RhoA-Dependent HGF and c-Met Mediate Gas6-Induced Inhibition of Epithelial–Mesenchymal Transition, Migration, and Invasion of Lung Alveolar Epithelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Culture
2.3. Isolation and Culture of Primary Cells
2.4. Incubation of Epithelial Cells
2.5. Transient Transfections
2.6. Immunoblot Analysis
2.7. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.8. RhoA Activity Assay
2.9. Enzyme-Linked Immunosorbent Assay (ELISA) Measurement
2.10. Migration and Invasion Assays
2.11. Statistical Analysis
3. Results
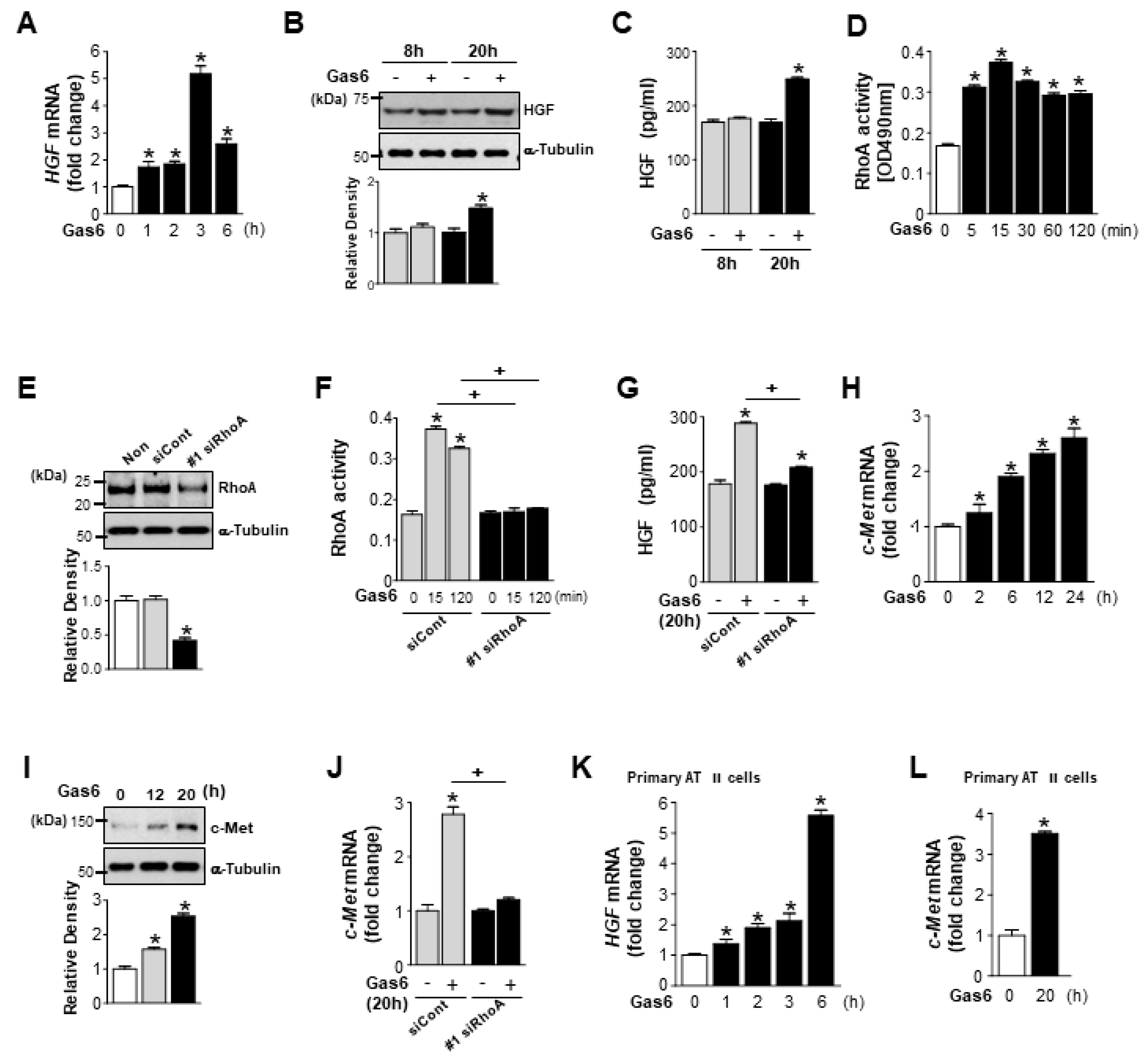
3.1. Growth Arrest-Specific Protein 6 (Gas6)/Axl or Mer Signaling Induces RhoA-Dependent Hepatocyte Growth Factor (HGF) Secretion and c-Met Expression in Lung Epithelial Cells
3.2. Axl and Mer Receptor Tyrosine Kinases Mediate Gas6-Induced RhoA Activity, and Expression of HGF and c-Met in LA-4 Cells
3.3. RhoA is Involved in the Gas6-Induced Inhibition of Epithelial–Mesenchymal Transition (EMT) Marker Changes in LA-4 Cells
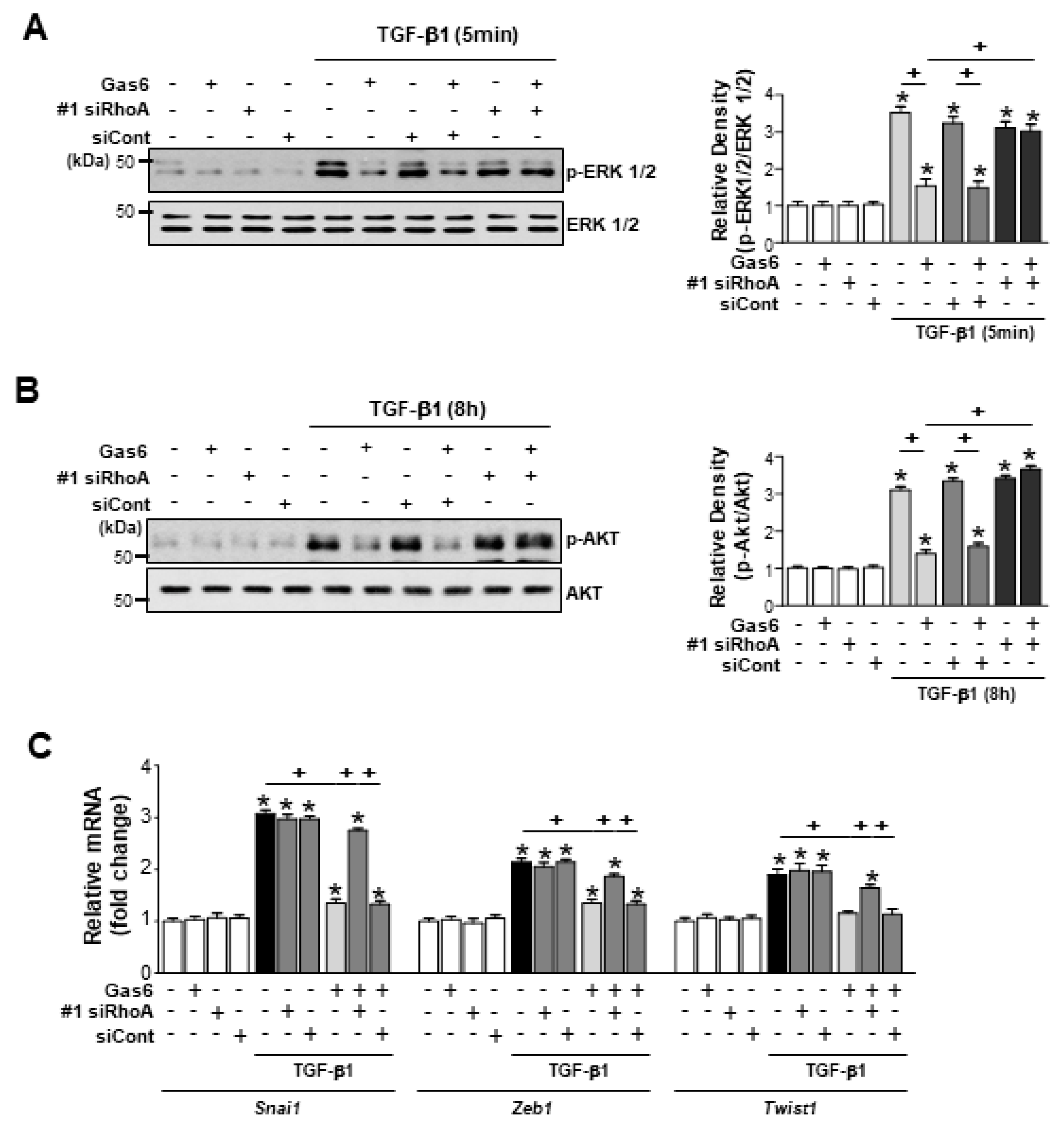
3.4. RhoA is Involved in the Inhibition of Phospho-Extracellular Signal-Related Kinase (ERK1/2) and AKT Activation and Reduction of EMT-Regulating Transcription Factor Expression by Gas6 in LA-4 Cells
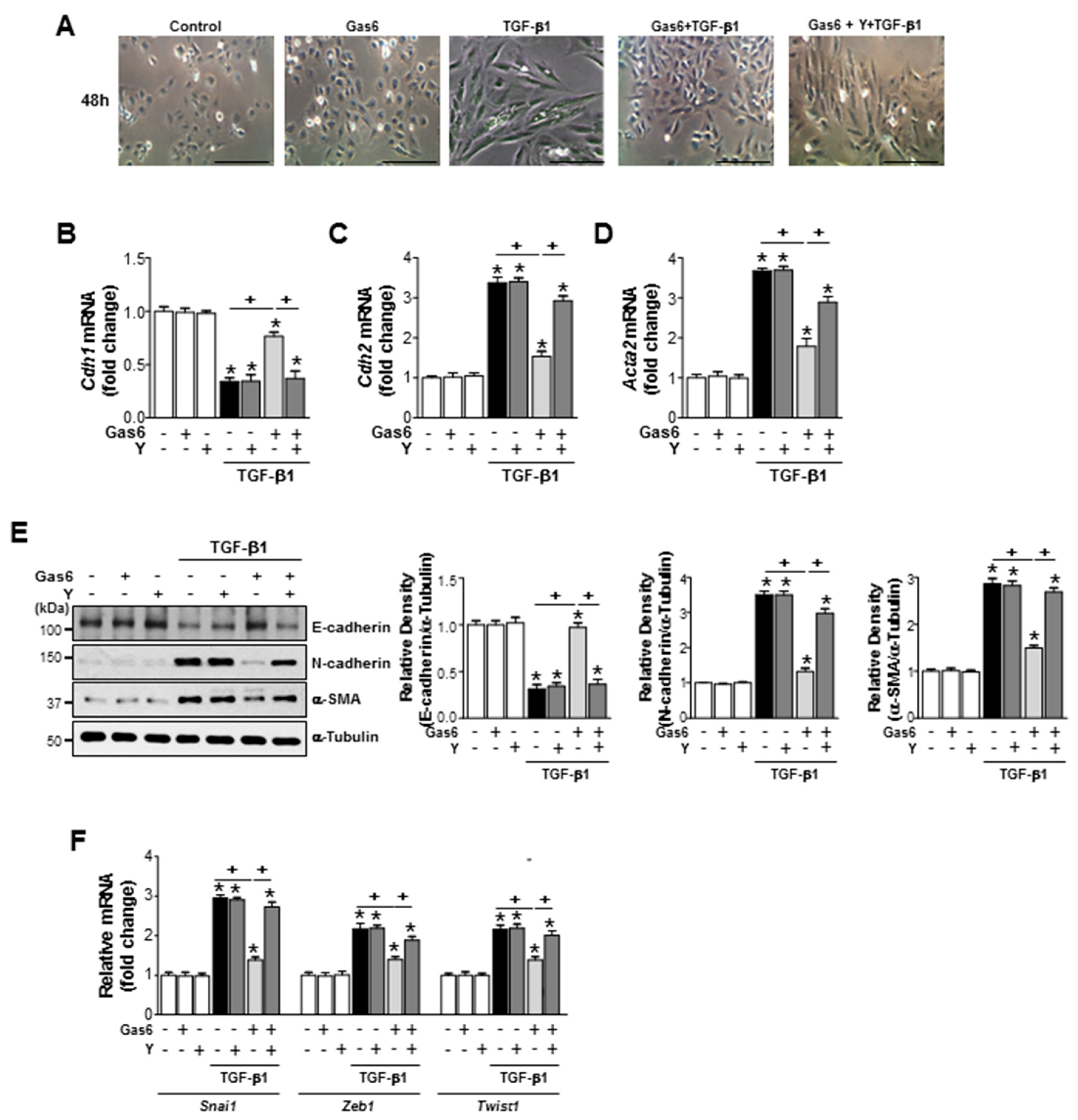
