
Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies

 ,
, 

 , ,
, ,  ,
, 
Abstract
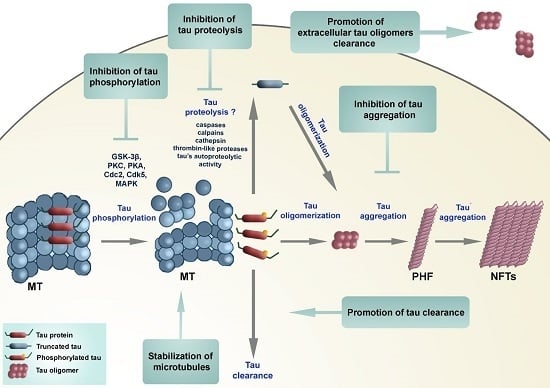
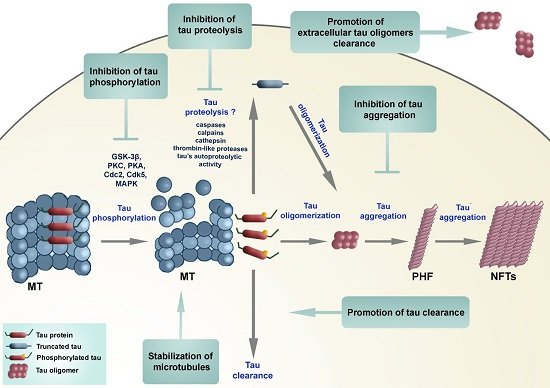
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
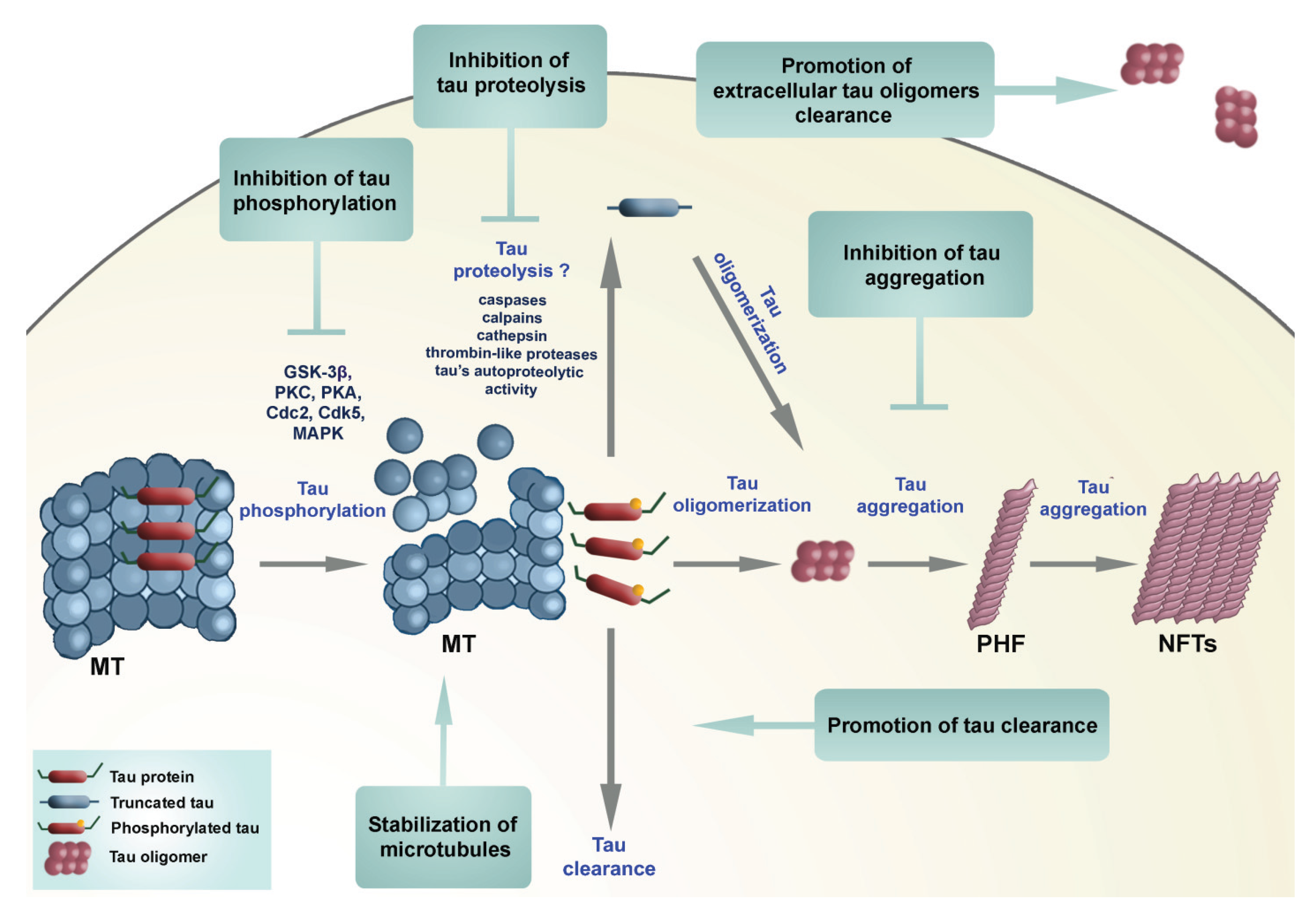{kind=link}
{kind=link}
1. Selective Overview of Major Discoveries on Tau Protein and Tauopathies
1.1. Neurofibrillary Tangles and Paired Helical Filaments
1.2. Tau Protein Isolation and Localization
1.3. Tau in Neurofibrillary Tangles
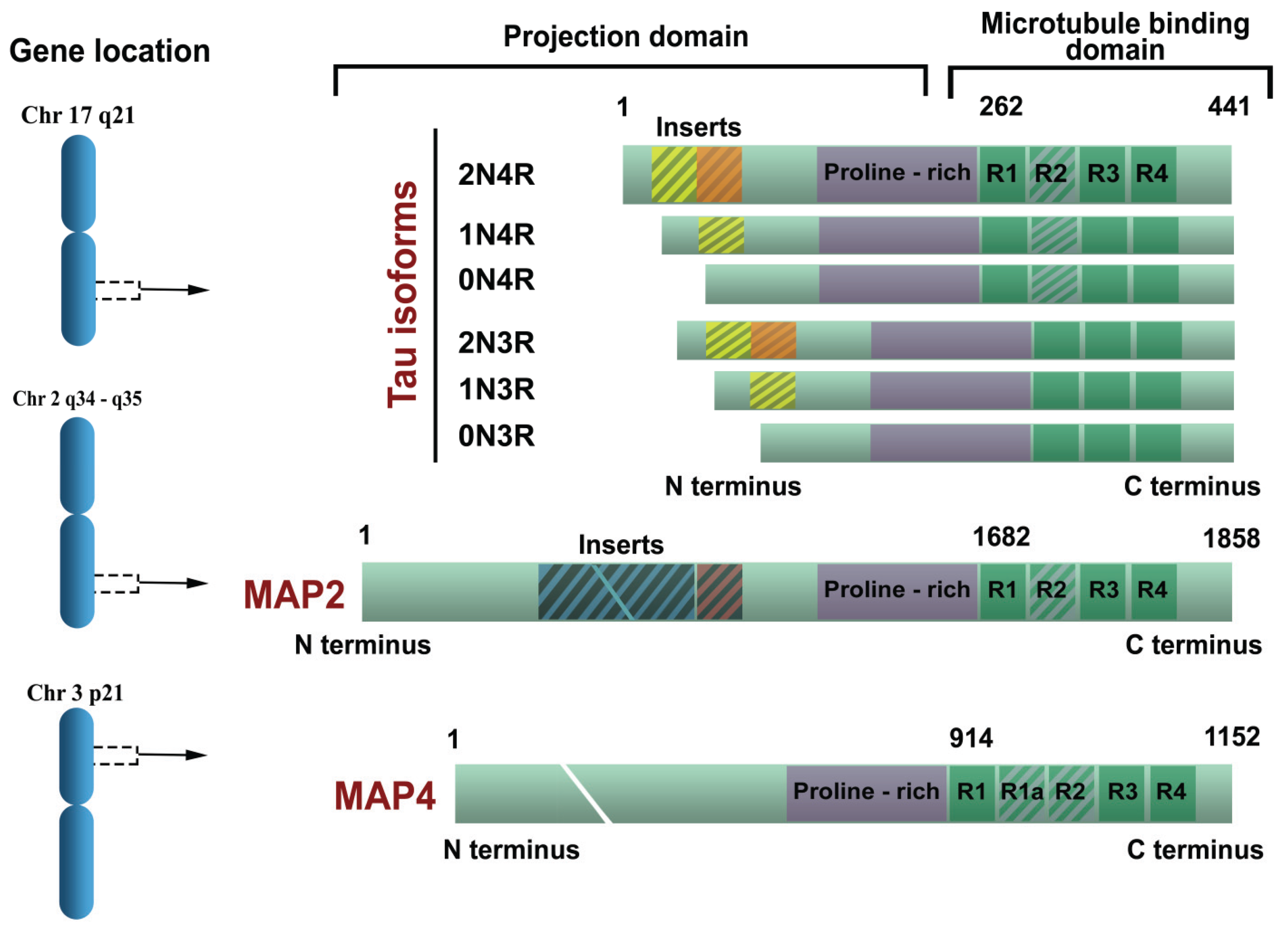
1.4. Tau Isoforms in the Central Nervous System

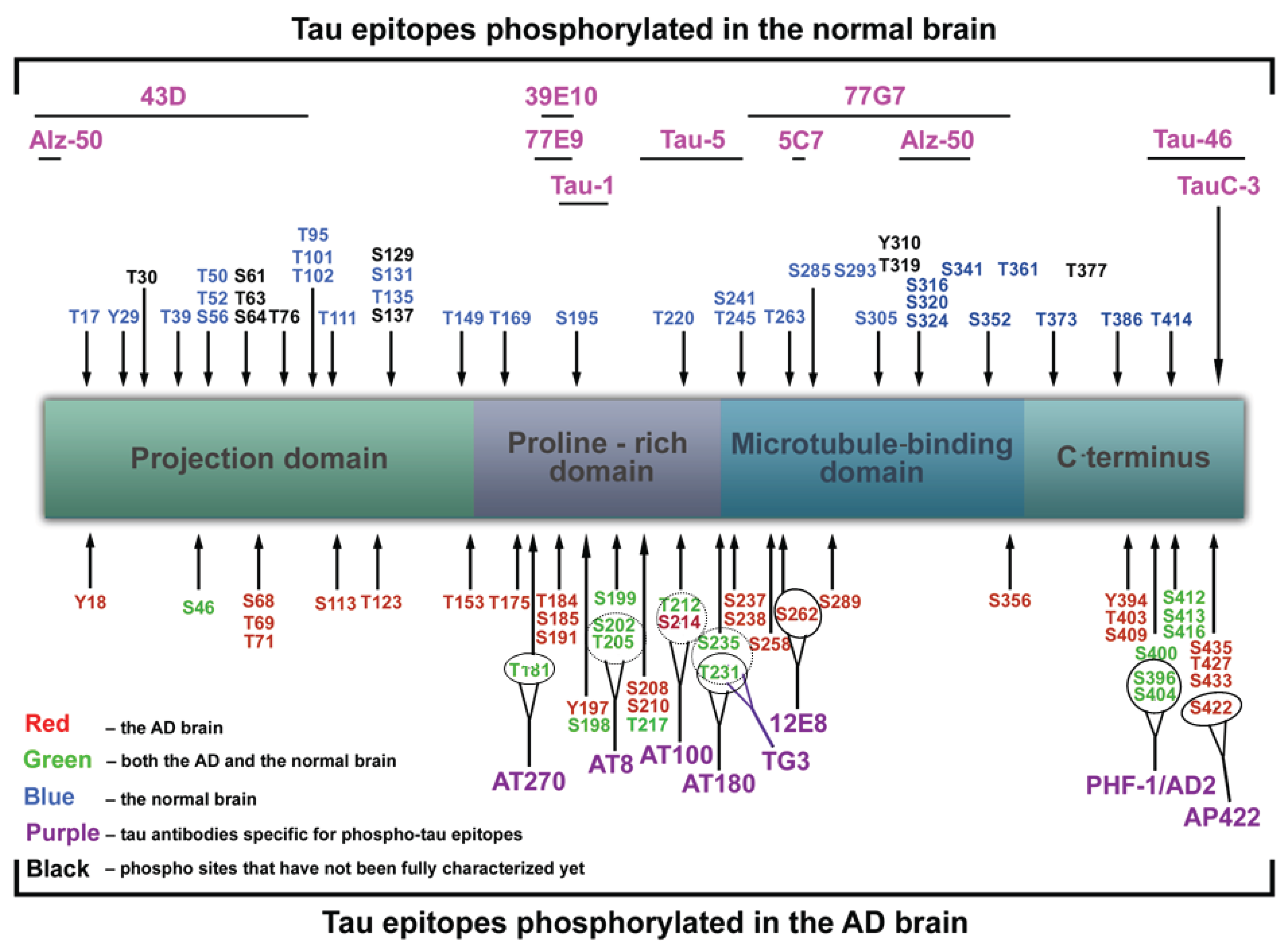
1.5. Functions of Tau Protein
1.6. Amyloid Cascade Theory
1.7. Staging of Tau Pathology
1.8. Mutations in MAPT Gene and Tauopathies
2. Tau Protein Pathological Changes in Primary and Secondary Tauopathies
2.1. Mechanisms
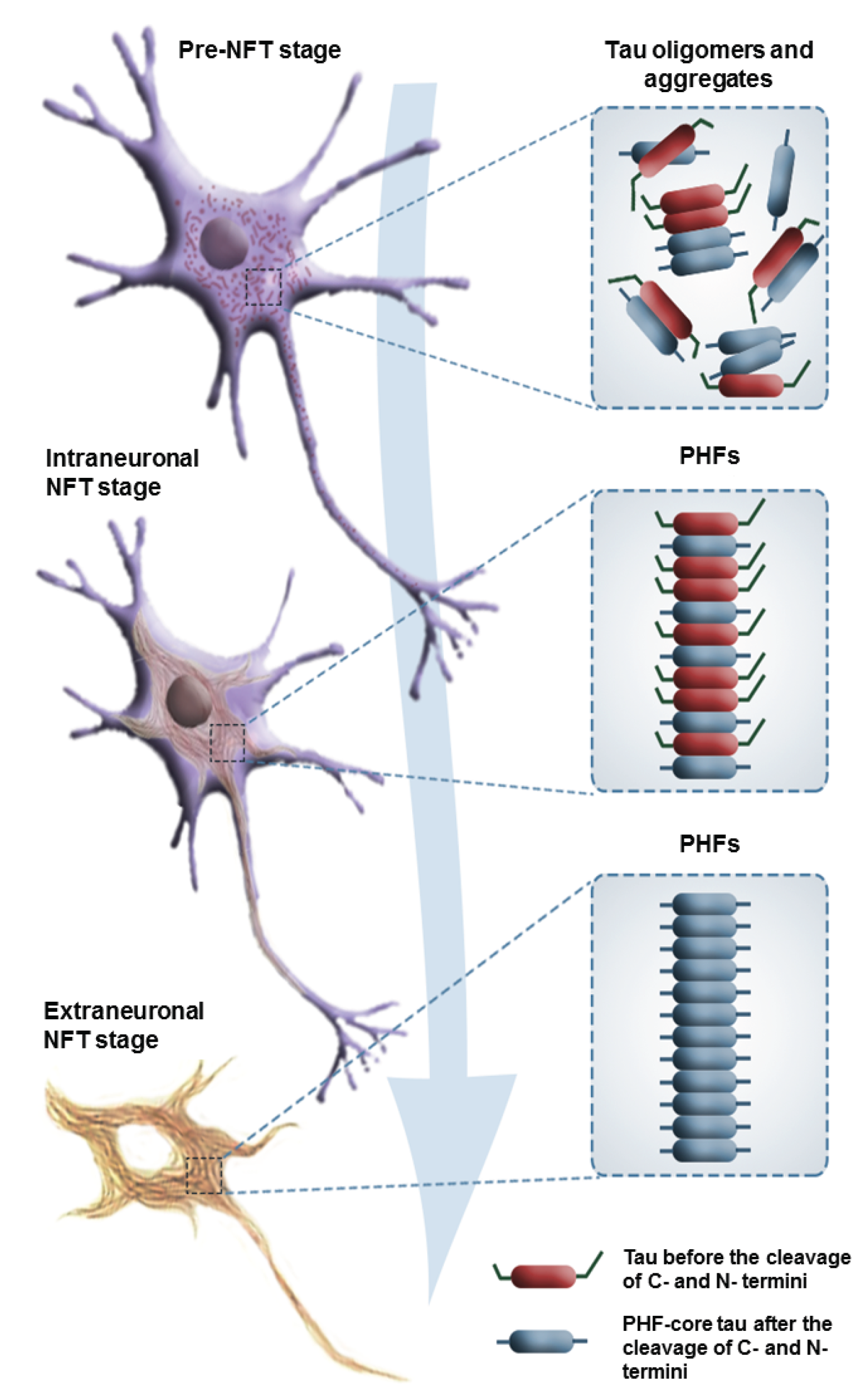
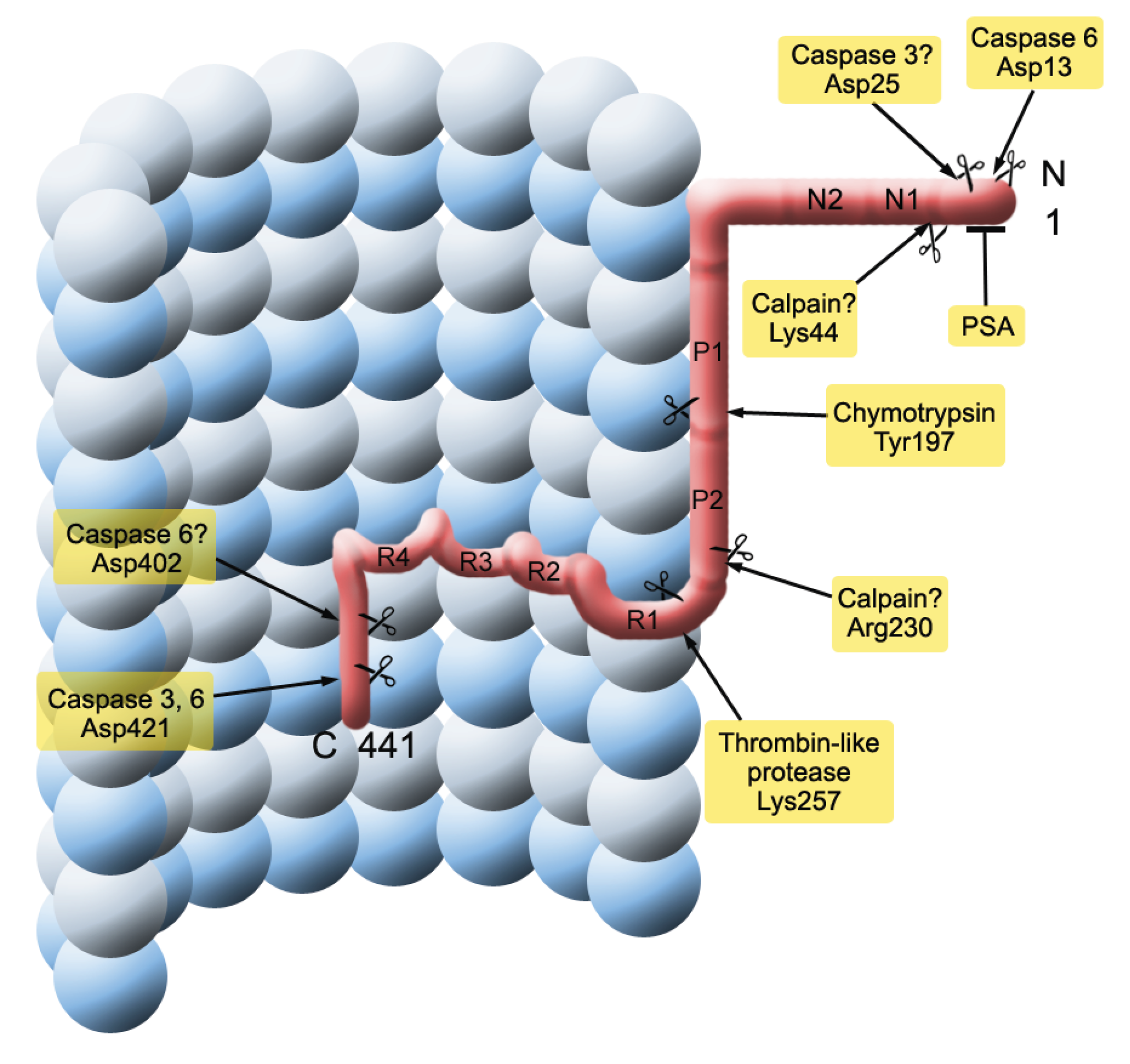
2.2. Seeding and Spreading of Tau Proteins
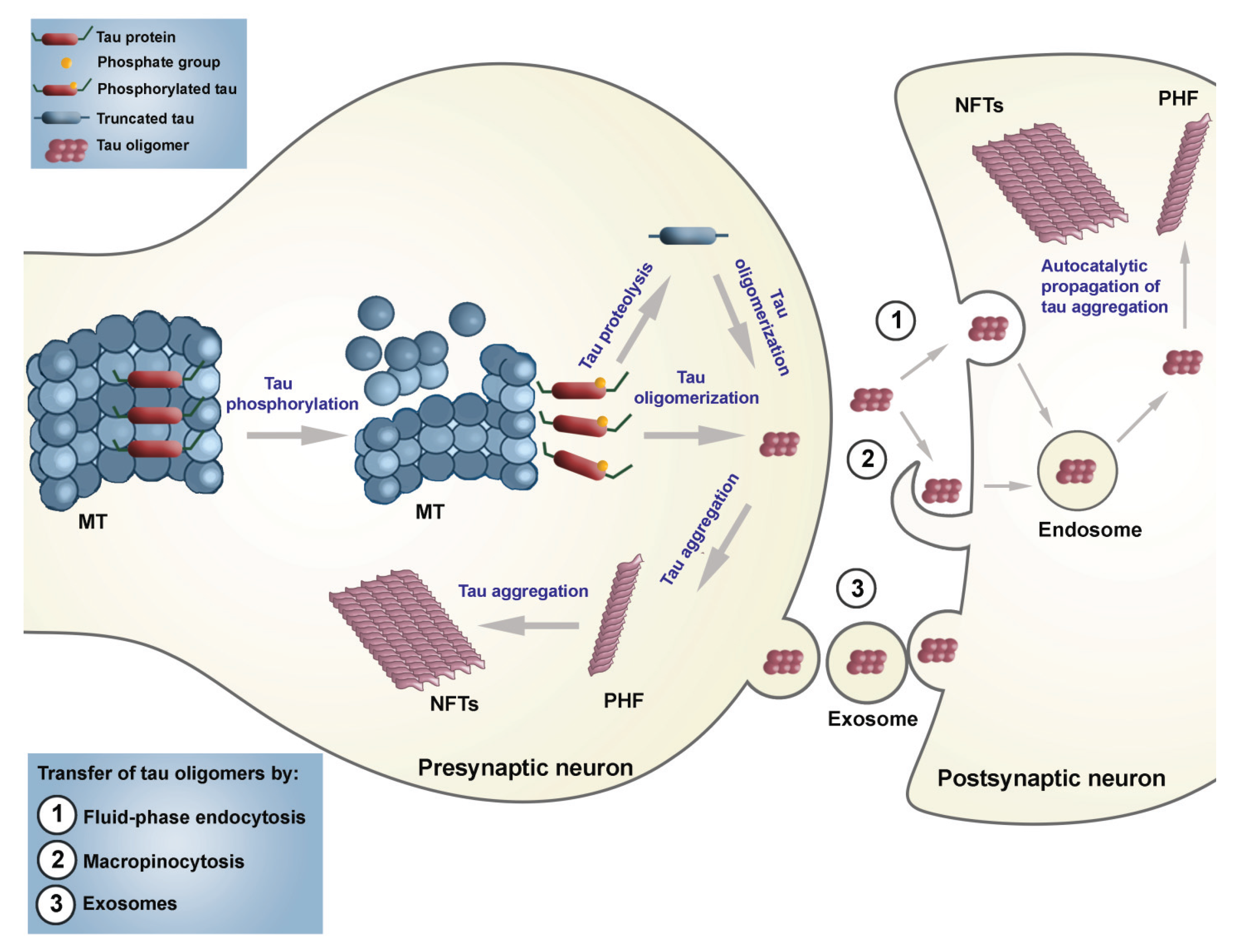
2.3. Therapeutic Approaches Targeting Tau Protein Processing in Tauopathies
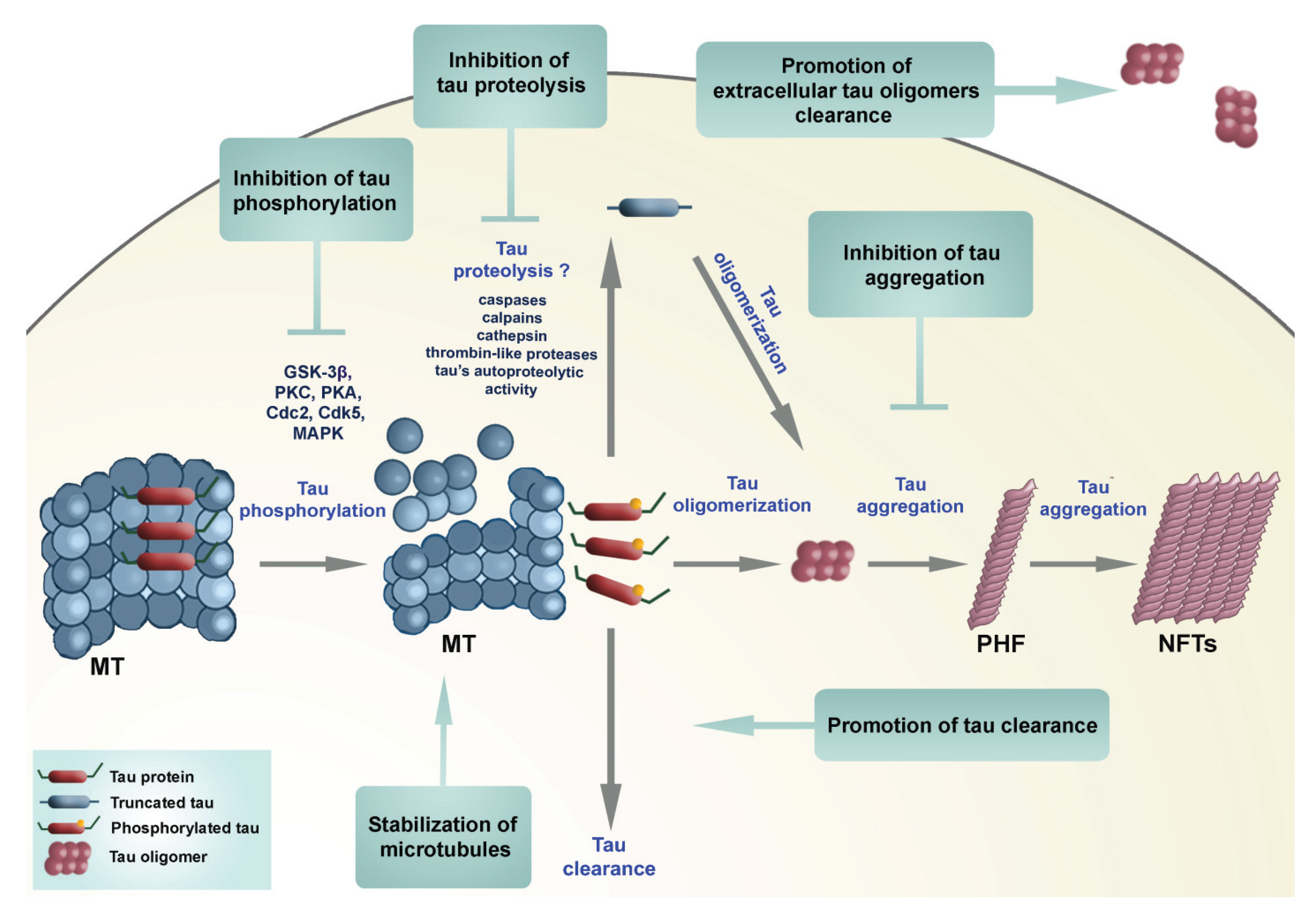
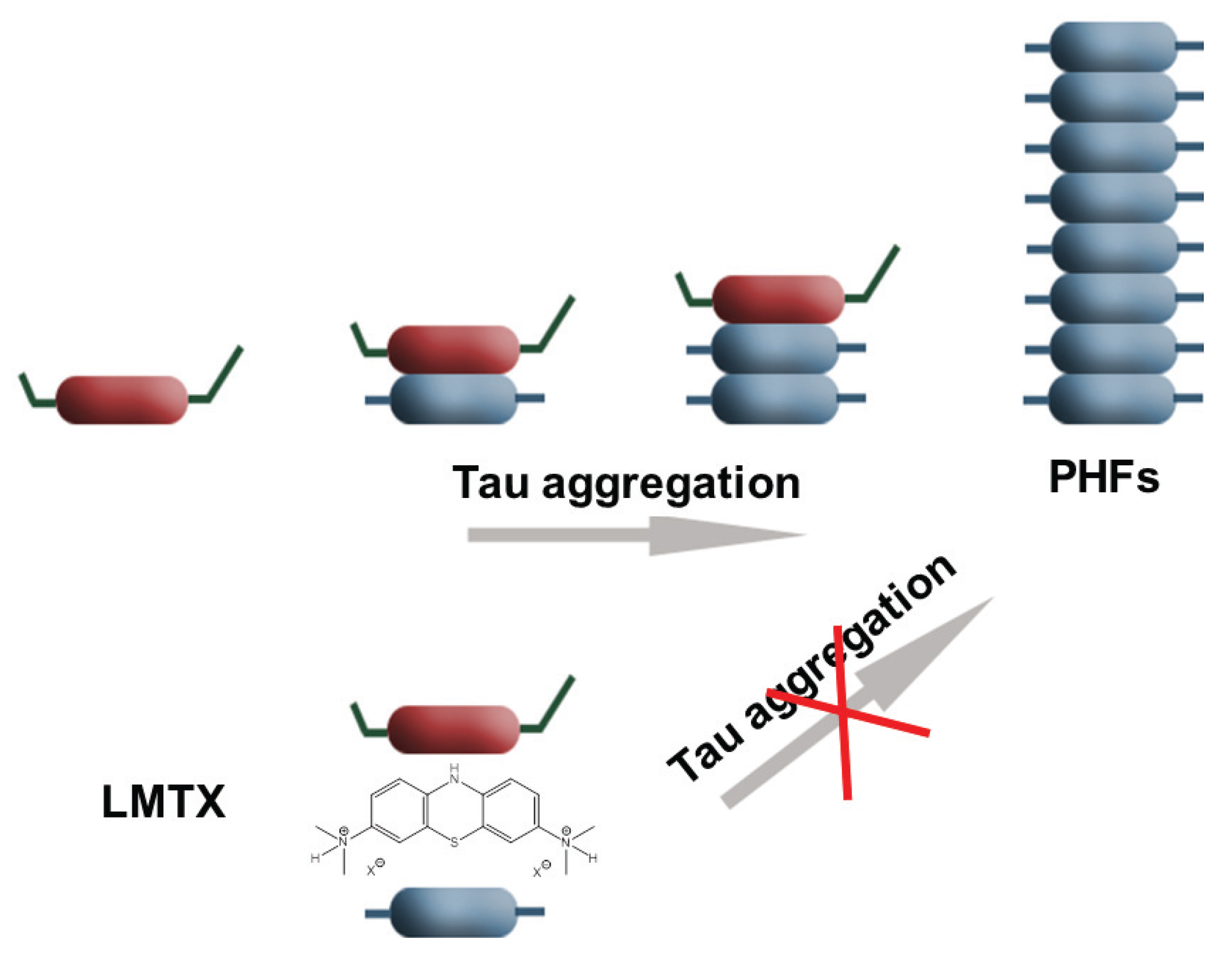
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 4R tau | tau isoforms with four microtubule-binding repeats |
| A | adenine |
| Aβ | amyloid β protein |
| AD | Alzheimer’s disease |
| AD2 | antibody specific for phospho-tau epitope Ser396 |
| ADAM10 | a disintegrin and metalloprotease domain 10 |
| AgD | argyrophilic grain disease |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| APH-1 | anterior pharynx-defective 1 |
| APOE | apolipoprotein E |
