Brusatol, a Nrf2 Inhibitor Targets STAT3 Signaling Cascade in Head and Neck Squamous Cell Carcinoma


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Culture Conditions
2.3. Preparation of Whole-Cell Lysates
2.4. Western Blot Analysis
2.5. Electrophoretic Mobility Shift Assay (EMSA)
2.6. Immunocytochemistry for the Distribution of STAT3
2.7. Monitoring of Cell Growth with the RTCA DP Instrument
2.8. Annexin V Assay
2.9. Measurement of Intracellular Reactive Oxygen Species
2.10. In Silico Analysis
2.11. Statistical Analysis
3. Results
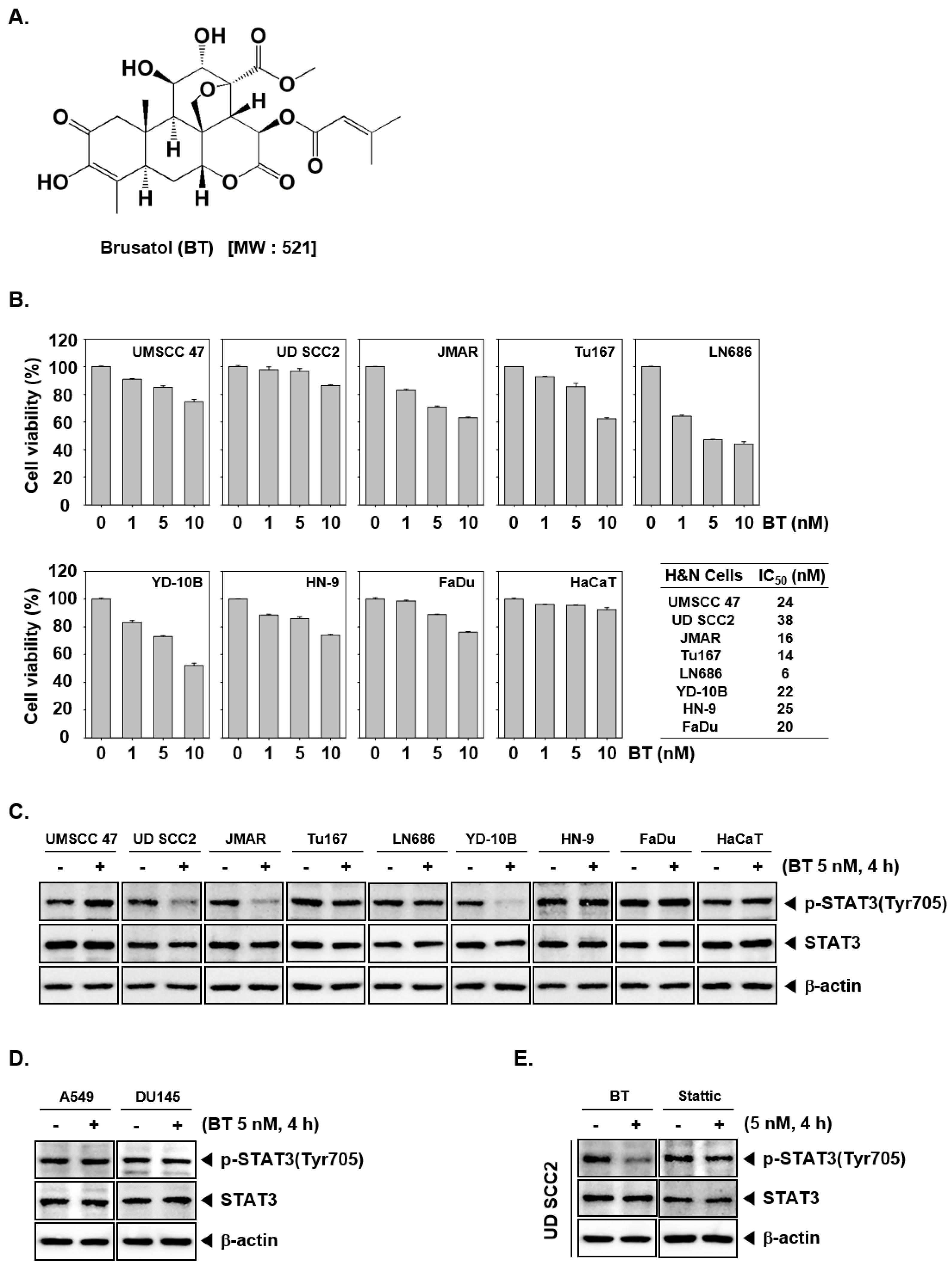
3.1. BT Reduced the Cell Viability of HNSCC Cells
3.2. BT Inhibited Constitutive STAT3 Phosphorylation in UD SCC2, JMAR, YD-10B Cells
3.3. BT Failed to Suppress STAT3 Activation in A549 and DU145 Cells
3.4. BT Mitigated STAT3 Activation More Effectively than Stattic in UD SCC2 Cells
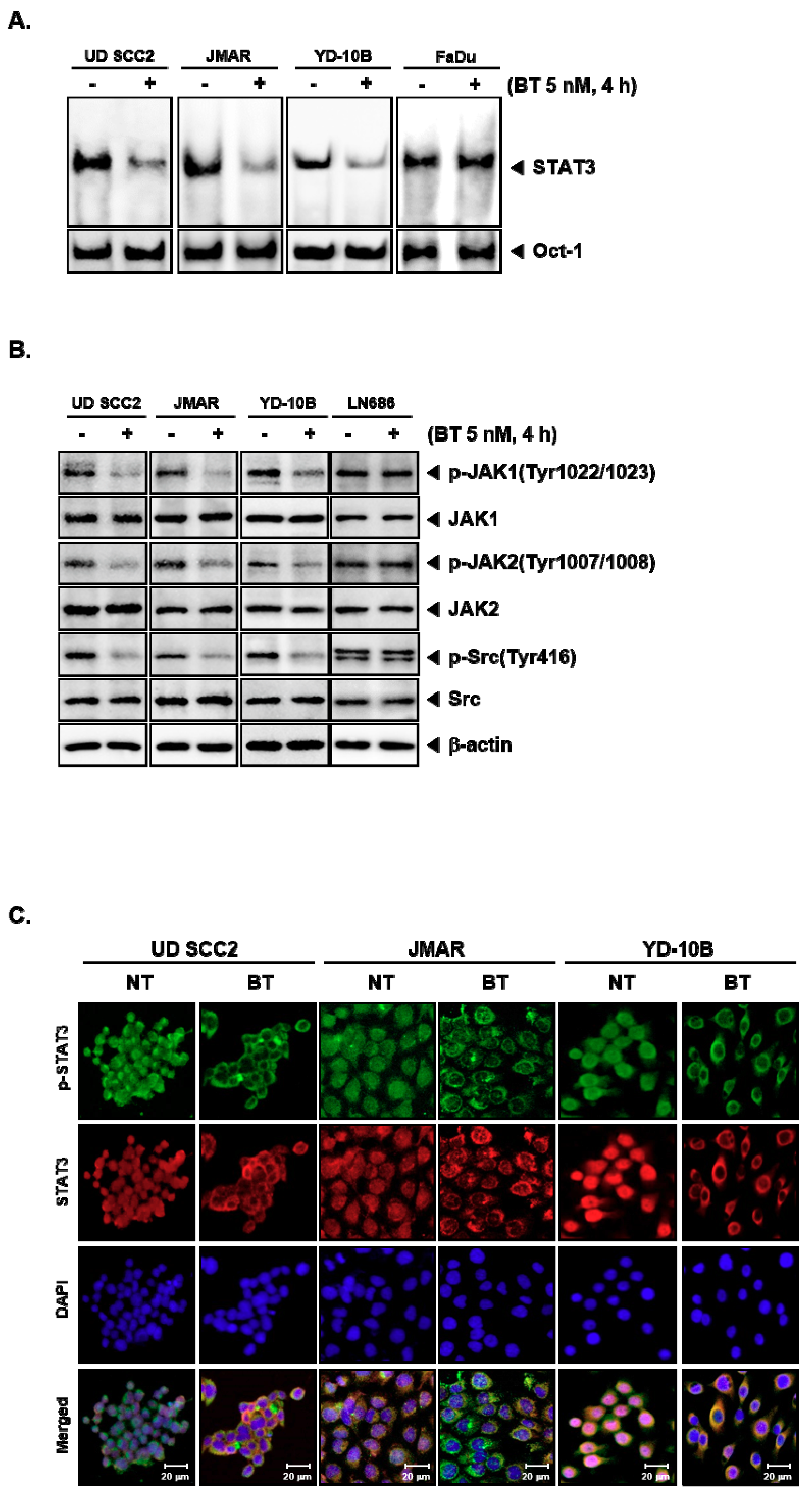
3.5. BT Abrogated DNA Binding Ability of STAT3
3.6. BT Repressed the Constitutive Phosphorylation of Upstream Kinases
3.7. BT Reduced the Nuclear STAT3 Level in HNSCC Cells
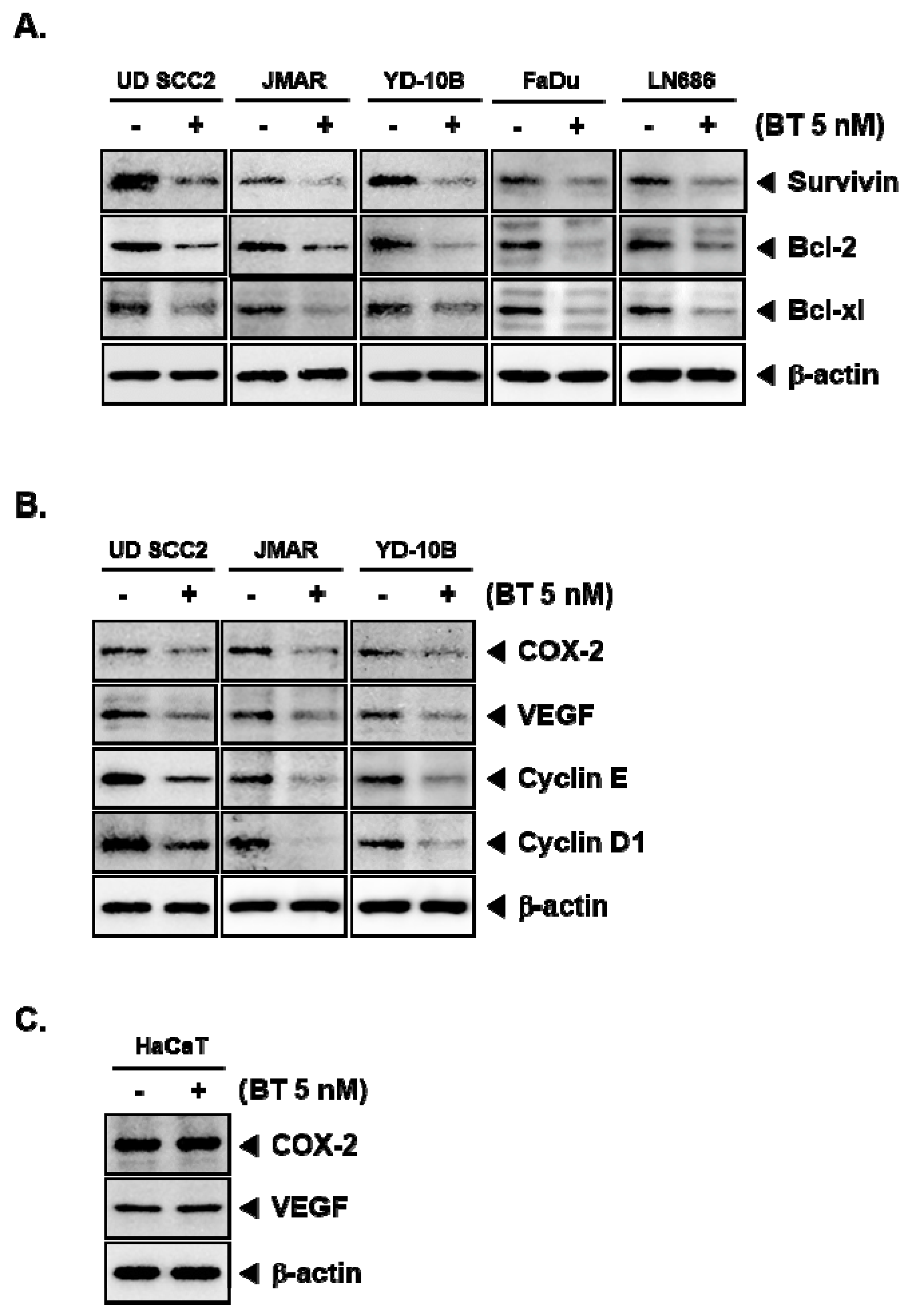
3.8. BT Downregulated the Protein Expression of STAT3-Driven Genes
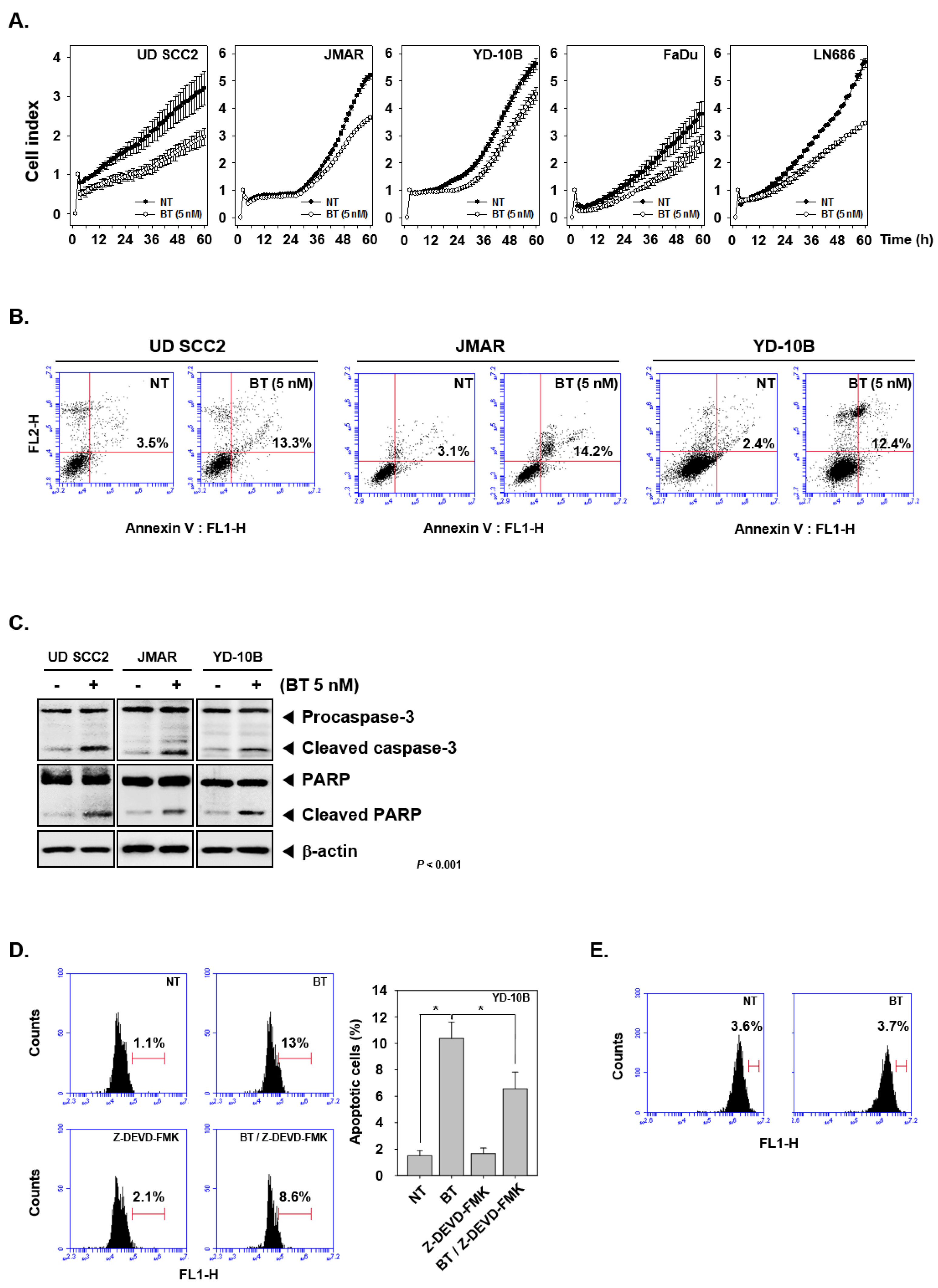
3.9. BT Abrogated the Growth of HNSCC Cells
3.10. BT Promoted Apoptotic Cell Death
3.11. BT Regulated Caspase-Mediated Apoptosis in HNSCC Cells
3.12. Specific Blockade of Caspase-3 Cleavage Abrogated BT-Induced Apoptosis
3.13. BT Induced STAT3 Inhibition May be Independent of Reactive Oxygen Species (ROS)
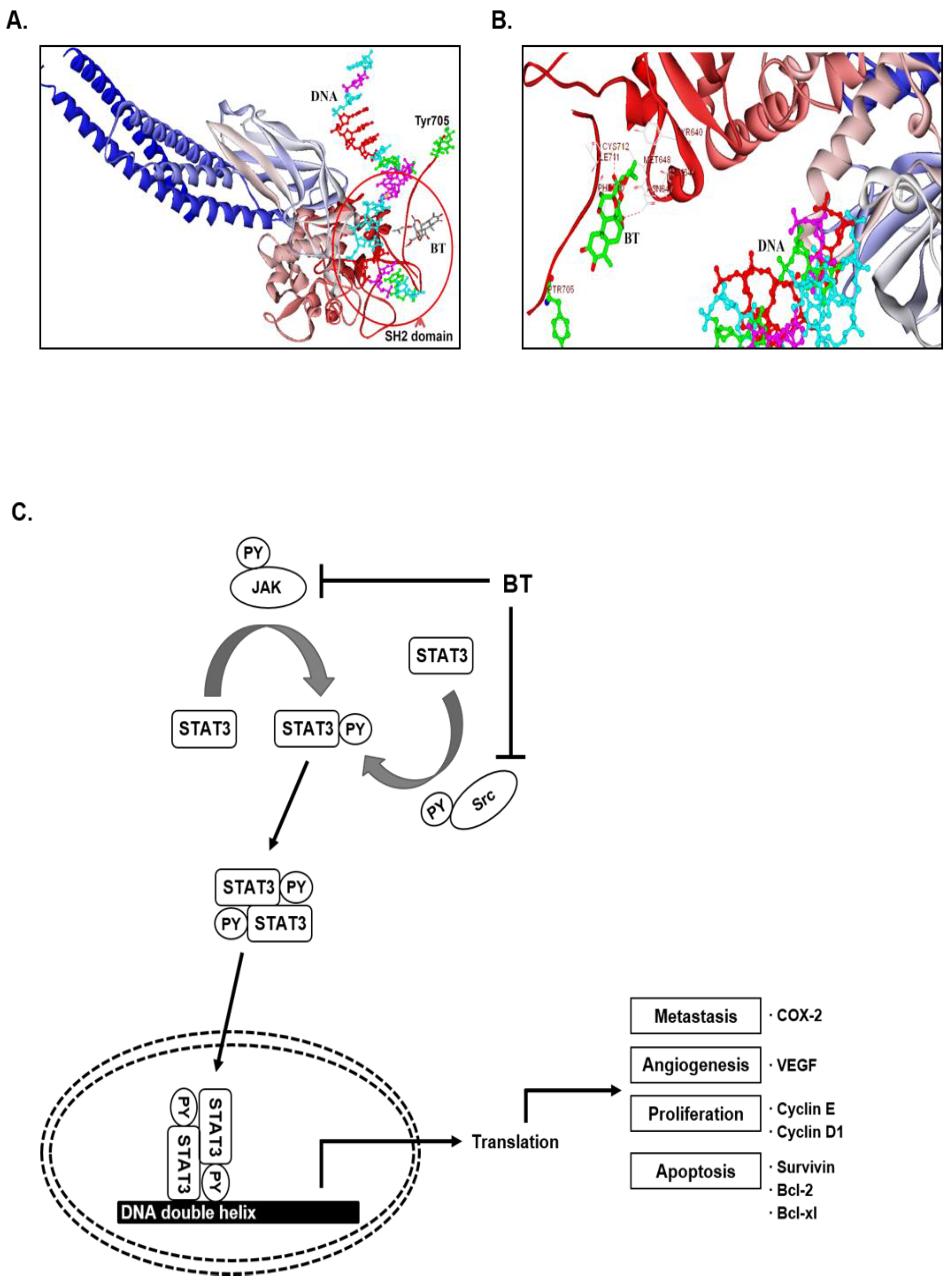
3.14. In Silico Interaction Studies between BT and SH2 Domain of STAT3
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Essid, N.; Chambard, J.C.; Elgaaïed, A.B. Induction of epithelial-mesenchymal transition (EMT) and Gli1 expression in head and neck squamous cell carcinoma (HNSCC) spheroid cultures. Bosn. J. Basic Med. Sci. 2018, 18, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Marur, S.; Forastiere, A.A. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin. Proc. 2016, 91, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.R.; Jefferson, G. Chapter 64—Head and Neck Cancer. In Genomic and Personalized Medicine, 2nd ed.; Ginsburg, G.S., Willard, H.F., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 742–748. [Google Scholar] [CrossRef]
- Gingerich, M.A.; Smith, J.D.; Michmerhuizen, N.L.; Ludwig, M.; Devenport, S.; Matovina, C.; Brenner, C.; Chinn, S.B. Comprehensive review of genetic factors contributing to head and neck squamous cell carcinoma development in low-risk, nontraditional patients. Head Neck 2018, 40, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Golusiński, W. Functional Organ Preservation Surgery in Head and Neck Cancer: Transoral Robotic Surgery and Beyond. Front. Oncol. 