3.5. The Rho Kinase is Involved in the Inhibition of EMT Marker Changes and Reduction of EMT-Regulating Transcription Factor Expression by Gas6 in Lung Epithelial Cells
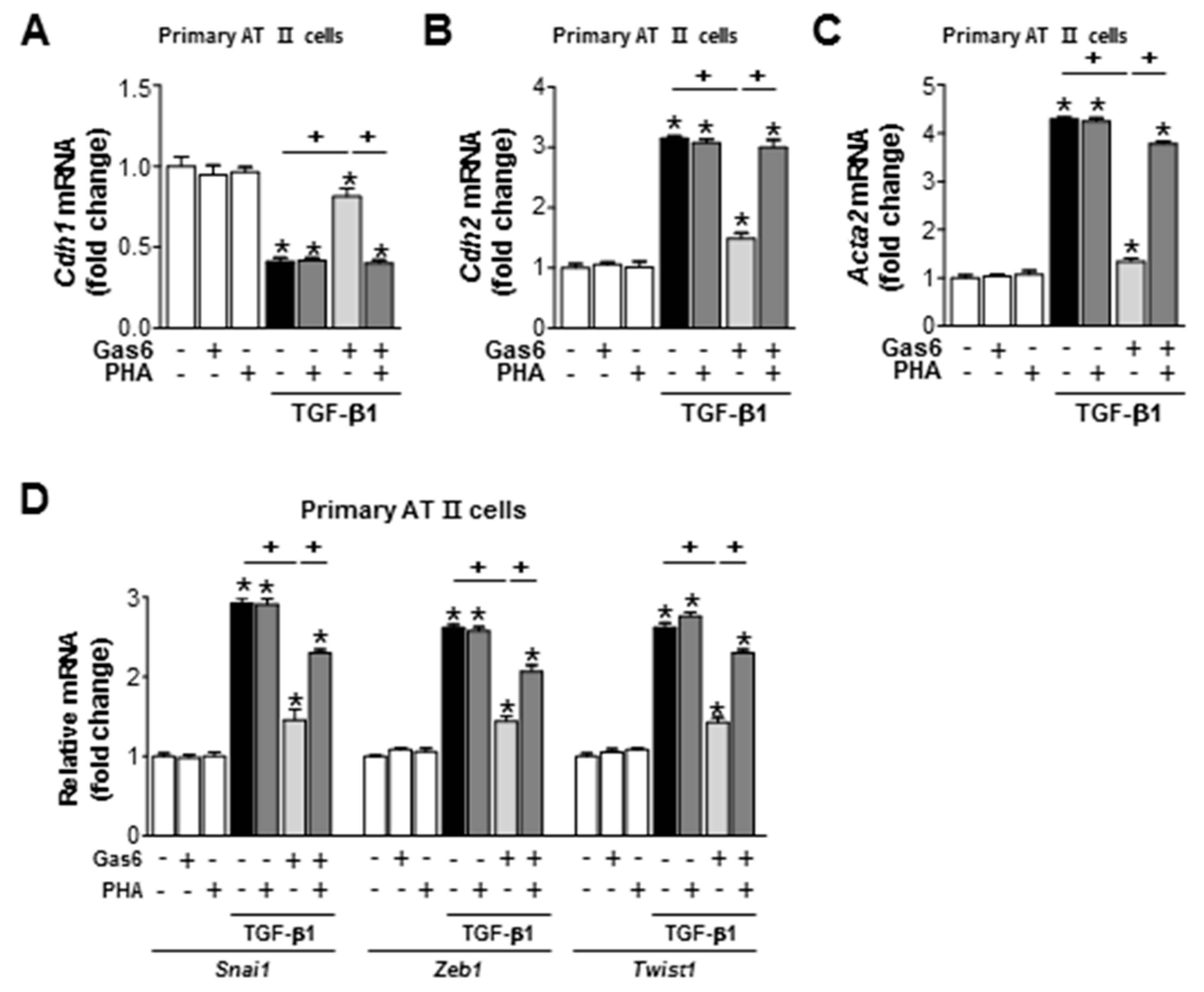
3.6. c-Met Signaling is Involved in the Gas6-Induced Inhibition of EMT in LA-4 Cells and Primary AT II Cells
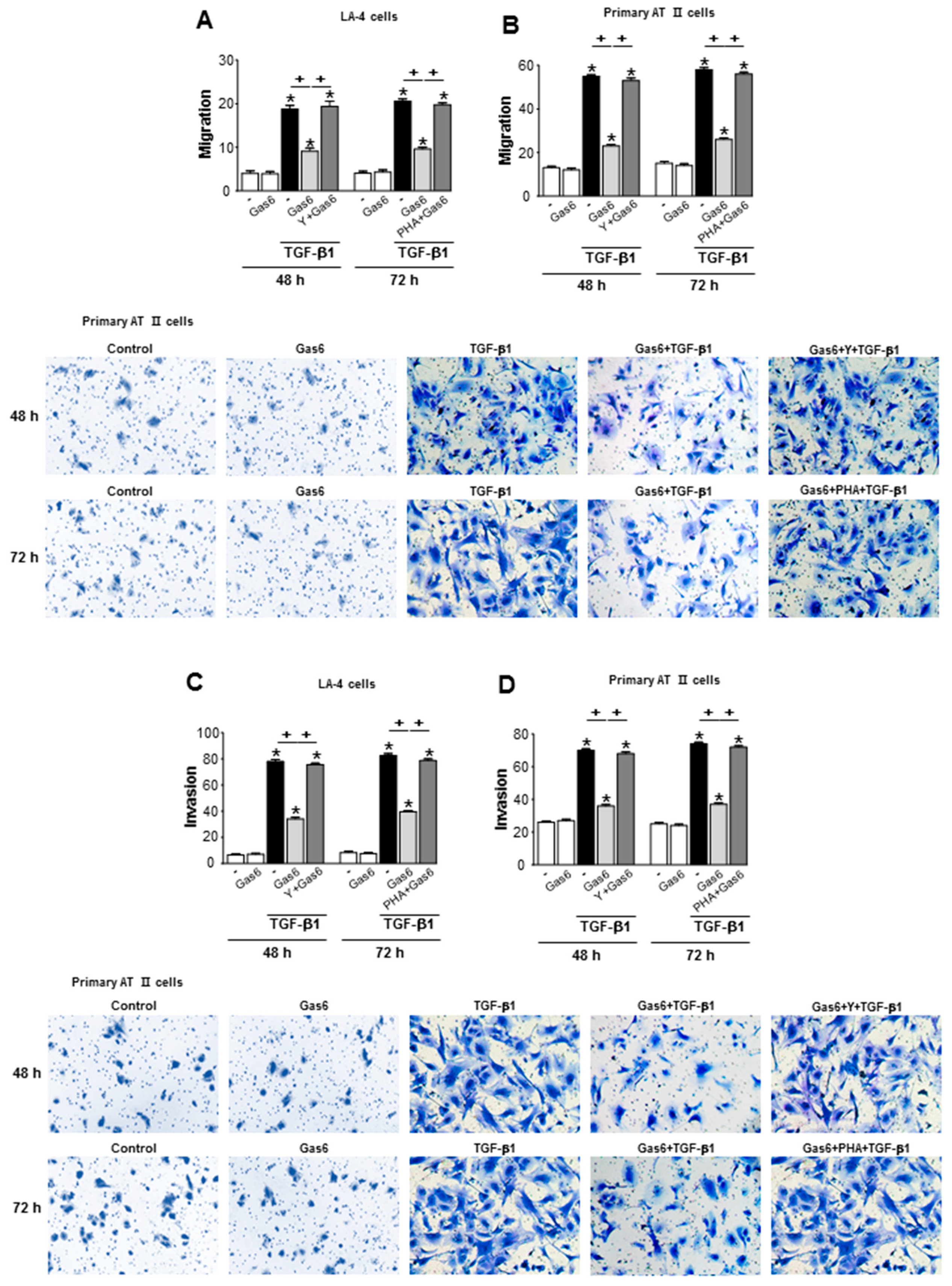
3.7. Treatment of Lung Epithelial Cells with Gas6 Inhibits the Migration and Invasion of Epithelial Cells via RhoA-Dependent HGF Secretion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Thannickal, V.J.; Toews, G.B.; White, E.S.; Lynch, J.P., III; Martinez, F.J. Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 2004, 5, 395–417. [Google Scholar] [CrossRef]
- Harari, S.; Caminati, A. IPF: New insight on pathogenesis and treatment. Allergy 2010, 5, 537–553. [Google Scholar] [CrossRef]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef]
- Quan, T.E.; Cowper, S.E.; Bucala, R. The role of circulating fibrocytes in fibrosis. Curr. Rheumatol. Rep. 2006, 8, 145–150. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Willis, B.C.; du Bois, R.M.; Borok, Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc. Am. Thorac. Soc. 2006, 3, 377–382. [Google Scholar] [CrossRef]
- Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Degryse, A.L.; Li, B.; Han, W.; Han, W.; Sherrill, T.P.; Plieth, D.; Neilson, E.G.; et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 657–665. [Google Scholar] [CrossRef]
