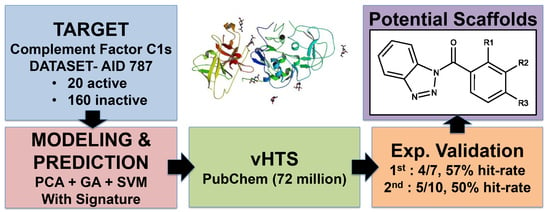
Pharmaceutical Machine Learning: Virtual High-Throughput Screens Identifying Promising and Economical Small Molecule Inhibitors of Complement Factor C1s


Abstract
:
1. Introduction
2. Results
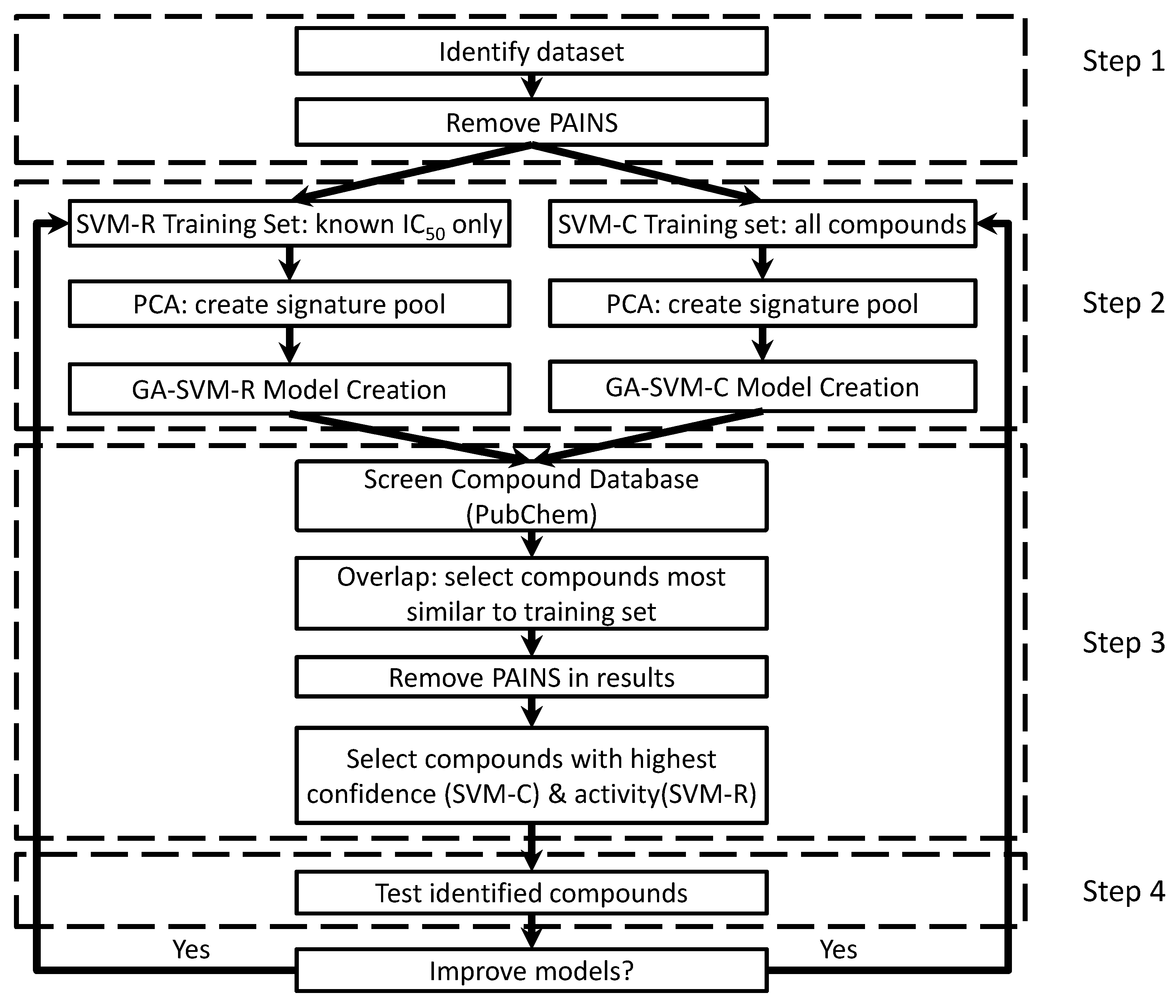
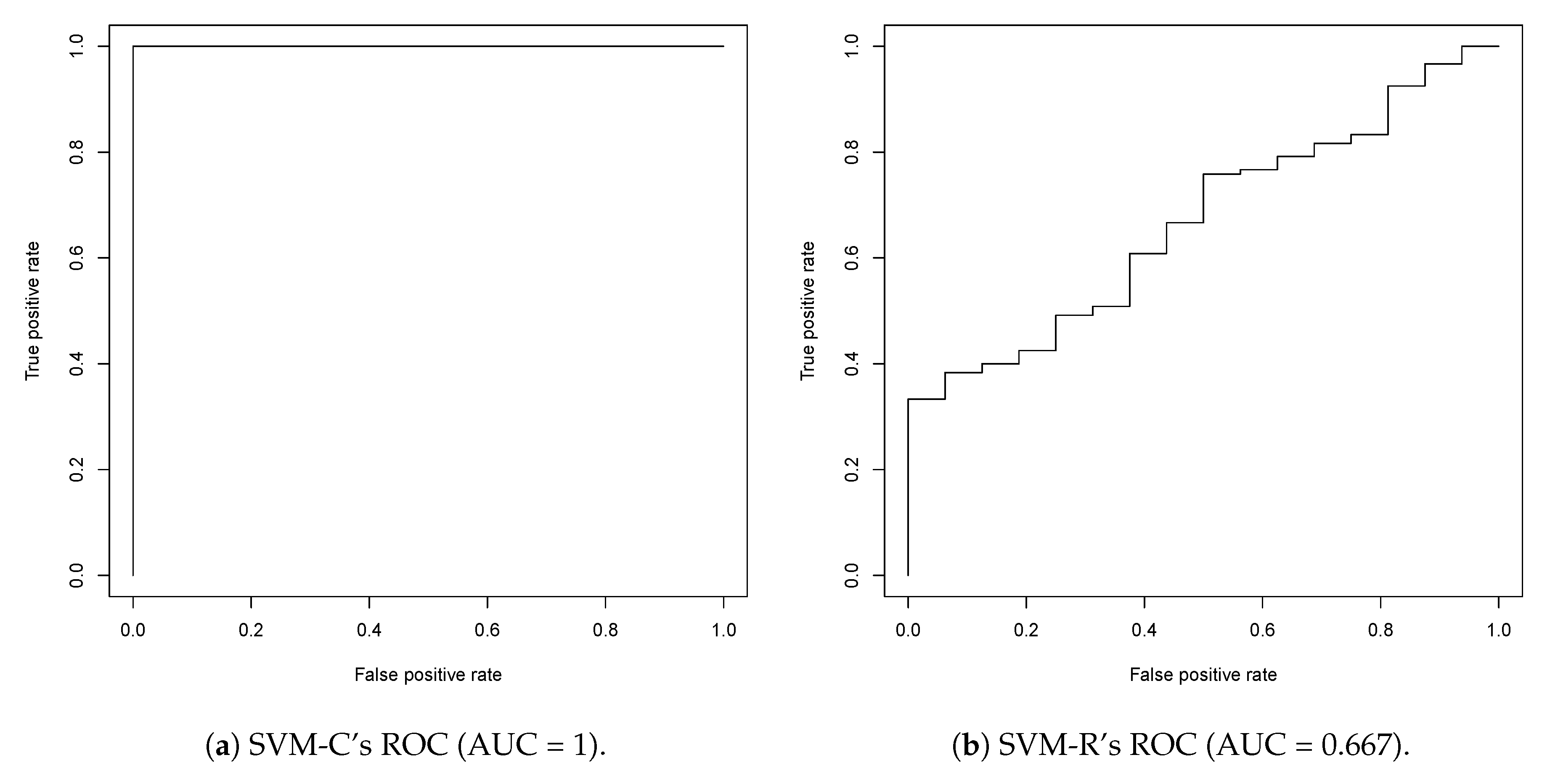
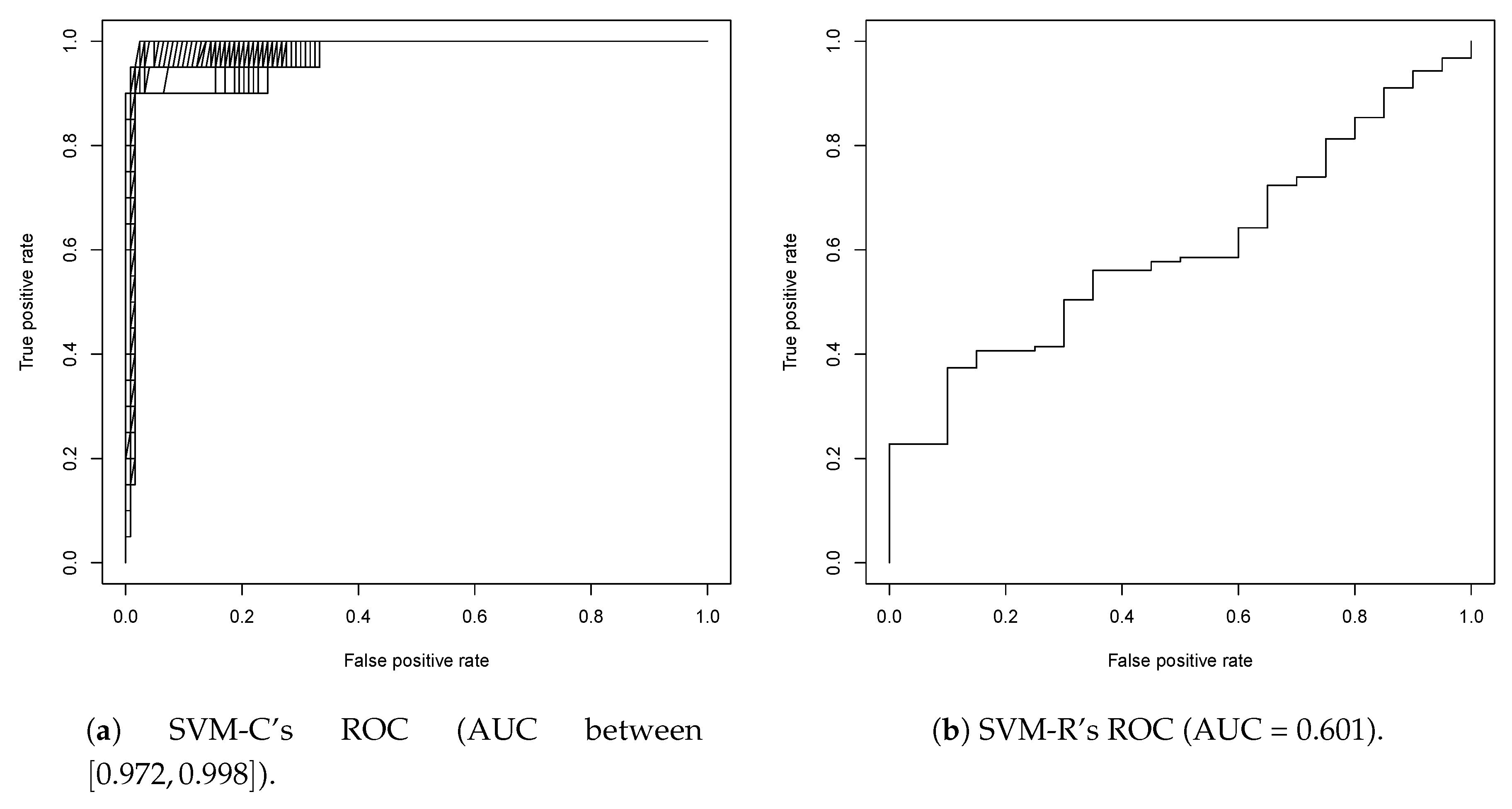