| APP | amyloid precursor protein |
| AT8 | antibody specific for phospho-tau epitopes Ser202 and Thr205 |
| BACE | β-site APP cleaving enzyme |
| BF | Bayes factor |
| CBD | corticobasal degeneration |
| CSF | cerebrospinal fluid |
| DDPAC | disinhibition-dementia-parkinsonism-amyotrophy complex |
| DM1 | myotonic dystrophy type I |
| EOAD | early-onset AD |
| fAD | familial AD |
| FTD | frontotemporal dementia |
| FTDP-17 | frontotemporal dementia and parkinsonism linked to chromosome 17 |
| FTLD | frontotemporal lobar degneration |
| G | guanine |
| Grb2 | growth factor receptor-bound protein 2 |
| HCHWA-D | hereditary cerebral hemorrhage with amyloidosis-Dutch type |
| HDAC6 | histone deacetylase 6 |
| LMTX | leucomethylthioninium |
| LOAD | late-onset AD |
| LTP | long-term potentiation |
| MAP2 | microtubule-associated protein 2 |
| MAPT | microtubule-associated protein tau |
| MB | Methylene blue |
| MCI | mild cognitive impairment |
| MN423 | antibody for PHF core |
| MSTD | multiple system tauopathy with presenile dementia |
| MT | microtubule |
| NFT | neurofibrillary tangles |
| NMDAR | N-methyl-d-aspartate receptors |
| NT | neuropil threads |
| O-GlcNAc | O-linked N-acetylglucosamine |
| PD | Parkinson’s disease |
| PEN-2 | presenilin enhancer 2 |
| PHF | paired helical filaments |
| PSA | puromycin-sensitive aminopeptidase |
| PSEN | presenilin |
| PSEN1 | presenilin 1 |
| PSEN2 | presenilin 2 |
| PSP | progressive supranuclear palsy |
| sAD | sporadic AD |
| SDS | sodium dodecyl sulfate |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SF | straight filaments |
| SP | senile plaques |
| TACE | tumor necrosis factor alpha converting enzyme |
