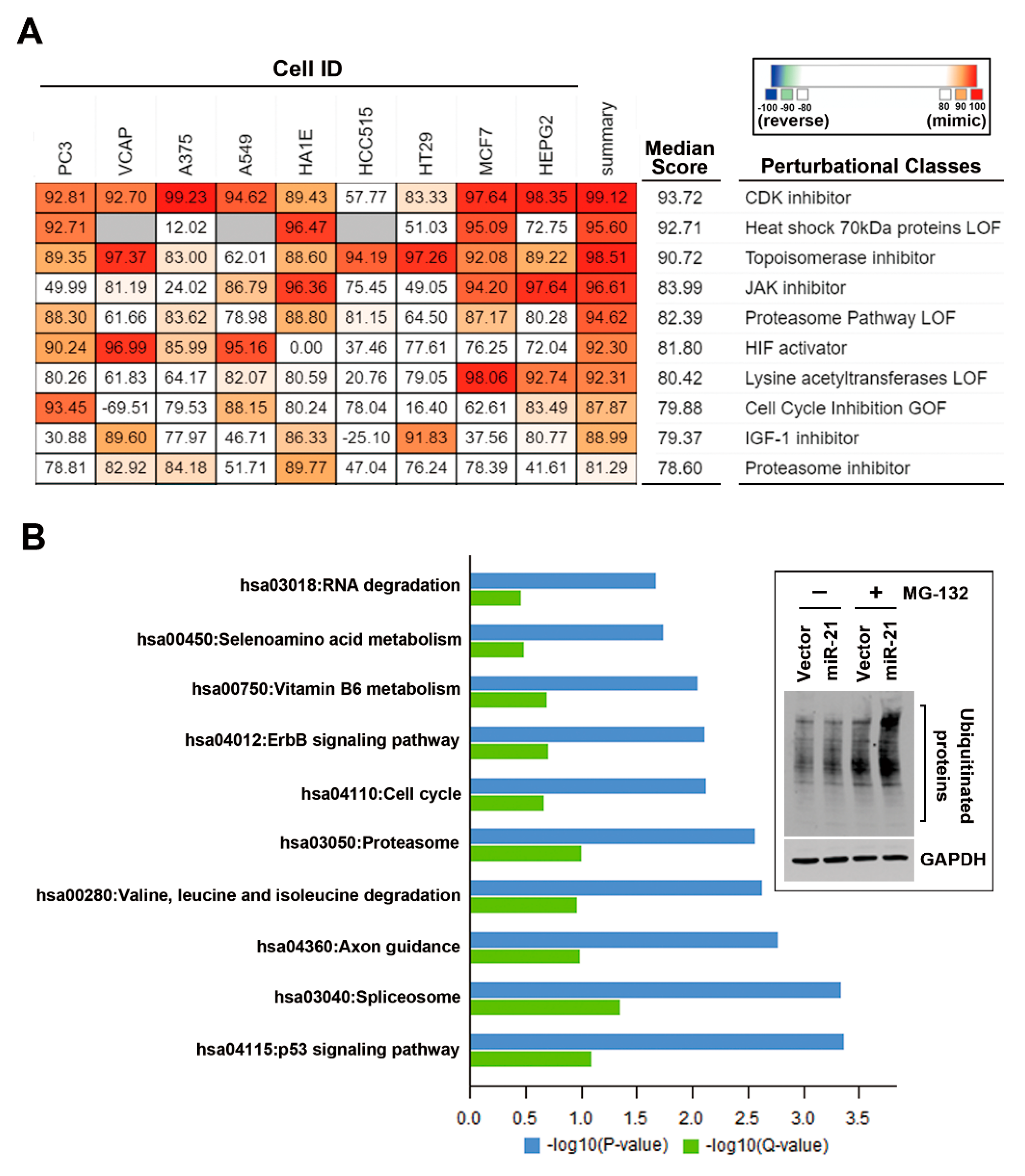
3.4. In Silico Analyses Reveal the Mechanistic Role of miR-21 Overexpression in the Development of Drug Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Waldner, M.J.; Neurath, M.F. The molecular therapy of colorectal cancer. Mol. Asp. Med. 2010, 31, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Kasinski, A.L.; Slack, F.J. Epigenetics and genetics. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Pekow, J.R.; Kwon, J.H. MicroRNAs in inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selcuklu, S.D.; Donoghue, M.T.; Spillane, C. miR-21 as a key regulator of oncogenic processes. Biochem. Soc. Trans. 2009, 37, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Krichevsky, A.M.; Gabriely, G. miR-21: A small multi-Faceted RNA. J. Cell. Mol. Med. 2009, 13, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Rajapakse, V.N.; Luna, A.; Yamade, M.; Loman, L.; Varma, S.; Sunshine, M.; Iorio, F.; Sousa, F.G.; Elloumi, F.; Aladjem, M.I.; et al. CellMinerCDB for Integrative Cross-Database Genomics and Pharmacogenomics Analyses of Cancer Cell Lines. iScience 2018, 10, 247–264. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, C.P.; Nakshatri, H. PROGmiR: A tool for identifying prognostic miRNA biomarkers in multiple cancers using publicly available data. J. Clin. Bioinform. 2012, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Goswami, C.P.; Nakshatri, H. PROGgeneV2: Enhancements on the existing database. BMC Cancer 2014, 14, 970. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Nebozhyn, M.V.; Watters, J.W.; Buser, C.A.; Shaw, P.M.; Huang, P.S.; Van’t Veer, L.; Tollenaar, R.A.; Jackson, D.B.; Agrawal, D.; et al. EMT is the dominant program in human colon cancer. BMC Med. Genom. 2011, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Rasheed, S.A.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-Transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Tanaka, K.; Endou, I.; Ichikawa, Y. Treatment for colorectal liver metastases: A review. Langenbecks Arch. Surg. 2009, 394, 973–983. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Cancer Cell Line Encyclopedia Consortium; Genomics of Drug Sensitivity in Cancer Consortium. Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015, 528, 84–87. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Tsukahara, S.; Oh-hara, T.; Isoe, T.; Tsuruo, T. Decreased expression of DNA topoisomerase I in camptothecin-Resistant tumor cell lines as determined by a monoclonal antibody. Cancer Res. 1990, 50, 6925–6930. [Google Scholar] [PubMed]
- Sugimoto, Y.; Tsukahara, S.; Oh-hara, T.; Liu, L.F.; Tsuruo, T. Elevated expression of DNA topoisomerase II in camptothecin-Resistant human tumor cell lines. Cancer Res. 1990, 50, 7962–7965. [Google Scholar] [PubMed]