2019, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, C.; Lee, J.; Um, J.Y.; Sethi, G.; Ahn, K.S. Arctiin is a pharmacological inhibitor of STAT3 phosphorylation at tyrosine 705 residue and potentiates bortezomib-induced apoptotic and anti-angiogenic effects in human multiple myeloma cells. Phytomedicine 2019, 55, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Hirpara, J.L.; Eu, J.Q.; Sethi, G.; Wang, L.; Goh, B.C.; Wong, A.L. Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biol. 2018, 101073. [Google Scholar] [CrossRef]
- Singh, S.S.; Yap, W.N.; Arfuso, F.; Kar, S.; Wang, C.; Cai, W.; Dharmarajan, A.M.; Sethi, G.; Kumar, A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015, 21, 12261–12273. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Ko, J.H.; Jung, Y.Y.; Jung, S.H.; Kim, E.; Kong, M.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; et al. Casticin inhibits growth and enhances ionizing radiation-induced apoptosis through the suppression of STAT3 signaling cascade. J. Cell. Biochem. 2019, 120, 9787–9798. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Baek, S.H.; Ko, J.H.; Lee, S.G.; Yang, W.M.; Um, J.Y.; Sethi, G.; Ahn, K.S. Capsazepine inhibits JAK/STAT3 signaling, tumor growth, and cell survival in prostate cancer. Oncotarget 2017, 8, 17700–17711. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Lee, S.-G.; Sethi, G.; Ahn, K.S. Ophiopogonin D, a Steroidal Glycoside Abrogates STAT3 Signaling Cascade and Exhibits Anti-Cancer Activity by Causing GSH/GSSG Imbalance in Lung Carcinoma. Cancers 2018, 10, 427. [Google Scholar] [CrossRef]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta 2014, 1845, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H.; Ko, J.H.; Lee, H.; Jung, J.; Kong, M.; Lee, J.W.; Lee, J.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; et al. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomedicine 2016, 23, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Lee, J.H.; Kim, C.; Ko, J.H.; Ryu, S.H.; Lee, S.G.; Yang, W.M.; Um, J.Y.; Chinnathambi, A.; Alharbi, S.A.; et al. Ginkgolic Acid C 17:1, Derived from Ginkgo biloba Leaves, Suppresses Constitutive and Inducible STAT3 Activation through Induction of PTEN and SHP-1 Tyrosine Phosphatase. Molecules 2017, 22, 276. [Google Scholar] [CrossRef] [PubMed]
- Chai, E.Z.; Siveen, K.S.; Shanmugam, M.K.; Arfuso, F.; Sethi, G. Analysis of the intricate relationship between chronic inflammation and cancer. Biochem. J. 2015, 468, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.Y.; Arya, A.; Naema, A.F.; Wong, W.F.; Sethi, G.; Looi, C.Y. Signal Transducer and Activator of Transcription (STATs) Proteins in Cancer and Inflammation: Functions and Therapeutic Implication. Front. Oncol. 2019, 9, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, E.Z.; Shanmugam, M.K.; Arfuso, F.; Dharmarajan, A.; Wang, C.; Kumar, A.P.; Samy, R.P.; Lim, L.H.; Wang, L.; Goh, B.C.; et al. Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol. Ther. 2016, 162, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, C.; Sethi, G.; Ahn, K.S. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget 2015, 6, 6386–6405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ahn, K.S.; Kim, C.; Shanmugam, M.K.; Siveen, K.S.; Arfuso, F.; Samym, R.P.; Deivasigamanim, A.; Lim, L.H.; Wang, L.; et al. Nimbolide-Induced Oxidative Stress Abrogates STAT3 Signaling Cascade and Inhibits Tumor Growth in Transgenic Adenocarcinoma of Mouse Prostate Model. Antioxid. Redox Signal. 2016, 24, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Li, F.; Shanmugam, M.K.; Kannaiyan, R.; Goh, J.N.; Wong, K.F.; Wang, W.; Khin, E.; Tergaonkar, V.; Kumar, A.P.; et al. Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev. Res. 2012, 5, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Rajendran, P.; Li, F.; Kim, C.; Sikka, S.; Siveen, K.S.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Abrogation of STAT3 signaling cascade by zerumbone inhibits proliferation and induces apoptosis in renal cell carcinoma xenograft mouse model. Mol. Carcinog. 2015, 54, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Arora, L.; Kumar, A.P.; Arfuso, F.; Chng, W.J.; Sethi, G. The Role of Signal Transducer and Activator of Transcription 3 (STAT3) and Its Targeted Inhibition in Hematological Malignancies. Cancers 2018, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.J.; Liu, Y.; Han, S.; Yang, C. Brusatol, an NRF2 inhibitor for future cancer therapeutic. Cell Biosci. 2019, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Ren, D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y. Brusatol enhances the chemotherapy efficacy of gemcitabine in pancreatic cancer via the Nrf2 signalling pathway. Oxid. Med. Cell. Longev. 2018, 2018, 2360427. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H. Resistance to gefitinib and cross-resistance to irreversible EGFR-TKIs mediated by disruption of the Keap1-Nrf2 pathway in human lung cancer cells. FASEB J. 2018, 32, 5862–5873. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, Q.; Wang, Y.; Du, L.; Xu, C.; Liu, Q. Brusatol Enhances the Radiosensitivity of A549 Cells by Promoting ROS Production and Enhancing DNA Damage. Int. J. Mol. Sci. 2016, 17, 997. [Google Scholar] [CrossRef]
- Vartanian, S. Application of mass spectrometry profiling to Establish Brusatol as an inhibitor of global protein synthesis. Mol. Cell. Proteom. 2016, 15, 1220–1231. [Google Scholar] [CrossRef]
- Oh, E.T. Brusatol-mediated inhibition of c-Myc increases HIF-1α degradation and causes cell death in colorectal cancer under hypoxia. Theranostics 2017, 7, 3415. [Google Scholar] [CrossRef]
- Xiang, Y. Brusatol inhibits growth and induces apoptosis in pancreatic cancer cells via JNK/p38 MAPK/NF-kappab/Stat3/Bcl-2 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 487, 820–826. [Google Scholar] [CrossRef]