| Tau | tubulin-associated unit |
References
- Bielschowsky, M. Die Silberimprägnation der Achsenzylinder. Neurol. Zentralb. (Leipzig) 1902, 13, 579–584. (In German) [Google Scholar]
- Alzheimer, A. Uber eine eigenartige Erkrankung der Hirnrinde. Allg Zeits Psychiatry Psych. Med. 1907, 64, 146–148. (In German) [Google Scholar]
- Jucker, M.; Beyreuther, K.; Haass, C.; Nitsch, R.; Christen, Y. Alzheimer: 100 Years and Beyond; Springer: Berlin, Germany, 2006. [Google Scholar]
- Kidd, M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D. The fine structure of neurofibrillary tangles in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1963, 22, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A.; Wischik, C.M. Image reconstruction of the Alzheimer paired helical filament. EMBO J. 1985, 4, 3661–3665. [Google Scholar] [PubMed]
- Wischik, C.M.; Crowther, R.A.; Stewart, M.; Roth, M. Subunit structure of paired helical filaments in Alzheimer’s disease. J. Cell Biol. 1985, 100, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc. Natl. Acad. Sci. USA 1991, 88, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Pierre, M.; Nunez, J. Multisite phosphorylation of tau proteins from rat brain. Biochem. Biophys. Res. Commun. 1983, 115, 212–219. [Google Scholar] [CrossRef]
- Jameson, L.; Frey, T.; Zeeberg, B.; Dalldorf, F.; Caplow, M. Inhibition of microtubule assembly by phosphorylation of microtubule-associated proteins. Biochemistry 1980, 19, 2472–2479. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [PubMed]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Papasozomenos, S.C.; Binder, L.I. Phosphorylation determines two distinct species of Tau in the central nervous system. Cell Motil. Cytoskeleton 1987, 8, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Migheli, A.; Butler, M.; Brown, K.; Shelanski, M.L. Light and electron microscope localization of the microtubule-associated tau protein in rat brain. J. Neurosci. 1988, 8, 1846–1851. [Google Scholar] [PubMed]
- Couchie, D.; Charrière-Bertrand, C.; Nunez, J. Expression of the mRNA for tau proteins during brain development and in cultured neurons and astroglial cells. J. Neurochem. 1988, 50, 1894–1899. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.; Passareiro, H.; Nunez, J.; Flament-Durand, J. Mise en évidence immunologique de la protéine tau au niveau des lésions de dégénérescence neurofibrillaire de la maladie d’Alzheimer. Arch. Biol. 1985, 95, 229–235. (In French) [Google Scholar]
- Anderton, B.H.; Breinburg, D.; Downes, M.J.; Green, P.J.; Tomlinson, B.E.; Ulrich, J.; Wood, J.N.; Kahn, J. Monoclonal antibodies show that neurofibrillary tangles and neurofilaments share antigenic determinants. Nature 1982, 298, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Nukina, N.; Miura, R.; Ogawara, M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem. 1986, 99, 1807–1810. [Google Scholar] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Multhaup, G.; Simms, G.; Pottgiesser, J.; Martins, R.N.; Beyreuther, K. Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985, 4, 2757–2763. [Google Scholar] [PubMed]