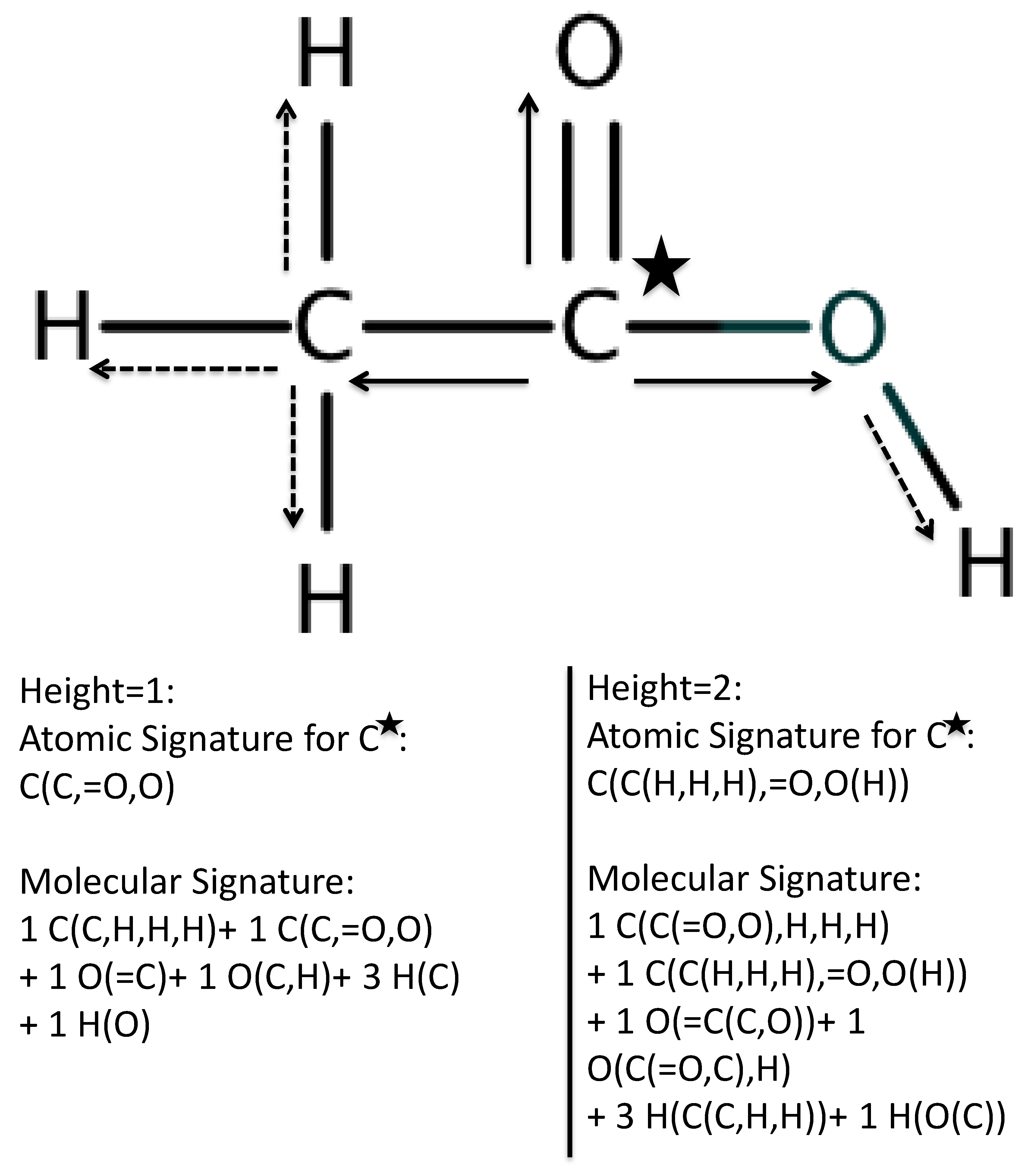
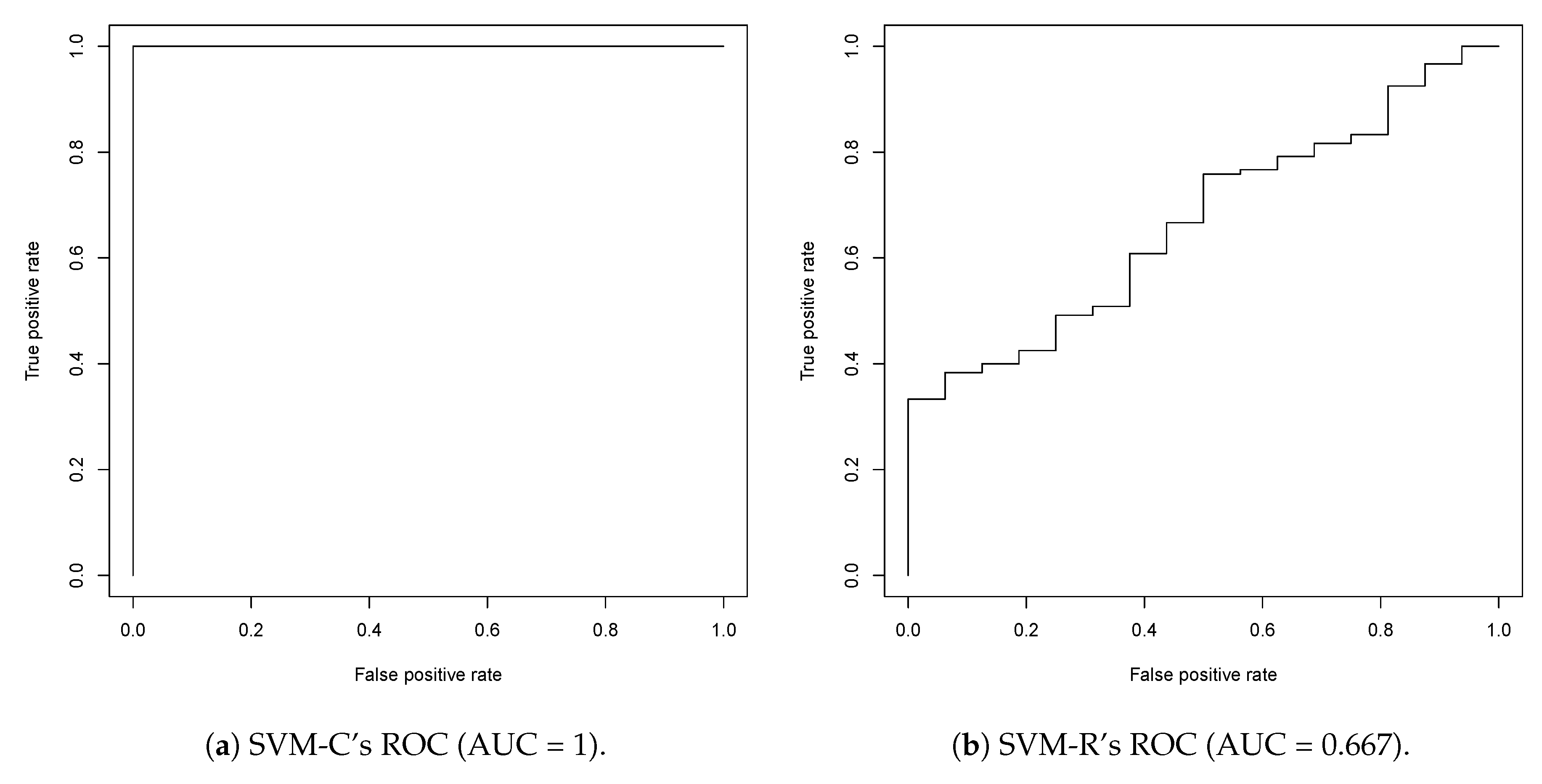
2.1. First Round Classification and QSAR Model Creation, vHTS and Validation Results
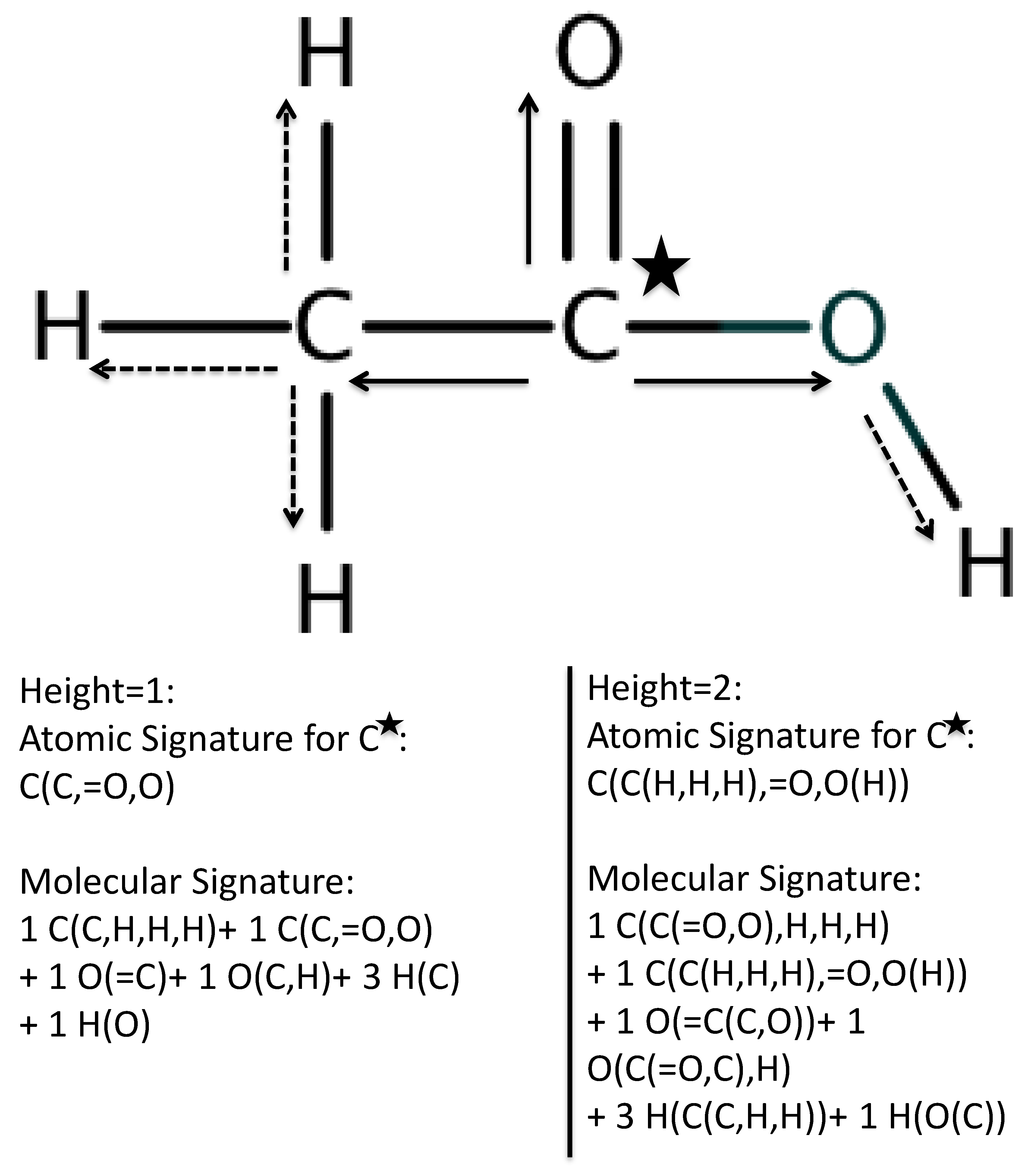
- Overlap = 1. All atomic Signatures in the candidate molecule are found in the training set.
- SVM-C score for all 115 SVM-C models. The models must