- Sebastian, A.; Pandey, V.; Mohan, C.D.; Chia, Y.T.; Rangappa, S.; Mathai, J.; Baburajeev, C.P.; Paricharak, S.; Mervin, L.H.; Bulusu, K.C.; et al. Novel Adamantanyl-Based Thiadiazolyl Pyrazoles Targeting EGFR in Triple-Negative Breast Cancer. ACS Omega 2016, 1, 1412–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baburajeev, C.P.; Mohan, C.D.; Rangappa, S.; Mason, D.J.; Fuchs, J.E.; Bender, A.; Barash, U.; Vlodavsky, I.; Basappa; Rangappa, K.S. Identification of Novel Class of Triazolo-Thiadiazoles as Potent Inhibitors of Human Heparanase and their Anticancer Activity. BMC Cancer 2017, 17, 235. [Google Scholar] [CrossRef]
- Chua, A.W.L.; Hay, H.S.; Rajendran, P.; Shanmugam, M.K.; Li, F.; Bist, P.; Koay, E.S.C.; Lim, L.H.K.; Kumar, A.P.; Sethi, G. Butein downregulates chemokine receptor CXCR4 expression and function through suppression of NF-κB activation in breast and pancreatic tumor cells. Biochem. Pharmacol. 2010, 80, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Bharathkumar, H.; Dukanya; Rangappa, S.; Shanmugam, M.K.; Chinnathambi, A.; Alharbi, S.A.; Alahmadi, T.A.; Bhattacharjee, A.; Lobie, P.E.; et al. N-Substituted Pyrido-1,4-Oxazin-3-Ones Induce Apoptosis of Hepatocellular Carcinoma Cells by Targeting NF-κB Signaling Pathway. Front. Pharmacol. 2018, 9, 1125. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Wang, B.; Mohan, C.D.; Raquib, A.R.; Rangappa, S.; Srinivasa, V.; Fuchs, J.E.; Girish, K.S.; Zhu, T.; Bender, A.; et al. Discovery of a small-molecule inhibitor of specific serine residue BAD phosphorylation. Proc. Natl. Acad. Sci. USA 2018, 115, E10505–E10514. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.S.; Shishodia, S.; Ahn, K.S.; Kunnumakkara, A.B.; Sethi, G.; Aggarwal, B.B. Deguelin, an Akt Inhibitor, Suppresses IκBα Kinase Activation Leading to Suppression of NF-κB-Regulated Gene Expression, Potentiation of Apoptosis, and Inhibition of Cellular Invasion. J. Immunol. 2006, 177, 5612–5622. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.Y.; Lee, J.H.; Nam, D.; Narula, A.S.; Namjoshi, O.A.; Blough, B.E.; Um, J.-Y.; Sethi, G.; Ahn, K.S. Anti-myeloma Effects of Icariin Are Mediated Through the Attenuation of JAK/STAT3-Dependent Signaling Cascade. Front. Pharmacol. 2018, 9, 531. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Lee, S.G.; Yang, W.M.; Arfuso, F.; Um, J.Y.; Kumar, A.P.; Bian, J.; Sethi, G.; Ahn, K.S. Formononetin-induced oxidative stress abrogates the activation of STAT3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer Lett. 2018, 431, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.; Lee, J.H.; Kim, H.S.; Kim, T.; Han, Y.T.; Suh, Y.-G.; Chun, J.; Kim, Y.S.; Ahn, K.S. Novel Galiellalactone Analogues Can Target STAT3 Phosphorylation and Cause Apoptosis in Triple-Negative Breast Cancer. Biomolecules 2019, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.C.; Hsu, A.; Kumar, A.P.; Sethi, G.; Tan, K.H.B. Thymoquinone Inhibits Tumor Growth and Induces Apoptosis in a Breast Cancer Xenograft Mouse Model: The Role of p38 MAPK and ROS. PLoS ONE 2013, 8, e75356. [Google Scholar] [CrossRef] [PubMed]