- Behr, J. The diagnosis and treatment of idiopathic pulmonary fibrosis. Dtsch. Arztebl. Int. 2013, 110, 875–881. [Google Scholar] [CrossRef]
- Kumar, A.; Kapnadak, S.G.; Girgis, R.E.; Raghu, G. Lung transplantation in idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 2018, 12, 375–385. [Google Scholar] [CrossRef]
- Hardie, W.D.; Glasser, S.W.; Hagood, J.S. Emerging concepts in the pathogenesis of lung fibrosis. Am. J. Pathol. 2009, 175, 3–16. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef] [PubMed]
- Saller, F.; Burnier, L.; Schapira, M.; Angelillo-Scherrer, A. Role of the growth arrest-specific gene 6 (gas6) product in thrombus stabilization. Blood Cells Mol. Dis. 2006, 36, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef]
- Godowski, P.J.; Mark, M.R.; Chen, J.; Sadick, M.D.; Raab, H.; Hammonds, R.G. Reevaluation of the roles of protein S and Gas6 as ligands for the receptor tyrosine kinase Rse/Tyro 3. Cell 1995, 82, 355–358. [Google Scholar] [CrossRef]
- Lemke, G. Biology of the TAM Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef]
- Goruppi, S.; Ruaro, E.; Schneider, C. Gas6, the ligand of Axl tyrosine kinase receptor, has mitogenic and survival activities for serum starved NIH3T3 fibroblasts. Oncogene 1996, 12, 471–480. [Google Scholar]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef]