- Mori, H.; Kondo, J.; Ihara, Y. Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science 1987, 235, 1641–1644. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Friedman, R.; Shaw, G.; Chau, V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc. Natl. Acad. Sci. USA 1987, 84, 3033–3036. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.H.; Gaskin, F.; Fu, S.M. Neurofibrillary tangles in senile dementia of the Alzheimer type share an antigenic determinant with intermediate filaments of the vimentin class. Am. J. Pathol. 1983, 113, 373–381. [Google Scholar] [PubMed]
- Yen, S.; Dickson, D.; Crowe, A. Alzheimer’s neurofibrillary tangles contain unique epitopes and epitopes in common with the heat-stable microtubule associated proteins tau and MAP2. Am. J. Pathol. 1987, 126, 81–91. [Google Scholar] [PubMed]
- Wischik, C.M.; Novak, M.; Thøgersen, H.C.; Edwards, P.C.; Runswick, M.J.; Jakes, R.; Walker, J.E.; Milstein, C.; Roth, M.; Klug, A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4506–4510. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Novak, M.; Edwards, P.C.; Klug, A.; Tichelaar, W.; Crowther, R.A. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Wischik, C.M.; Edwards, P.; Pannell, R.; Milstein, C. Characterisation of the first monoclonal antibody against the pronase resistant core of the Alzheimer PHF. Prog. Clin. Biol. Res. 1989, 317, 755–761. [Google Scholar] [PubMed]
- Novak, M.; Jakes, R.; Edwards, P.C.; Milstein, C.; Wischik, C.M. Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc. Natl. Acad. Sci. USA 1991, 88, 5837–5841. [Google Scholar] [CrossRef] [PubMed]
- Resch, J.F.; Lehr, G.S.; Wischik, C.M. Design and synthesis of a potential affinity/cleaving reagent for beta-pleated sheet protein structures. Bioorg. Med. Chem. Lett. 1991, 1, 519–522. [Google Scholar] [CrossRef]