unanimously agree that a molecule should be in the active class and above a certain threshold.
- Predicted IC < 50 M.
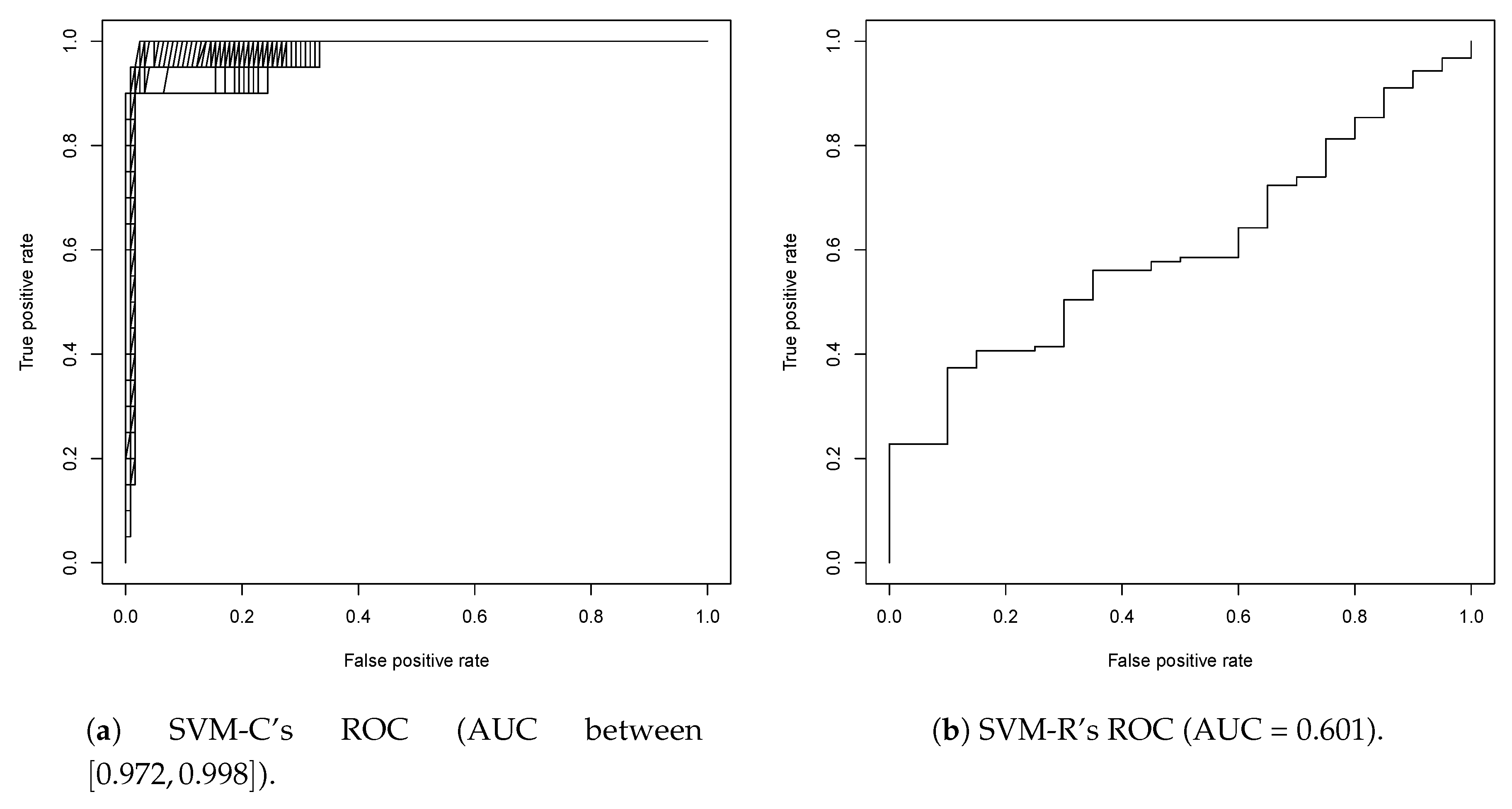
2.2. Second Round Classification and QSAR Model Creation, vHTS and Validation Results
- Overlap i.e., 90% of atomic Signatures in the candidate molecule are found in the training set (modified).