- Law, L.A.; Graham, D.K.; Di Paola, J.; Branchford, B.R. GAS6/TAM Pathway Signaling in Hemostasis and Thrombosis. Front. Med. 2018, 5, 137. [Google Scholar] [CrossRef] [Green Version]
- Gould, W.R.; Baxi, S.M.; Schroeder, R.; Peng, Y.W.; Leadley, R.J.; Peterson, J.T.; Perrin, L.A. Gas6 receptors Axl, Sky and Mer enhance platelet activation and regulate thrombotic responses. J. Thromb. Haemost. 2005, 3, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Melaragno, M.G.; Cavet, M.E.; Yan, C.; Tai, L.K.; Jin, Z.G.; Haendeler, J.; Berk, B.C. Gas6 inhibits apoptosis in vascular smooth muscle: Role of Axl kinase and Akt. J. Mol. Cell. Cardiol. 2004, 37, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loges, S.; Schmidt, T.; Tjwa, M.; van Geyte, K.; Lievens, D.; Lutgens, E.; Vanhoutte, D.; Borgel, D.; Plaisance, S.; Hoylaerts, M.; et al. Malignant cells fuel tumor growth by educating infiltrating leukocytes to produce the mitogen Gas6. Blood 2010, 115, 2264–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alciato, F.; Sainaghi, P.P.; Sola, D.; Castello, L.; Avanzi, G.C. TNF-alpha, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J. Leukoc. Biol. 2010, 87, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Eken, C.; Martin, P.J.; Sadallah, S.; Treves, S.; Schaller, M.; Schifferli, J.A. Ectosomes released by polymorphonuclear neutrophils induce a MerTK-dependent anti-inflammatory pathway in macrophages. J. Biol. Chem. 2010, 285, 39914–39921. [Google Scholar] [CrossRef]