- Baburajeev, C.P.; Mohan, C.D.; Patil, G.S.; Rangappa, S.; Pandey, V.; Sebastian, A.; Fuchs, J.E.; Bender, A.; Lobie, P.E.; Basappa, B.; et al. Nano-cuprous oxide catalyzed one-pot synthesis of a carbazole-based STAT3 inhibitor: A facile approach via intramolecular C–N bond formation reactions. RSC Adv. 2016, 6, 36775–36785. [Google Scholar] [CrossRef]
- Sulaiman, N.B.S.; Mohan, C.D.; Rangappa, S.; Bharathkumar, H.; Kumar, A.P.; Basappa; Pandey, V.; Lobie, P.E.; Rangappa, K.S. An azaspirane derivative suppresses growth and induces apoptosis of ER-positive and ER-negative breast cancer cells through the modulation of JAK2/STAT3 signaling pathway. Int. J. Oncol. 2016, 49, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A.; et al. Development of a Novel Azaspirane That Targets the Janus Kinase-Signal Transducer and Activator of Transcription (STAT) Pathway in Hepatocellular Carcinomain Vitroandin Vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Lee, J.H.; Chai, E.Z.P.; Kanchi, M.M.; Kar, S.; Arfuso, F.; Dharmarajan, A.; Kumar, A.P.; Ramar, P.S.; Looi, C.Y.; et al. Cancer prevention and therapy through the modulation of transcription factors by bioactive natural compounds. Semin. Cancer Biol. 2016, 40–41, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.-W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [Green Version]
- Ashwini, N.; Garg, M.; Mohan, C.D.; Fuchs, J.E.; Rangappa, S.; Anusha, S.; Swaroop, T.R.; Rakesh, K.S.; Kanojia, D.; Madan, V.; et al. Synthesis of 1,2-benzisoxazole tethered 1,2,3-triazoles that exhibit anticancer activity in acute myeloid leukemia cell lines by inhibiting histone deacetylases, and inducing p21 and tubulin acetylation. Bioorg. Med. Chem. 2015, 23, 6157–6165. [Google Scholar] [CrossRef] [PubMed]
- Somu, C.; Hegde, M.; Kumar, K.S.S.; Hanumappa, A.; Srivastava, M.; Harsha, K.B.; Mohan, C.D.; Ananthaswamy, K.; Raghavan, S.C.; Rangappa, K.S. Synthesis and Biological Evaluation of Novel Thiazol-2yl-amine Derivatives as Potential Anticancer Agents. Lett. Org. Chem. 2018, 15, 270–281. [Google Scholar] [CrossRef]
- Nirvanappa, A.C.; Mohan, C.D.; Rangappa, S.; Ananda, H.; Sukhorukov, A.Y.; Shanmugam, M.K.; Sundaram, M.S.; Nayaka, S.C.; Girish, K.S.; Chinnathambi, A.; et al. Novel Synthetic Oxazines Target NF-kappaB in Colon Cancer In Vitro and Inflammatory Bowel Disease In Vivo. PLoS ONE 2016, 11, e0163209. [Google Scholar] [CrossRef]
- Baburajeev, C.P.; Dhananjaya Mohan, C.; Ananda, H.; Rangappa, S.; Fuchs, J.E.; Jagadish, S.; Sivaraman Siveen, K.; Chinnathambi, A.; Alharbi, S.A.; Zayed, M.E.; et al. Development of Novel Triazolo-Thiadiazoles from Heterogeneous "Green" Catalysis as Protein Tyrosine Phosphatase 1B Inhibitors. Sci. Rep. 2015, 5, 14195. [Google Scholar] [CrossRef]