- Lee, G.; Cowan, N.; Kirschner, M. The primary structure and heterogeneity of tau protein from mouse brain. Science 1988, 239, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Wischik, C.M.; Crowther, R.A.; Walker, J.E.; Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. USA 1988, 85, 4051–4055. [Google Scholar] [CrossRef] [PubMed]
- Delacourte, A.; Defossez, A. Alzheimer’s disease: Tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J. Neurol. Sci. 1986, 76, 173–186. [Google Scholar] [CrossRef]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Nukina, N.; Ihara, Y. One of the antigenic determinants of paired helical filaments is related to tau protein. J. Biochem. 1986, 99, 1541–1544. [Google Scholar] [PubMed]
- Wood, J.G.; Mirra, S.S.; Pollock, N.J.; Binder, L.I. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. USA 1986, 83, 4040–4043. [Google Scholar] [CrossRef] [PubMed]
- Montejo de Garcini, E.; Serrano, L.; Avila, J. Self assembly of microtubule associated protein tau into filaments resembling those found in Alzheimer disease. Biochem. Biophys. Res. Commun. 1986, 141, 790–796. [Google Scholar] [CrossRef]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Mol. Brain Res. 1986, 1, 271–280. [Google Scholar] [CrossRef]
- Donlon, T.; Harris, P.; Neve, R. Localization of microtubule-associated protein tau (MTBT1) to chromosome 17q21 (Abstract). Cytogenet. Cell Genet. 1987, 46, 607. [Google Scholar]
- Goedert, M.; Spillantini, M.; Potier, M.; Ulrich, J.; Crowther, R. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [PubMed]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [PubMed]