- Lee, Y.J.; Park, H.J.; Woo, S.Y.; Park, E.M.; Kang, J.L. RhoA/phosphatidylinositol 3-kinase/protein kinase B/mitogen-activated protein kinase signaling after growth arrest-specific protein 6/mer receptor tyrosine kinase engagement promotes epithelial cell growth and wound repair via upregulation of hepatocyte growth factor in macrophages. J. Pharmacol. Exp. Ther. 2014, 350, 563–577. [Google Scholar]
- Yoon, Y.S.; Lee, Y.J.; Choi, Y.H.; Park, Y.M.; Kang, J.L. Macrophages programmed by apoptotic cells inhibit epithelial-mesenchymal transition in lung alveolar epithelial cells via PGE2, PGD2, and HGF. Sci. Rep. 2016, 6, 20992. [Google Scholar] [CrossRef] [Green Version]
- Ogunwobi, O.O.; Liu, C. Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial mesenchymal transition in hepatocellular carcinoma via Akt and COX 2 pathway. Clin. Exp. Metastasis 2011, 28, 721–731. [Google Scholar] [CrossRef]
- Previdi, S.; Maroni, P.; Matteucci, E.; Broggini, M.; Bendinelli, P.; Desiderio, M.A. Interaction between human breast cancer metastasis and bone microenvironment through activated hepatocyte growth factor/Met and β-catenin/Wnt pathways. Eur. J. Cancer 2010, 46, 1679–1691. [Google Scholar] [CrossRef]