- Roopashree, R.; Mohan, C.D.; Swaroop, T.R.; Jagadish, S.; Raghava, B.; Balaji, K.S.; Jayarama, S.; Basappa; Rangappa, K.S. Novel synthetic bisbenzimidazole that targets angiogenesis in Ehrlich ascites carcinoma bearing mice. Bioorg. Med. Chem. Lett. 2015, 25, 2589–2593. [Google Scholar] [CrossRef]
- Anusha, S.; Mohan, C.D.; Ananda, H.; Baburajeev, C.; Rangappa, S.; Mathai, J.; Fuchs, J.E.; Li, F.; Shanmugam, M.K.; Bender, A.; et al. Adamantyl-tethered-biphenylic compounds induce apoptosis in cancer cells by targeting Bcl homologs. Bioorg. Med. Chem. Lett. 2016, 26, 1056–1060. [Google Scholar] [CrossRef]
- Mohan, C.D.; Anilkumar, N.C.; Rangappa, S.; Shanmugam, M.K.; Mishra, S.; Chinnathambi, A.; Alharbi, S.A.; Bhattacharjee, A.; Sethi, G.; Kumar, A.P.; et al. Novel 1,3,4-Oxadiazole Induces Anticancer Activity by Targeting NF-kappaB in Hepatocellular Carcinoma Cells. Front. Oncol. 2018, 8, 42. [Google Scholar] [CrossRef]
- Dai, X.; Ahn, K.S.; Kim, C.; Siveen, K.S.; Ong, T.H.; Shanmugam, M.K.; Li, F.; Shi, J.; Kumar, A.P.; Wang, L.Z.; et al. Ascochlorin, an isoprenoid antibiotic inhibits growth and invasion of hepatocellular carcinoma by targeting STAT3 signaling cascade through the induction of PIAS3. Mol. Oncol. 2015, 9, 818–833. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, C.; Kim, S.H.; Sethi, G.; Ahn, K.S. Farnesol inhibits tumor growth and enhances the anticancer effects of bortezomib in multiple myeloma xenograft mouse model through the modulation of STAT3 signaling pathway. Cancer Lett. 2015, 360, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, A.; Shanmugam, M.K.; Ong, T.H.; Li, F.; Perumal, E.; Chen, L.; Vali, S.; Abbasi, T.; Kapoor, S.; Ahn, K.S.; et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. Br. J. Pharmacol. 2013, 170, 807–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, A.; Shanmugam, M.K.; Perumal, E.; Li, F.; Nachiyappan, A.; Dai, X.; Swamy, S.N.; Ahn, K.S.; Kumar, A.P.; Tan, B.K.; et al. Potential role of signal transducer and activator of transcription (STAT)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochim. Biophys. Acta 2013, 1835, 46–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, G.; Chatterjee, S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Wong, K.F.; Kumar, A.P.; Senapati, P.; Behera, A.K.; Hui, K.M.; et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer 2014, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Lee, J.H.; Sethi, G.; Kim, C.; Baek, S.H.; Nam, D.; Chung, W.-S.; Kim, S.-H.; Shim, B.S.; Ahn, K.S. Bergamottin, a natural furanocoumarin obtained from grapefruit juice induces chemosensitization and apoptosis through the inhibition of STAT3 signaling pathway in tumor cells. Cancer Lett. 2014, 354, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Rajendran, P.; Sethi, G. Thymoquinone inhibits proliferation, induces apoptosis and chemosensitizes human multiple myeloma cells through suppression of signal transducer and activator of transcription 3 activation pathway. Br. J. Pharmacol. 