- Goedert, M.; Spillantini, M.G.; Crowther, R.A. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 1983–1987. [Google Scholar] [CrossRef] [PubMed]
- Jovanov-Milošević, N.; Petrović, D.; Sedmak, G.; Vukšić, M.; Hof, P.R.; Šimić, G. Human fetal tau protein isoform: Possibilities for Alzheimer’s disease treatment. Int. J. Biochem. Cell Biol. 2012, 44, 1290–1294. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.G.; Davies, P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA 1990, 87, 5827–5831. [Google Scholar] [CrossRef] [PubMed]
- Mulot, S.F.; Hughes, K.; Woodgett, J.R.; Anderton, B.H.; Hanger, D.P. PHF-tau from Alzheimer’s brain comprises four species on SDS-PAGE which can be mimicked by in vitro phosphorylation of human brain tau by glycogen synthase kinase-3 beta. FEBS Lett. 1994, 349, 359–364. [Google Scholar] [CrossRef]
- Wolozin, B.L.; Pruchnicki, A.; Dickson, D.W.; Davies, P. A neuronal antigen in the brains of Alzheimer patients. Science 1986, 232, 648–650. [Google Scholar] [CrossRef] [PubMed]
- Flament, S.; Delacourte, A. Abnormal tau species are produced during Alzheimer’s disease neurodegenerating process. FEBS Lett. 1989, 247, 213–216. [Google Scholar] [CrossRef]
- Lee, V.M.; Balin, B.J.; Otvos, L.; Trojanowski, J.Q. A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.Y.; Gertz, H.N.; Wischik, D.J.; Xuereb, J.H.; Mukaetova-Ladinska, E.B.; Harrington, C.R.; Edwards, P.C.; Mena, R.; Paykel, E.S.; Brayne, C.; et al. Examination of phosphorylated tau protein as a PHF-precursor at early stage Alzheimer’s disease. Neurobiol. Aging 1995, 16, 433–445. [Google