- SVM-C score for all 1224 SVM-C models. The models must unanimously agree a molecule should be in the active class (modified).
- Predicted IC M.
3. Discussion
3.1. Model Discussion
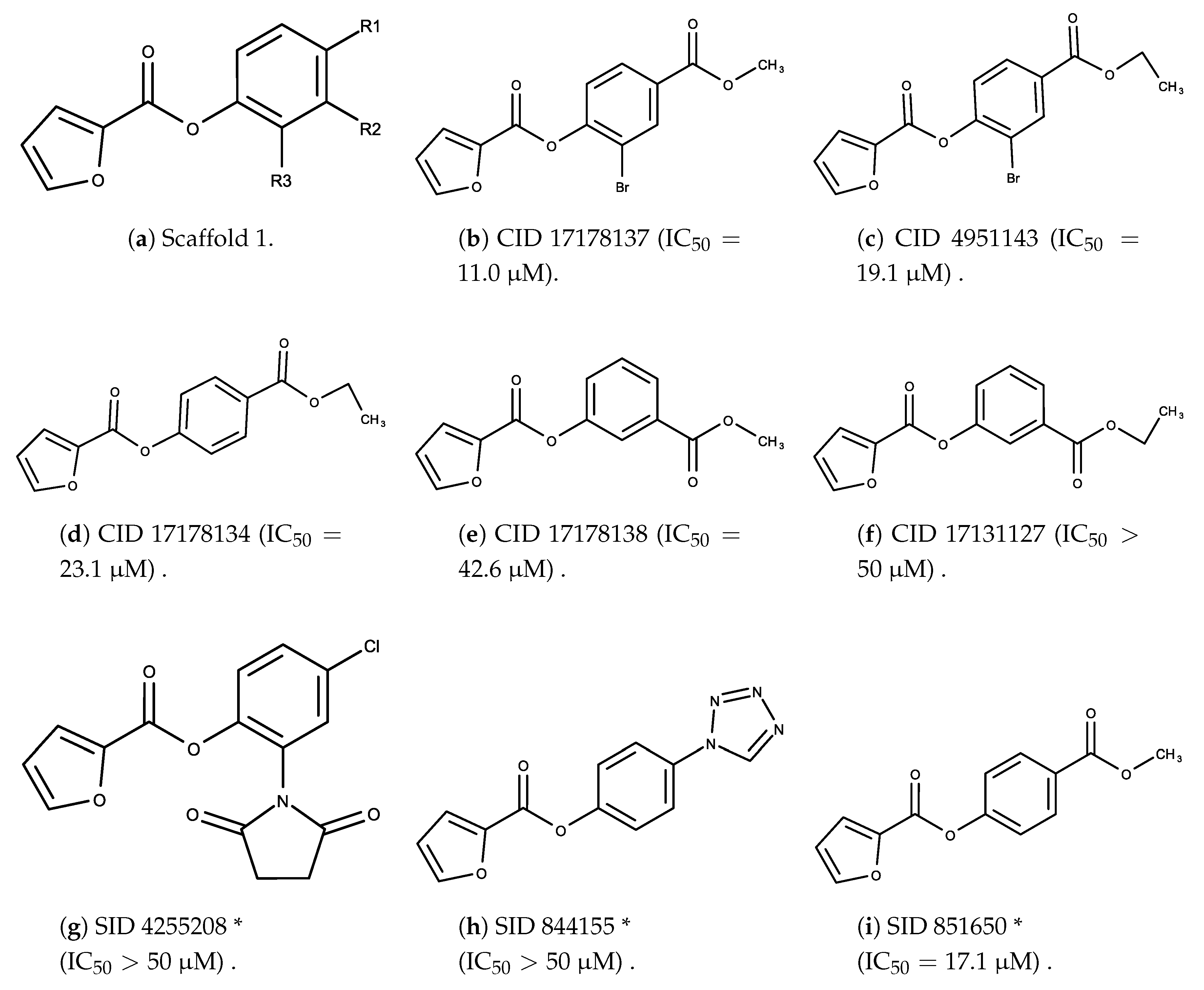
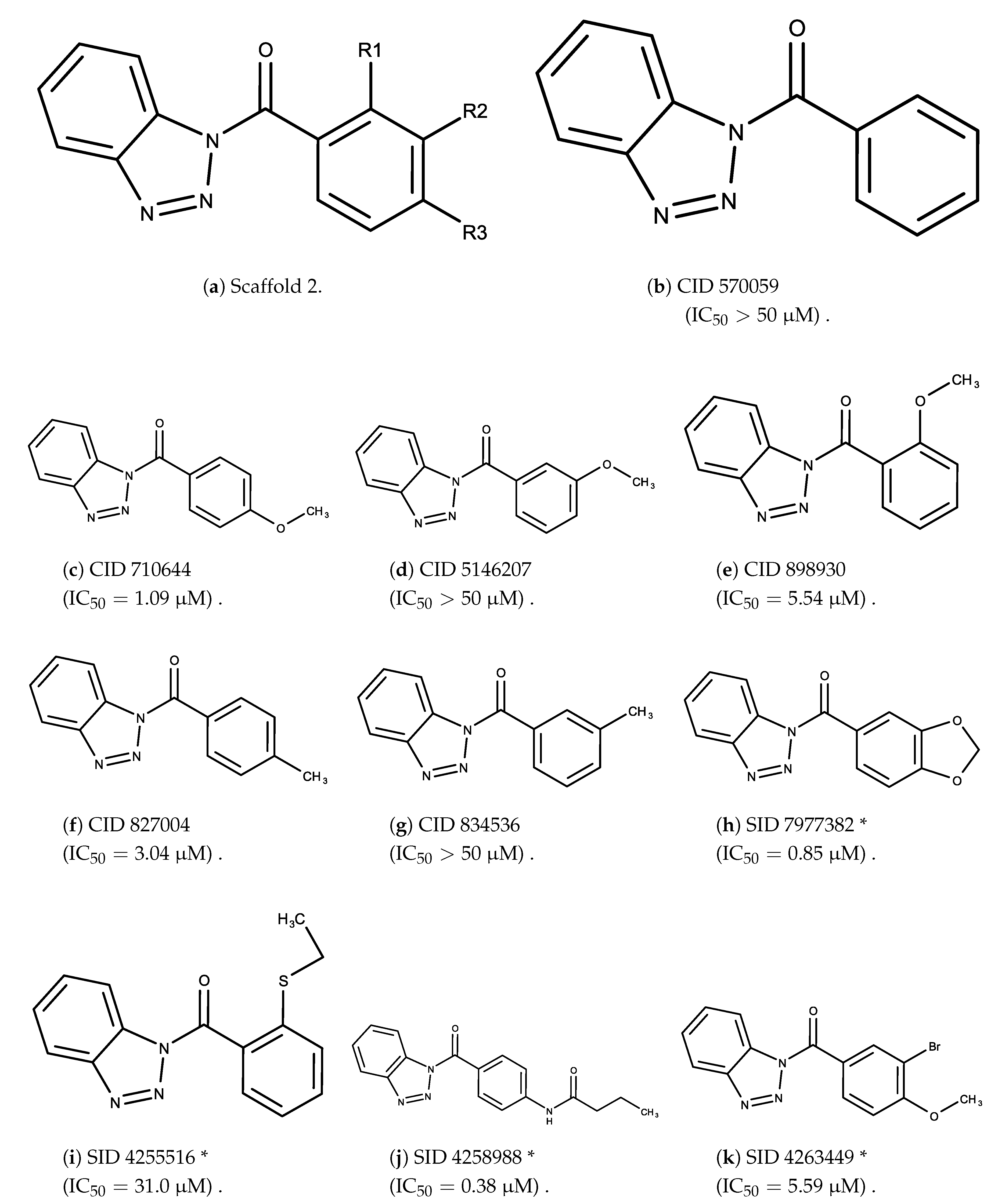
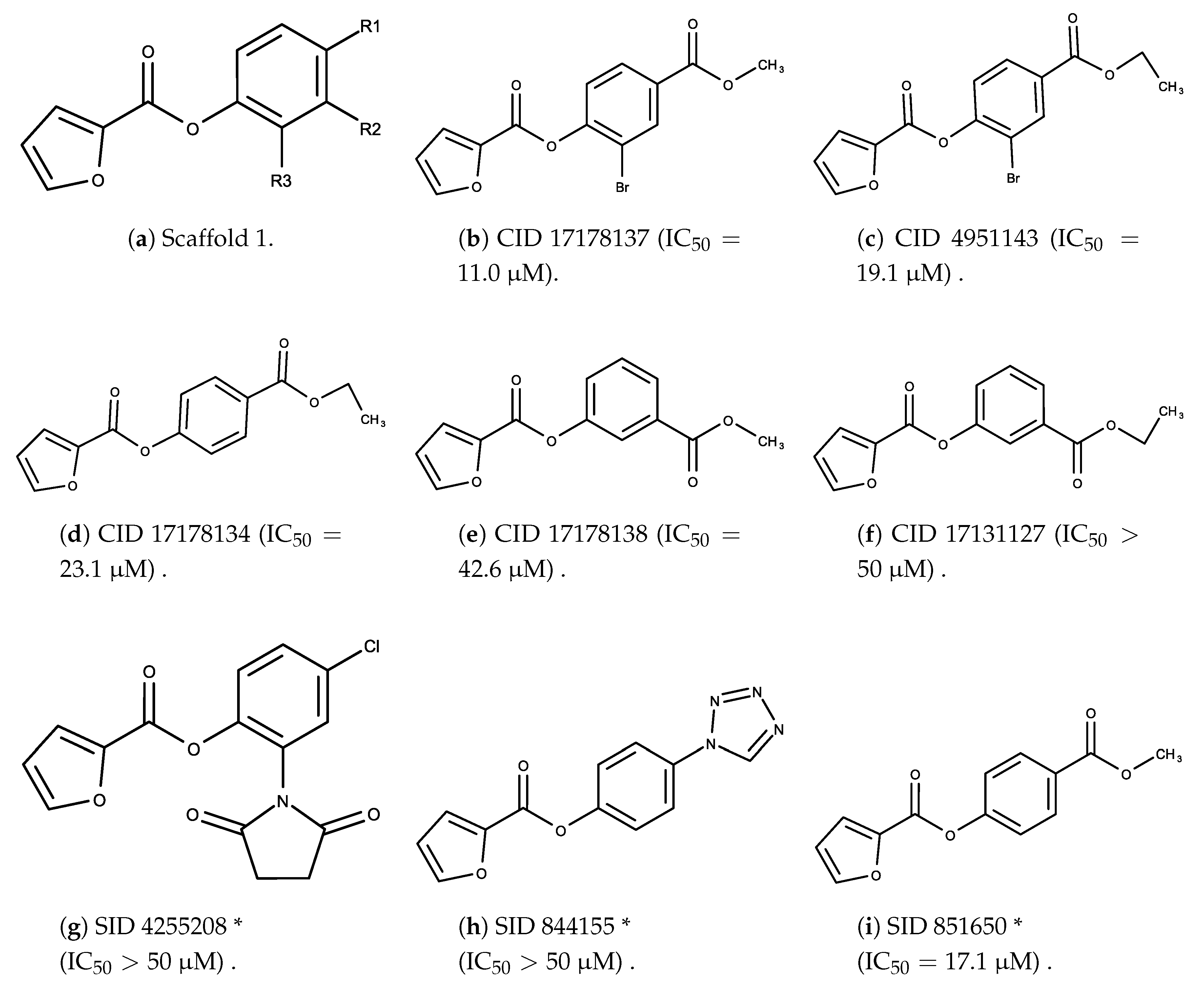
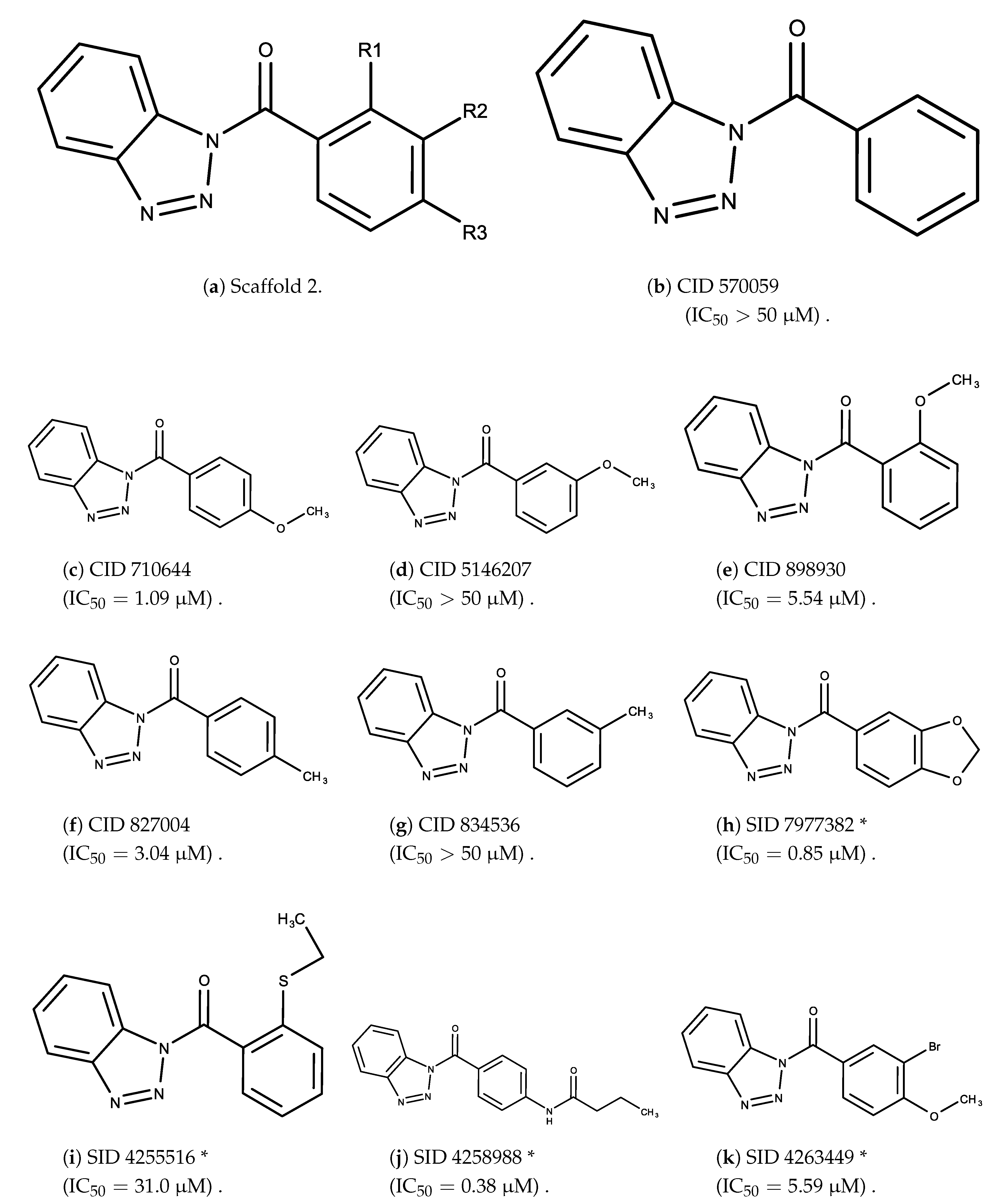
3.2. Complement Factor C1s Inhibitors
- The bromine group in the ortho position seems to have a smaller impact on activity. Structures shown in Figure 5c,d are identical except for the bromine group and the IC values are similar as well ( M vs. M, respectively). A similar analysis can be made for the structures shown in Figure 5b,i ( M vs. M, respectively).
- The position of the larger functional groups has a larger impact on activity. Structures in Figure 5d,f are the same except the ester group is shifted from the para to the meta position. However, this is the difference between active and inactive. A similar analysis can also be made using the structures in Figure 5b,e. If the Br group is assumed to have a small or minimal effect (as mentioned above), then the difference in activity can be accounted by the shift of the ester group from the para to the meta position. Thus, perhaps the para position is preferable to the meta position.
- The identity of the functional group is important. PubChem Substance ID (SID) 4255208 and 844155, shown in Figure 5g,h, have very different functional groups than the rest of the structures shown in Figure 5. The previous conjecture for functional group placement at the ortho and para positions suggests that the inactivity is due to the identity of the functional groups rather than the position of them.
- The bromine group in the meta position seems to have a smaller impact on activity. Structures shown in Figure 6c,k are identical except for the bromine group and the IC values are similar as well ( M vs. M, respectively).
- The position of the large functional group greatly affects activity. Structures in Figure 6c–e are the same structure with the ether group at para, meta, and ortho positions. The ether group at the para position is most active, then ortho and, finally, meta. A similar respective pattern can also be seen with structures in Figure 6f,g and mirrors the pattern seen with scaffold 1. As a reference, Figure 6b, which is scaffold 2 with H at all substitution positions, is inactive so any activity and variance in activity are due primarily to the presence and position of the functional group and not the scaffold.
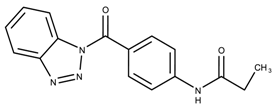
- The identity of the functional group is important. CID 898930 and SID 4255516, shown in Figure 6e,i have functional groups that occupy the the same ortho position. However, there is a big difference in activity ( M vs. M, respectively). The difference in activity is likely due to the identity of the functional group. A similar analysis can be done of CID 710644 and SID 4258988, shown in Figure 6c,j. While the difference in activity is not a big ( M vs. M, respectively), the observed difference is likely due to the identity of the functional group at the para position.