- Jung, J.; Lee, Y.J.; Choi, Y.H.; Park, E.M.; Kim, H.S.; Kang, J.L. Gas6 prevents epithelial-mesenchymal transition in alveolar epithelial cells via production of PGE2, PGD2 and their receptors. Cells 2019, 8, 643. [Google Scholar] [CrossRef] [PubMed]
- Corti, M.; Brody, A.R.; Harrison, J.H. Isolation and primary culture of murine alveolar type II cells. Am. J. Respir. Cell. Mol. Biol. 1996, 14, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Lazar, M.H.; Christensen, P.J.; Du, M.; Yu, B.; Subbotina, N.M.; Hanson, K.E.; Hansen, J.M.; White, E.S.; Simon, R.H.; Sisson, T.H. Plasminogen activator inhibitor-1 impairs alveolar epithelial repair by binding to vitronectin. Am. J. Respir. Cell. Mol. Biol. 2004, 31, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, M.J.; Yoon, Y.S.; Choi, Y.H.; Kim, H.S.; Kang, J.L. Simvastatin treatment boosts benefits of apoptotic cell infusion in murine lung fibrosis. Cell Death Dis. 2017, 8, e2860. [Google Scholar] [CrossRef]
- Brown, J.R.; Goldblatt, D.; Buddle, J.; Morton, L.; Thrasher, A.J. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD). J. Leukoc. Biol. 2003, 73, 591–599. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Dai, C.; Liu, Y. Hepatocyte growth factor gene therapy and angiotensin II blockade synergistically attenuate renal interstitial fibrosis in mice. J. Am. Soc. Nephrol. 2002, 13, 2464–2477. [Google Scholar] [CrossRef]