- Hsiang, Y.H.; Liu, L.F. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer Res. 1988, 48, 1722–1726. [Google Scholar] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-Eating and self-Killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Lamb, J. The Connectivity Map: A new tool for biomedical research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-Expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Delgado, M.E.; Dyck, L.; Laussmann, M.A.; Rehm, M. Modulation of apoptosis sensitivity through the interplay with autophagic and proteasomal degradation pathways. Cell Death Dis. 2014, 5, 1011. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Kouzminova, N.; Lu, T.; Lin, A.Y. Molecular basis of colorectal cancer. New Engl. J. Med. 2010, 362, 1245–1246. [Google Scholar] [CrossRef] [PubMed]
- Reissfelder, C.; Rahbari, N.N.; Koch, M.; Ulrich, A.; Pfeilschifter, I.; Waltert, A.; Muller, S.A.; Schemmer, P.; Buchler, M.W.; Weitz, J. Validation of prognostic scoring systems for patients undergoing resection of colorectal cancer liver metastases. Ann. Surg. Oncol. 2009, 16, 3279–3288. [Google Scholar] [CrossRef]
- Schee, K.; Lorenz, S.; Worren, M.M.; Gunther, C.C.; Holden, M.; Hovig, E.; Fodstad, O.; Meza-Zepeda, L.A.; Flatmark, K. Deep Sequencing the MicroRNA Transcriptome in Colorectal Cancer. PLoS ONE 2013, 8, 66165. [Google Scholar] [CrossRef]
- Sun, G.; Cheng, Y.W.; Lai, L.; Huang, T.C.; Wang, J.; Wu, X.; Wang, Y.; Huang, Y.; Wang, J.; Zhang, K.; et al. Signature miRNAs in colorectal cancers were revealed using a bias reduction small RNA deep sequencing protocol. Oncotarget 2016, 7, 3857–3872. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, Y.; Zhao, G. Emerging role of microRNA-21 in colorectal cancer. Cancer Biomark. 2015, 15, 219–226. [Google Scholar] [CrossRef]
- Hong, L.; Han, Y.; Zhang, Y.; Zhang, H.; Zhao, Q.; Wu, K.; Fan, D. MicroRNA-21: A therapeutic target for reversing drug resistance in cancer. Expert Opin. Ther. Targets 2013, 17, 1073–1080. [Google Scholar] [CrossRef]
- Javanmardi, S.; Aghamaali, M.R.; Abolmaali, S.S.; Mohammadi, S.; Tamaddon, A.M. miR-21, An Oncogenic Target miRNA for Cancer Therapy: Molecular Mechanisms and Recent Advancements in Chemo and Radio-resistance. Curr. Gene Ther. 2017, 16, 375–389. [Google Scholar] [CrossRef]
- Pan, X.; Wang, Z.X.; Wang, R. MicroRNA-21: A novel therapeutic target in human cancer. Cancer Biol. Ther. 2010, 10, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Kanwar, S.S.; Patel, B.B.; Oh, P.S.; Nautiyal, J.; Sarkar, F.H.; Majumdar, A.P. MicroRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (TGFbetaR2) in colon cancer cells. Carcinogenesis 2012, 33, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Tanahashi, T.; Iima, T.; Yamamoto, Y.; Akaike, Y.; Nishida, K.; Masuda, K.; Kuwano, Y.; Murakami, Y.; Fukushima, M.; et al. Role of miR-19b and its target mRNAs in 5-Fluorouracil resistance in colon cancer cells. J. Gastroenterol. 