- There may be a compensatory effect of having functional groups at the meta and para positions. When methoxy groups at the meta and para positions fuse into a dioxol ring as seen in SID 7977382 from the training set (Figure 6h), the resulting compound is also active. This suggests a compensatory effect of having a ring or having groups at both the meta and para positions instead of just methoxy groups at the para or meta positions (as in Figure 6c,d, respectively.)
4. Materials and Methods
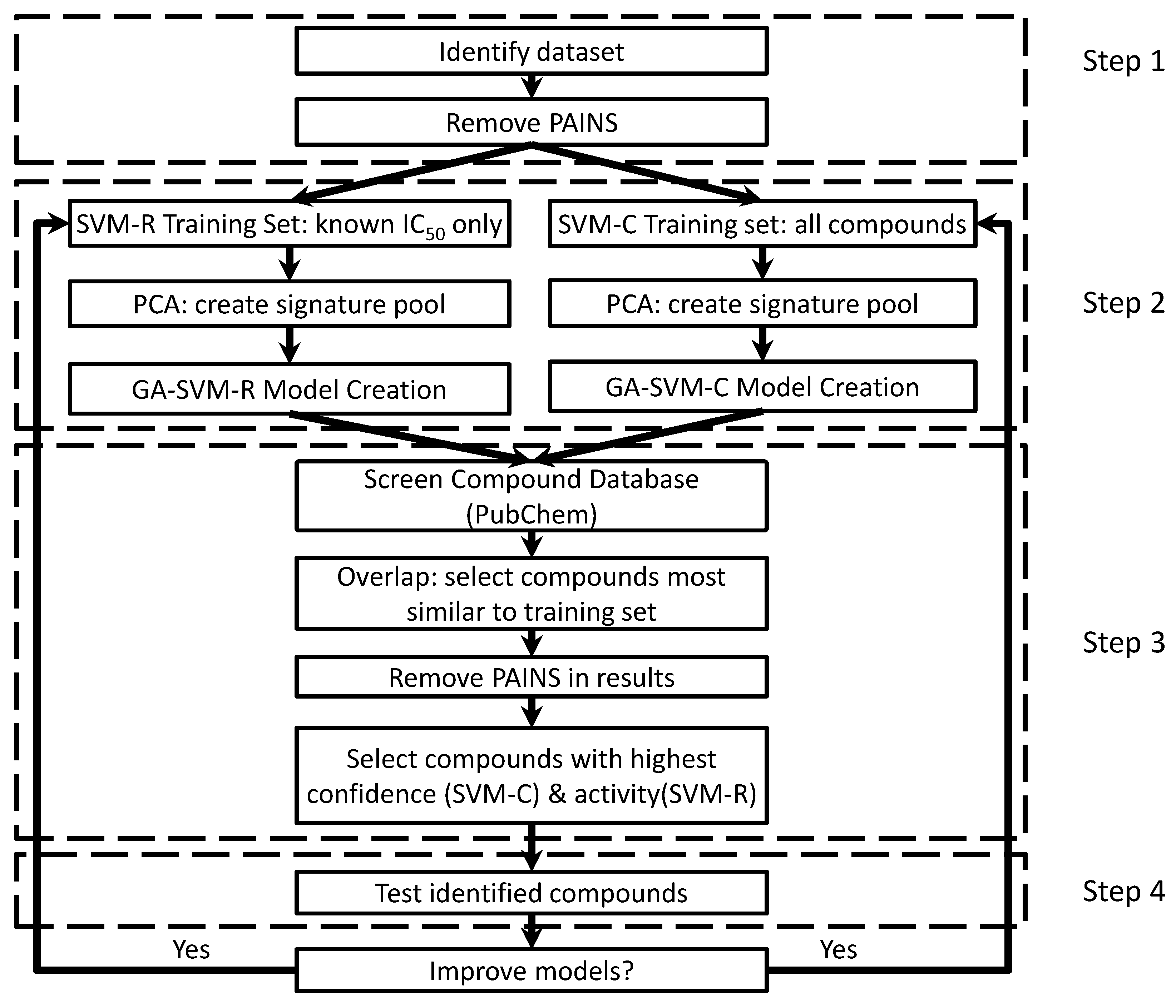
4.1. Creating/Training Predictive Models
4.2. Screening with Predictive Models
4.3. Model Generation Parameters
4.4. Fluorescent Complement Factor C1s Inhibitor Screening Assay
4.4.1. Assay Materials
4.4.2. Assay Protocol
- Serial dilute identified compounds at 50× concentration in DMSO: eight four-fold dilutions from 2.5 mM to 152.6 nM. Final testing concentration from 50 M to 3.05 nM.
- Fill 96 well plate with 50 L substrate solution except column 10.
- Add 2 L of the compound from step 1 to corresponding wells in the plate.
- Add 50 L enzyme to all wells with compound added and column 10.
- Control: Add 50 L enzyme to column 12.
- Blank: Add 50 L buffer to column 11.
- Protein Check: Add 50 L buffer to column 10.
- Incubate for 2.5 h at room temperature.
- Read fluorescence (excitation 355, emission 460) on Tecan M200.
4.4.3. Percent Inhibition Determination
5. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ziccardi, R.J. The first component of human complement (C1): Activation and control. In Springer Seminars in Immunopathology; Springer: Berlin, Germany, 1983; Volume 6, pp. 213–230. [Google Scholar]
- Morris, K.; Bing, D.; Andrews, J.; Silverstein, L.; Shohet, R.; Attisano, C.; Goran, P. Biosynthesis of the Subcomponents of C1 by twelve human established Cell Lines. J. Immunol. 1978, 120, 1786–1787. [Google Scholar]
- Müller, W.; Hanauske-Abel, H.; Loos, M. Biosynthesis of the first component of complement by human and guinea pig peritoneal macrophages: Evidence for an independent production of the C1 subunits. J. Immunol. 1978, 121, 1578–1584. [Google Scholar] [PubMed]
- Gulati, P.; Lemercier, C.; Guc, D.; Lappin, D.; Whaley, K. Regulation of the synthesis of C1 subcomponents and C1-inhibitor. Behring Inst. Mitt. 1993, 93, 196–203. [Google Scholar]
- Goldberger, G.; Bruns, G.; Rits, M.; Edge, M.; Kwiatkowski, D. Human complement factor I: Analysis of cDNA-derived primary structure and assignment of its gene to chromosome 4. J. Biol. Chem. 1987, 262, 10065–10071. [Google Scholar] [PubMed]
- Wallis, R.; Mitchell, D.A.; Schmid, R.; Schwaeble, W.J.; Keeble, A.H. Paths reunited: Initiation of the classical and lectin pathways of complement activation. Immunobiology 2010, 215, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gaboriaud, C.; Thielens, N.M.; Gregory, L.A.; Rossi, V.; Fontecilla-Camps, J.C.; Arlaud, G.J. Structure and activation of the C1 complex of complement: Unraveling the puzzle. Trends Immunol. 2004, 25, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Bokisch, V.A.; Müller-Eberhard, H.J.; Cochrane, C.G. Isolation of a fragment (C3a) of the third component of human complement containing anaphylatoxin and chemotactic activity and description of an anaphylatoxin inactivator of human serum. J. Exp. Med. 1969, 129, 1109–1130. [Google Scholar] [CrossRef] [PubMed]
- Hugli, T.E. Biochemistry and biology of anaphylatoxins. Complement 1986, 3, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Müller-Eberhard, H.J. The killer molecule of complement. J. Investig. Dermatol. 1985, 85, S47–S52. [Google Scholar] [CrossRef]
- Ratnoff, O.D.; Lepow, I.H. Some properties of an esterase derived from preparations of the first component of complement. J. Exp. Med. 1957, 106, 327–343. [Google Scholar] [CrossRef] [PubMed]
- Sim, R.B.; Arlaud, G.J.; Colomb, M.G. C1 inhibitor-dependent dissociation of human complement component C1 bound to immune complexes. Biochem. J. 1979, 179, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Ziccardi, R.J.; Cooper, N.R. Active disassembly of the first complement component, C1, by C1 inactivator. J. Immunol. 1979, 123, 788–792. [Google Scholar] [PubMed]
- Ziccardi, R. Activation of the early components of the classical complement pathway under physiologic conditions. J. Immunol. 1981, 126, 1769–1773. [Google Scholar] [PubMed]
- Ziccardi, R. Demonstration of the interaction of native C1 with monomeric immunoglobulins and C1 inhibitor. J. Immunol. 1985, 134, 2559–2563. [Google Scholar] [PubMed]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.J.; Anderson, A.J.; Barnum, S.R.; Stevens, B.; Tenner, A.J. The complement cascade: Yin–Yang in neuroinflammation–neuro-protection and-degeneration. J. Neurochem. 2008, 107, 1169–1187. [Google Scholar] [CrossRef] [PubMed]
- Gompels, M.; Lock, R.; Abinun, M.; Bethune, C.; Davies, G.; Grattan, C.; Fay, A.; Longhurst, H.; Morrison, L.; Price, A.; et al. C1 inhibitor deficiency: Consensus document. Clin. Exp. Immunol. 2005, 139, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Kawalec, P.; Holko, P.; Paszulewicz, A. Cost-utility analysis of Ruconest®(conestat alfa) compared to Berinert® P (human C1 esterase inhibitor) in the treatment of acute, life-threatening angioedema attacks in patients with hereditary angioedema. Adv. Dermatol. Allergol. 2013, 30, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Craig, T.J.; Levy, R.J.; Wasserman, R.L.; Bewtra, A.K.; Hurewitz, D.; Obtułowicz, K.; Reshef, A.; Ritchie, B.; Moldovan, D.; Shirov, T.; et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J. Allergy Clin. Immunol. 2009, 124, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Lunn, M.; Santos, C.; Craig, T. Cinryze™ as the first approved C1 inhibitor in the USA for the treatment of hereditary angioedema: Approval, efficacy and safety. J. Blood Med. 2010, 1, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Longhurst, H. Rhucin, a recombinant C1 inhibitor for the treatment of hereditary angioedema and cerebral ischemia. Curr. Opin. Investig. Drugs 2008, 9, 310–323. [Google Scholar] [PubMed]
- Cruz, M.P. Conestat alfa (ruconest): First recombinant c1 esterase inhibitor for the treatment of acute attacks in patients with hereditary angioedema. Pharm. Ther. 2015, 40, 109. [Google Scholar]
- Wilson, D.A.; Bork, K.; Shea, E.P.; Rentz, A.M.; Blaustein, M.B.; Pullman, W.E. Economic costs associated with acute attacks and long-term management of hereditary angioedema. Ann. Allergy Asthma Immunol. 2010, 104, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Petraroli, A.; Squeglia, V.; Di Paola, N.; Barbarino, A.; Bova, M.; Spanò, R.; Marone, G.; Triggiani, M. Home therapy with plasma-derived C1 inhibitor: A strategy to improve clinical outcomes and costs in hereditary angioedema. Int. Arch. Allergy Immunol. 2015, 166, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Buerke, M.; Schwertz, H.; Seitz, W.; Meyer, J.; Darius, H. Novel small molecule inhibitor of C1s exerts cardioprotective effects in ischemia-reperfusion injury in rabbits. J. Immunol. 2001, 167, 5375–5380. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Rose, E.; Hussain, S.; Tom, S.; Strober, W.; Sloan, S.R.; Parry, G.; Stagliano, N. TNT009, a classical complement pathway specific inhibitor, prevents complement dependent hemolysis induced by cold agglutinin disease patient autoantibodies. Blood 2013, 122, 42. [Google Scholar]