- Shukla, M.N.; Rose, J.L.; Ray, R.; Lathrop, K.L.; Ray, A.; Ray, P. Hepatocyte growth factor inhibits epithelial to myofibroblast transition in lung cells via Smad7. Am. J. Respir. Cell. Mol. Biol. 2009, 40, 643–653. [Google Scholar] [CrossRef]
- Li, Y.; Yang, J.; Dai, C.; Wu, C.; Liu, Y. Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J. Clin. Investig. 2003, 112, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Nagai, T.; Arao, T.; Furuta, K.; Sakai, K.; Kudo, K.; Kaneda, H.; Tamura, D.; Aomatsu, K.; Kimura, H.; Fujita, Y.; et al. Sorafenib Inhibits the Hepatocyte Growth Factor–Mediated Epithelial Mesenchymal Transition in Hepatocellular Carcinoma. Mol. Cancer Ther. 2011, 10, 169–177. [Google Scholar] [CrossRef]
- Gao, D.; Vahdat, L.T.; Wong, S.; Chang, J.C.; Mittal, V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res. 2012, 72, 4883–4889. [Google Scholar] [CrossRef] [PubMed]
- Gentile, A.; Trusolino, L.; Comoglio, P.M. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008, 27, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tsao, M.S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, O.; Diebold, D.; Fan, D.; Peterson, M.; Nho, R.S.; Bitterman, P.B.; Henke, C.A. Fibrotic myofibroblasts manifest genome-wide derangements of translational control. PLoS ONE 2008, 3, e3220. [Google Scholar] [CrossRef] [PubMed]