2012, 47, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pang, Y.; Gao, X.; Zhao, M.; Zhang, X.; Zhang, H.; Xuan, B.; Wang, Y. MicroRNA-137 chemosensitizes colon cancer cells to the chemotherapeutic drug oxaliplatin (OXA) by targeting YBX1. Cancer Biomark. 2017, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Zhao, L.; Zhang, P.; Wang, J.; Xu, N.; Mi, W.; Jiang, X.; Zhang, C.; Qu, J. MicroRNA-195 chemosensitizes colon cancer cells to the chemotherapeutic drug doxorubicin by targeting the first binding site of BCL2L2 mRNA. J. Cell. Physiol. 2015, 230, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Hewish, M.; Lord, C.J.; Martin, S.A.; Cunningham, D.; Ashworth, A. Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nat. Rev. Clin. Oncol. 2010, 7, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Meyers, M.; Wagner, M.W.; Mazurek, A.; Schmutte, C.; Fishel, R.; Boothman, D.A. DNA mismatch repair-Dependent response to fluoropyrimidine-Generated damage. J. Biol. Chem. 2005, 280, 5516–5526. [Google Scholar] [CrossRef]
- Valeri, N.; Gasparini, P.; Braconi, C.; Paone, A.; Lovat, F.; Fabbri, M.; Sumani, K.M.; Alder, H.; Amadori, D.; Patel, T.; et al. MicroRNA-21 induces resistance to 5-Fluorouracil by down-Regulating human DNA MutS homolog 2 (hMSH2). Proc. Natl. Acad. Sci. USA 2010, 107, 21098–21103. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Nicolaides, N.C.; Liu, B.; Parsons, R.; Lengauer, C.; Palombo, F.; D’Arrigo, A.; Markowitz, S.; Willson, J.K.; Kinzler, K.W.; et al. Mutations of GTBP in genetically unstable cells. Science 1995, 268, 1915–1917. [Google Scholar] [CrossRef]
- Chen, C.; Lu, L.; Yan, S.; Yi, H.; Yao, H.; Wu, D.; He, G.; Tao, X.; Deng, X. Autophagy and doxorubicin resistance in cancer. Anti-Cancer Drugs 2018, 29, 1–9. [Google Scholar] [CrossRef]
- Biasoli, D.; Kahn, S.A.; Cornelio, T.A.; Furtado, M.; Campanati, L.; Chneiweiss, H.; Moura-Neto, V.; Borges, H.L. Retinoblastoma protein regulates the crosstalk between autophagy and apoptosis, and favors glioblastoma resistance to etoposide. Cell Death Dis. 2013, 4, 767. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Lee, N.H.; Ho, T.J.; Hsu, H.H.; Kuo, C.H.; Kuo, W.W.; Lin, Y.M.; Tsai, F.J.; Tsai, C.H.; Huang, C.Y. Resistance to irinotecan (CPT-11) activates epidermal growth factor receptor/nuclear factor kappa B and increases cellular metastasis and autophagy in LoVo colon cancer cells. Cancer Lett. 2014, 349, 51–60. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Dong, X.; Zhai, B.; Jiang, X.; Dong, D.; Li, B.; Jiang, H.; Xu, S.; Sun, X. MiR-21 mediates sorafenib resistance of hepatocellular carcinoma cells by inhibiting autophagy via the PTEN/Akt pathway. Oncotarget 2015, 6, 28867–28881. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Liu, S.; Zhang, L.; Yao, H.; Gao, F.; Xu, D.; Li, Q. MiR-21 modulates radiosensitivity of cervical cancer through inhibiting autophagy via the PTEN/Akt/HIF-1alpha feedback loop and the Akt-mTOR signaling pathway. Tumour Biol. 2016, 37, 12161–12168. [Google Scholar] [CrossRef]