- Van Vlasselaer, P.; Parry, G.; Stagliano, N.; Panicker, S. Anti-Complement C1s Antibodies and Methods of Inhibiting Complement C1s Activity, 2017. US Patent 9,562,106, 7 February 2017. [Google Scholar]
- Szalai, A.J.; Digerness, S.B.; Agrawal, A.; Kearney, J.F.; Bucy, R.P.; Niwas, S.; Kilpatrick, J.M.; Babu, Y.S.; Volanakis, J.E. The Arthus reaction in rodents: Species-specific requirement of complement. J. Immunol. 2000, 164, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Subasinghe, N.L.; Ali, F.; Illig, C.R.; Rudolph, M.J.; Klein, S.; Khalil, E.; Soll, R.M.; Bone, R.F.; Spurlino, J.C.; DesJarlais, R.L.; et al. A novel series of potent and selective small molecule inhibitors of the complement component C1s. Bioorg. Med. Chem. Lett. 2004, 14, 3043–3047. [Google Scholar] [CrossRef] [PubMed]
- Subasinghe, N.L.; Travins, J.M.; Ali, F.; Huang, H.; Ballentine, S.K.; Marugán, J.J.; Khalil, E.; Hufnagel, H.R.; Bone, R.F.; DesJarlais, R.L.; et al. A novel series of arylsulfonylthiophene-2-carboxamidine inhibitors of the complement component C1s. Bioorg. Med. Chem. Lett. 2006, 16, 2200–2204. [Google Scholar] [CrossRef] [PubMed]
- Travins, J.M.; Ali, F.; Huang, H.; Ballentine, S.K.; Khalil, E.; Hufnagel, H.R.; Pan, W.; Gushue, J.; Leonard, K.; Bone, R.F.; et al. Biphenylsulfonyl-thiophene-carboxamidine inhibitors of the complement component C1s. Bioorg. Med. Chem. Lett. 2008, 18, 1603–1606. [Google Scholar] [CrossRef] [PubMed]
- Subasinghe, N.L.; Khalil, E.; Travins, J.M.; Ali, F.; Ballentine, S.K.; Hufnagel, H.R.; Pan, W.; Leonard, K.; Bone, R.F.; Soll, R.M.; et al. Design and synthesis of polyethylene glycol-modified biphenylsulfonyl-thiophene-carboxamidine inhibitors of the complement component C1s. Bioorg. Med. Chem. Lett. 2012, 22, 5303–5307. [Google Scholar] [CrossRef] [PubMed]
- Van Drie, J.H. Computer-aided drug design: The next 20 years. J. Comput. Aided Mol. Des. 2007, 21, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Vulpetti, A.; Randl, S.; Rüdisser, S.; Ostermann, N.; Erbel, P.; Mac Sweeney, A.; Zoller, T.; Salem, B.; Gerhartz, B.; Cumin, F.; et al. Structure-based library design and fragment screening for the identification of reversible complement Factor D protease inhibitors. J. Med. Chem. 2017, 60, 1946–1958. [Google Scholar] [CrossRef] [PubMed]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Dobson, C.M. Chemical space and biology. Nature 2004, 432, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.A.; Williams, J.A. Origin and evolution of high throughput screening. Br. J. Pharmacol. 2007, 152, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Drews, J. Drug Discovery: A Historical Perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Triggle, D.J. Drug discovery and delivery in the 21st century. Med. Princ. Pract. 2007, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Suzek, T.; Zhang, J.; Wang, J.; He, S.; Cheng, T.; Shoemaker, B.A.; Gindulyte, A.; Bryant, S.H. PubChem BioAssay: 2014 update. Nucleic Acids Res. 2014, 42, D1075–D1082. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; Overington, J.P. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; King, F.J.; He, Y.; Caldwell, J.S.; Zhou, Y. Learning from the data: Mining of large high-throughput screening databases. J. Chem. Inf. Model. 2006, 46, 2381–2395. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.F.; McCammon, J.A. Protein flexibility and computer-aided drug design. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Computer-aided drug-discovery techniques that account for receptor flexibility. Curr. Opin. Pharmacol. 2010, 10, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Douguet, D.; Munier-Lehmann, H.; Labesse, G.; Pochet, S. LEA3D: A computer-aided ligand design for structure-based drug design. J. Med. Chem. 2005, 48, 2457–2468. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-Based Virtual Screening for Drug Discovery: A Problem-Centric Review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-mMolecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Wu, X. Alzheimer’s disease drug development based on computer-aided drug Design. Eur. J. Med. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sinko, W.; Lindert, S.; McCammon, J.A. Accounting for receptor flexibility and enhanced sampling methods in computer-aided drug design. Chem. Biol. Drug Des. 2013, 81, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.H.; da Silva, V.B.; Resende, J.; Rodrigues, P.F.; Bononi, F.C.; Benevenuto, C.G.; Taft, C.A. Computer-aided drug design and ADMET predictions for identification and evaluation of novel potential farnesyltransferase inhibitors in cancer therapy. J. Mol. Graph. Model. 2010, 28, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Alvarsson, J.; Eklund, M.; Andersson, C.; Carlsson, L.; Spjuth, O.; Wikberg, J.E.S. Benchmarking study of parameter variation when using signature fingerprints together with support vector machines. J. Chem. Inf. Model. 2014, 54, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Alvarsson, J.; Eklund, M.; Engkvist, O.; Spjuth, O.; Carlsson, L.; Wikberg, J.E.S.; Noeske, T. Ligand-based target prediction with signature fingerprints. J. Chem. Inf. Model. 2014, 54, 2647–2653. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Mussa, H.Y.; Glen, R.C.; Reiling, S. Similarity searching of chemical databases using atom environment Descriptors (MOLPRINT 2D): Evaluation of Performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Li, M.; Wang, J.; Pan, Y. HybridDock: A hybrid protein-ligand docking protocol integrating protein- and ligand-based approaches. J. Chem. Inf. Model. 2015, 56, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.F.; Visco, D.P., Jr. Developing an in silico pipeline for faster drug candidate discovery: Virtual high throughput screening with the signature molecular descriptor using support vector machine models. Chem. Eng. Sci. 2016, 159, 31–42. [Google Scholar] [CrossRef]
- Chen, J.J.F.; Visco, D.P., Jr. Identifying novel factor XIIa inhibitors with PCA-GA-SVM developed vHTS models. European J. Med. Chem. 2017, 140, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Faulon, J.L.; Churchwell, C.J.; Visco, D.P.J. The signature molecular descriptor. 2. Enumerating molecules from their extended valence sequences. J. Chem. Inf. Comput. Sci. 2003, 43, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Visco, D.P.J.; Pophale, R.S.; Rintoul, M.D.; Faulon, J.L. Developing a methodology for an inverse quantitative structure–activity relationship using the signature molecular descriptor. J. Mol. Graph. Model. 2002, 20, 429–438. [Google Scholar] [CrossRef]