2010, 161, 541–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannaiyan, R.; Hay, H.S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P.; et al. Celastrol inhibits proliferation and induces chemosensitization through down-regulation of NF-κB and STAT3 regulated gene products in multiple myeloma cells. Br. J. Pharmacol. 2011, 164, 1506–1521. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Jove, R. STAT proteins: Novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Li, F.; Manu, K.A.; Shanmugam, M.K.; Loo, S.Y.; Kumar, A.P.; Sethi, G. γ-Tocotrienol is a novel inhibitor of constitutive and inducible STAT3 signalling pathway in human hepatocellular carcinoma: Potential role as an antiproliferative, pro-apoptotic and chemosensitizing agent. Br. J. Pharmacol. 2011, 163, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Ahn, K.S.; Kumar, A.P.; Sethi, G. Honokiol inhibits signal transducer and activator of transcription-3 signaling, proliferation, and survival of hepatocellular carcinoma cells via the protein tyrosine phosphatase SHP-1. J. Cell. Physiol. 2012, 227, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, V.; Mohan, C.D.; Baburajeev, C.; Rangappa, S.; Jagadish, S.; Fuchs, J.E.; Sukhorukov, A.Y.; Chandra; Mason, D.J.; Kumar, K.S.S.; et al. Synthesis and characterization of novel oxazines and demonstration that they specifically target cyclooxygenase 2. Bioorg. Med. Chem. Lett. 2015, 25, 2931–2936. [Google Scholar] [CrossRef] [PubMed]
- Cuendet, M.; Pezzuto, J.M. Antitumor activity of bruceantin: An old drug with new promise. J. Nat. Prod. 2004, 67, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.A.; Grandis, J.R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef]
- Aoki, Y.; Feldman, G.M.; Tosato, G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003, 101, 1535. [Google Scholar] [CrossRef]
- Bharathkumar, H.; Paricharak, S.; Dinesh, K.R.; Siveen, K.S.; Fuchs, J.E.; Rangappa, S.; Mohan, C.D.; Mohandas, N.; Kumar, A.P.; Sethi, G.; et al. Synthesis, biological evaluation and in silico and in vitro mode-of-action analysis of novel dihydropyrimidones targeting PPAR-γ. RSC Adv. 2014, 4, 45143–45146. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- Chaitanya, G.V.; Alexander, J.S.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Sehara, Y.; Sawicka, K.; Hwang, J.-Y.; Latuszek-Barrantes, A.; Etgen, A.M.; Zukin, R.S. Survivin Is a transcriptional target of STAT3 critical to estradiol neuroprotection in global ischemia. J. Neurosci. 2013, 33, 12364–12374. [Google Scholar] [CrossRef] [PubMed]