- Liu, X.; Luo, F.; Ling, M.; Lu, L.; Shi, L.; Lu, X.; Xu, H.; Chen, C.; Yang, Q.; Xue, J.; et al. MicroRNA-21 activation of ERK signaling via PTEN is involved in arsenite-Induced autophagy in human hepatic L-02 cells. Toxicol. Lett. 2016, 252, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Yang, T.L.; Zhang, Z.; Li, X.G.; Zhong, Q.Q.; Zhao, T.T.; Gong, L. MicroRNA-21 suppresses ox-LDL-Induced human aortic endothelial cells injuries in atherosclerosis through enhancement of autophagic flux: Involvement in promotion of lysosomal function. Exp. Cell Res. 2017, 359, 374–383. [Google Scholar] [CrossRef]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, M.L.; Eckardt, J.R.; Kuhn, J.G.; Burris, H.A., 3rd; Nelson, J.; Hilsenbeck, S.G.; Rodriguez, G.I.; Thurman, A.M.; Smith, L.S.; Eckhardt, S.G.; et al. Phase II trial of irinotecan in patients with progressive or rapidly recurrent colorectal cancer. J. Clin. Oncol. 1996, 14, 1128–1135. [Google Scholar] [CrossRef]
- Sparreboom, A.; Planting, A.S.; Jewell, R.C.; van der Burg, M.E.; van der Gaast, A.; de Bruijn, P.; Loos, W.J.; Nooter, K.; Chandler, L.H.; Paul, E.M.; et al. Clinical pharmacokinetics of doxorubicin in combination with GF120918, a potent inhibitor of MDR1 P-Glycoprotein. Anti-Cancer Drugs 1999, 10, 719–728. [Google Scholar] [CrossRef]
- Van Zuylen, L.; Schellens, J.H.; Goey, S.H.; Pronk, L.C.; de Boer-Dennert, M.M.; Loos, W.J.; Ma, J.; Stoter, G.; Verweij, J. Phase I and pharmacologic study of the arotinoid Ro 40-8757 in combination with cisplatin and etoposide in patients with non-Small cell lung cancer. Anti-Cancer Drugs 1999, 10, 361–368. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-C.; Hsieh, Y.-Y.; Lo, H.-L.; Li, A.; Chou, C.-J.; Yang, P.-M. In Vitro and In Silico Mechanistic Insights into miR-21-5p-Mediated Topoisomerase Drug Resistance in Human Colorectal Cancer Cells. Biomolecules 2019, 9, 467. https://doi.org/10.3390/biom9090467
Chen J-C, Hsieh Y-Y, Lo H-L, Li A, Chou C-J, Yang P-M. In Vitro and In Silico Mechanistic Insights into miR-21-5p-Mediated Topoisomerase Drug Resistance in Human Colorectal Cancer Cells. Biomolecules. 2019; 9(9):467. https://doi.org/10.3390/biom9090467
Chicago/Turabian StyleChen, Jung-Chien, Yao-Yu Hsieh, Hsiang-Ling Lo, Albert Li, Chia-Jung Chou, and Pei-Ming Yang. 2019. "In Vitro and In Silico Mechanistic Insights into miR-21-5p-Mediated Topoisomerase Drug Resistance in Human Colorectal Cancer Cells" Biomolecules 9, no. 9: 467. https://doi.org/10.3390/biom9090467