- Faulon, J.L. Stochastic generator of chemical structure. 1. Application to the structure elucidation of large molecules. J. Chem. Inf. Comput. Sci. 1994, 34, 1204–1218. [Google Scholar] [CrossRef]
- Li, H.; Visco, D.P.; Leipzig, N.D. Confirmation of predicted activity for factor XIa inhibitors from a virtual screening approach. AIChE J. 2014, 60, 2741–2746. [Google Scholar] [CrossRef]
- Faulon, J.L.; Visco, D.P.J.; Pophale, R.S. The signature molecular descriptor. 1. Using extended valence sequences in QSAR and QSPR studies. J. Chem. Inf. Comput. Sci. 2003, 43, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Roe, D.; Faulon, J.L. Predicting protein–protein interactions using signature products. Bioinformatics 2005, 21, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Churchwell, C.J.; Rintoul, M.D.; Martin, S.; Visco, D.P.J.; Kotu, A.; Larson, R.S.; Sillerud, L.O.; Brown, D.C.; Faulon, J.L. The signature molecular descriptor. 3. Inverse-quantitative structure–activity relationship of ICAM-1 inhibitory peptides. J. Mol. Graph. Model. 2004, 22, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Weis, D.C.; Faulon, J.L.; LeBorne, R.C.; Visco, D.P. The signature molecular descriptor. 5. The design of hydrofluoroether foam blowing agents using inverse-QSAR. Ind. Eng. Chem. Res. 2005, 44, 8883–8891. [Google Scholar] [CrossRef]
- Dev, V.A.; Chemmangattuvalappil, N.G.; Eden, M.R. Structure generation of candidate reactants using signature descriptors. In Computer Aided Chemical Engineering; Elsevier: New York, NY, USA, 2014; Volume 33, pp. 151–156. [Google Scholar]
- Chemmangattuvalappil, N.G.; Eden, M.R. A novel methodology for property-based molecular design using multiple topological indices. Ind. Eng. Chem. Res. 2013, 52, 7090–7103. [Google Scholar] [CrossRef]
- Weis, D.C.; Visco, D.P.J. Computer-aided molecular design using the Signature molecular descriptor: Application to solvent selection. Comput. Chem. Eng. 2010, 34, 1018–1029. [Google Scholar] [CrossRef]
- Chemmangattuvalappil, N.G.; Solvason, C.C.; Bommareddy, S.; Eden, M.R. Reverse problem formulation approach to molecular design using property operators based on signature descriptors. Comput. Chem. Eng. 2010, 34, 2062–2071. [Google Scholar] [CrossRef]
- Diamond, S.L. AID 787-Complement Factor C1s IC150 from Mixture Screen. 2008. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/787 (accessed on 23 June 2017).
- Kayello, H.M.; Tadisina, N.K.R.; Shlonimskaya, N.; Biernacki, J.J.; Visco, D.P. An Application of computer-aided molecular design (CAMD) dsing the dignature molecular descriptor—Part 1. Identification of surface tension reducing agents and the search for shrinkage reducing admixtures. J. Am. Ceram. Soc. 2014, 97, 365–377. [Google Scholar] [CrossRef]
- Weis, D.C.; Visco, D.P.J.; Faulon, J.L. Data mining PubChem using a support vector machine with the Signature molecular descriptor: Classification of factor XIa inhibitors. J. Mol. Graph. Model. 2008, 27, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.F.; Schmucker, L.N.; Visco, D.P., Jr. Identifying new clotting factor XIa inhibitors in virtual high-throughput screening using PCA-GA-SVM models and signature. Biotechnol. Prog. 2018. in review. [Google Scholar]
- Whitley, D. A genetic algorithm tutorial. Stat. Comput. 1994, 4, 65–85. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Chen, X.; Reynolds, C.H. Performance of similarity measures in 2D fragment-based similarity searching: Comparison of structural descriptors and similarity coefficients. J. Chem. Inf. Comput. Sci. 2002, 42, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Scrucca, L. GA: A Package for genetic algorithms in R. J. Stat. Softw. 2013, 53, 1–37. [Google Scholar] [CrossRef]
- Karatzoglou, A.; Smola, A.; Hornik, K. kernlab—An S4 package for kernel methods in R. J. Stat. Softw. 2004, 11, 1–22. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SVM-C | SVM-R | |
| Training set NO PAINS | 136 compounds (16 active, 120 inactive) | 16 compounds (all active) |
| Training set atomic Signatures | 11 h = 0; 136 h = 1; 925 h = 2; 1072 total | 8 h = 0; 57 h = 1; 165 h = 2; 230 total |
| Principal Component Analysis results | 159 of 1072 atomic Signatures | 230 of 230 atomic Signatures |
| Models Created | 115 | 1 |
| Training Error | 0 | 0 |
| Cross-Validation Error | 0.007 | 0.090 |
| Structure | CID | Predicted IC[M] | Experimental IC[M] |
|---|---|---|---|
 | 17178137 | 21.9 | |
 | 4951143 | 6.77 | |
 | 2986934 | 1.18 | |
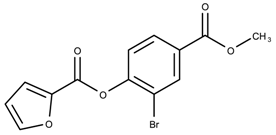
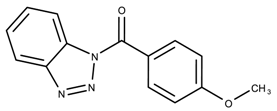 | 710644 | 4.36 | |
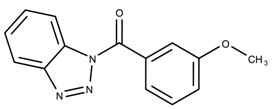 | 5146207 | 10.9 | >50 * |
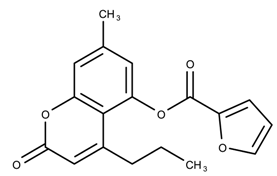 | 807111 | 8.88 | >50 * |
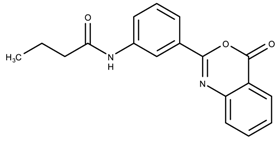 | 1107361 | 11.2 | >50 |
| SVM-C | SVM-R | |
| Training set NO PAINS | 143 compounds (20 active, 123 inactive) | 20 compounds (all active) |
| Training set Signatures | 11 h = 0; 136 h = 1; 925 h = 2; 1072 total | 8 h = 0; 57 h = 1; 165 h = 2; 230 total |
| Principal Component Analysis results | 164 of 1072 atomic Signatures | 186 of 230 atomic Signatures |
| Models Created | 1224 | 1 |
| Training Error | 0.021 | 0.108 |
| Cross-Validation Error | 0.020 | 0.162 |
| Structure | CID | Predicted IC[M] | Experimental IC[M] |
|---|---|---|---|
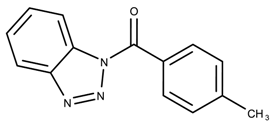 | 827004 | 0.30 | |
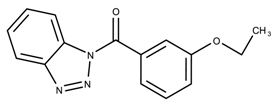 | 4957387 | 4.27 | |
 | 898930 | 26.01 | |
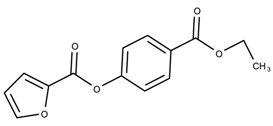 | 17178134 | 7.21 | |
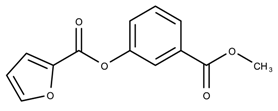 | 17178138 | 33.44 | |
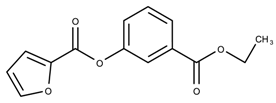 | 17131127 | 23.71 | >50 * |
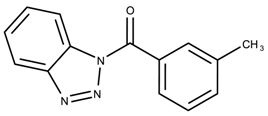 | 834536 | 1.66 | >50 * |
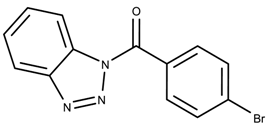 | 693001 | 0.43 | >50 * |
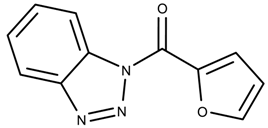 | 792914 | 2.05 | >50 * |
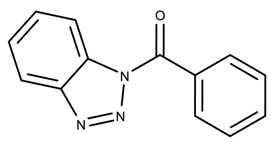 | 570059 | 4.20 | >50 * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.J.; Schmucker, L.N.; Visco, D.P., Jr. Pharmaceutical Machine Learning: Virtual High-Throughput Screens Identifying Promising and Economical Small Molecule Inhibitors of Complement Factor C1s. Biomolecules 2018, 8, 24. https://doi.org/10.3390/biom8020024
Chen JJ, Schmucker LN, Visco DP Jr. Pharmaceutical Machine Learning: Virtual High-Throughput Screens Identifying Promising and Economical Small Molecule Inhibitors of Complement Factor C1s. Biomolecules. 2018; 8(2):24. https://doi.org/10.3390/biom8020024
Chicago/Turabian StyleChen, Jonathan J., Lyndsey N. Schmucker, and Donald P. Visco, Jr. 2018. "Pharmaceutical Machine Learning: Virtual High-Throughput Screens Identifying Promising and Economical Small Molecule Inhibitors of Complement Factor C1s" Biomolecules 8, no. 2: 24. https://doi.org/10.3390/biom8020024