- Keerthy, H.K.; Garg, M.; Mohan, C.D.; Madan, V.; Kanojia, D.; Shobith, R.; NanjundaSwamy, S.; Mason, D.J.; Bender, A.; Koeffler, H.P.; et al. Synthesis and Characterization of Novel 2-Amino-Chromene-Nitriles that Target Bcl-2 in Acute Myeloid Leukemia Cell Lines. PLoS ONE 2014, 9, e107118. [Google Scholar] [CrossRef] [PubMed]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, K.S.; Jagadish, S.; Balaji, K.S.; Zameer, F.; Swaroop, T.R.; Mohan, C.D.; Jayarama, S.; Rangappa, K.S. 3,5-Disubstituted Isoxazole Derivatives: Potential Inhibitors of Inflammation and Cancer. Inflammation 2016, 39, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, K.S.; Jagadish, S.; Swaroop, T.R.; Mohan, C.D.; Ashwini, N.; Harsha, K.B.; Zameer, F.; Girish, K.S.; Rangappa, K.S. Anti-Cancer Activity of 2,4-Disubstituted Thiophene Derivatives: Dual Inhibitors of Lipoxygenase and Cyclooxygenase. Med. Chem. 2015, 11, 462–472. [Google Scholar] [CrossRef]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar] [CrossRef]
- Mohan, C.D.; Hari, S.; Preetham, H.D.; Rangappa, S.; Barash, U.; Ilan, N.; Nayak, S.C.; Gupta, V.K.; Basappa; Vlodavsky, I.; et al. Targeting Heparanase in Cancer: Inhibition by Synthetic, Chemically Modified, and Natural Compounds. iScience 2019, 15, 360–390. [Google Scholar] [CrossRef] [Green Version]
- Gilandoust, M.; Harsha, K.B.; Mohan, C.D.; Raquib, A.R.; Rangappa, S.; Pandey, V.; Lobie, P.E.; Basappa; Rangappa, K.S. Synthesis, characterization and cytotoxicity studies of 1,2,3-triazoles and 1,2,4-triazolo [1,5-a] pyrimidines in human breast cancer cells. Bioorg. Med. Chem. Lett. 2018, 28, 2314–2319. [Google Scholar] [CrossRef]
- Gray-Bablin, J.; Zalvide, J.; Fox, M.P.; Knickerbocker, C.J.; DeCaprio, J.A.; Keyomarsi, K. Cyclin E, a redundant cyclin in breast cancer. Proc. Natl. Acad. Sci. USA 1996, 93, 15215–15220. [Google Scholar] [CrossRef] [Green Version]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Shanmugam, M.K.; Chen, L.; Chatterjee, S.; Basha, J.; Kumar, A.P.; Kundu, T.K.; Sethi, G. Garcinol, a polyisoprenylated benzophenone modulates multiple pro-inflammatory signaling cascades leading to suppression of growth and survival of head and neck carcinoma. Cancer Prev. Res. 2013, 6, 843–854. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Rangappa, S.; Mohan, C.D.; Basappa; Sethi, G.; Lin, Z.-X.; Rangappa, K.S.; Ahn, K.S. Brusatol, a Nrf2 Inhibitor Targets STAT3 Signaling Cascade in Head and Neck Squamous Cell Carcinoma. Biomolecules 2019, 9, 550. https://doi.org/10.3390/biom9100550
Lee JH, Rangappa S, Mohan CD, Basappa, Sethi G, Lin Z-X, Rangappa KS, Ahn KS. Brusatol, a Nrf2 Inhibitor Targets STAT3 Signaling Cascade in Head and Neck Squamous Cell Carcinoma. Biomolecules. 2019; 9(10):550. https://doi.org/10.3390/biom9100550
Chicago/Turabian StyleLee, Jong Hyun, Shobith Rangappa, Chakrabhavi Dhananjaya Mohan, Basappa, Gautam Sethi, Zhi-Xiu Lin, Kanchugarakoppal S. Rangappa, and Kwang Seok Ahn. 2019. "Brusatol, a Nrf2 Inhibitor Targets STAT3 Signaling Cascade in Head and Neck Squamous Cell Carcinoma" Biomolecules 9, no. 10: 550. https://doi.org/10.3390/biom9100550