- White, E.S.; Lazar, M.H.; Thannickal, V.J. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J. Pathol. 2003, 201, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.Y.; Youn, Y.S.; Lee, Y.J.; Choi, Y.H.; Woo, S.Y.; Kang, J.L. Interaction of apoptotic cells with macrophages upregulates COX-2/PGE2 and HGF expression via a positive feedback loop. Mediators Inflamm. 2014, 2014, 463524. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, J.; Yang, K.; Kim, H.-J.; Lee, Y.-J.; Kim, M.; Choi, Y.-H.; Kang, J.L. RhoA-Dependent HGF and c-Met Mediate Gas6-Induced Inhibition of Epithelial–Mesenchymal Transition, Migration, and Invasion of Lung Alveolar Epithelial Cells. Biomolecules 2019, 9, 565. https://doi.org/10.3390/biom9100565
Jung J, Yang K, Kim H-J, Lee Y-J, Kim M, Choi Y-H, Kang JL. RhoA-Dependent HGF and c-Met Mediate Gas6-Induced Inhibition of Epithelial–Mesenchymal Transition, Migration, and Invasion of Lung Alveolar Epithelial Cells. Biomolecules. 2019; 9(10):565. https://doi.org/10.3390/biom9100565
Chicago/Turabian StyleJung, Jihye, Kyungwon Yang, Hee-Ja Kim, Ye-Ji Lee, Minsuk Kim, Youn-Hee Choi, and Jihee Lee Kang. 2019. "RhoA-Dependent HGF and c-Met Mediate Gas6-Induced Inhibition of Epithelial–Mesenchymal Transition, Migration, and Invasion of Lung Alveolar Epithelial Cells" Biomolecules 9, no. 10: 565. https://doi.org/10.3390/biom9100565