Scholar] [CrossRef]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Gertz, H.N.; Xuereb, J.H.; Paykel, E.S.; Brayne, C.; Huppert, F.A.; Mukaetova-Ladinska, E.B.; Mena, R. Quantitative analysis of tau protein in paired helical filament preparations: Implications for the role of tau protein phosphorylation in PHF assembly in Alzheimer’s disease. Neurobiol. Aging 1995, 16, 409–417, discussion 418–431. [Google Scholar] [CrossRef]
- Jakes, R.; Novak, M.; Davison, M.; Wischik, C.M. Identification of 3- and 4-repeat tau isoforms within the PHF in Alzheimer’s disease. EMBO J. 1991, 10, 2725–2729. [Google Scholar] [PubMed]
- Wischik, C.; Lai, R.; Harrington, C. Modelling prion-like processing of tau protein in Alzheimer’s disease for pharmaceutical development. In Brain Microtubule Associated Proteins: Modifications in Disease; Avila, J., Brandt, R., Kosik, K., Eds.; Harwood Academic Publishers: Amsterdam, The Nederland, 1997; pp. 185–241. [Google Scholar]
- Gu, Y.; Oyama, F.; Ihara, Y. Tau is widely expressed in rat tissues. J. Neurochem. 1996, 67, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Yu, W.; Andreadis, A.; Luo, M.; Baas, P.W. Tau protects microtubules in the axon from severing by katanin. J. Neurosci. 2006, 26, 3120–3129. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Teng, J.; Harada, A.; Hirokawa, N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol. 2000, 150, 989–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Yoshida, H.; Watanabe, A.; Titani, K.; Ihara, Y. Hyperphosphorylation of tau in PHF. Neurobiol. Aging 1995, 16, 365–371, discussion 371–380. [Google Scholar] [CrossRef]
- Bretteville, A.; Ando, K.; Ghestem, A.; Loyens, A.; Bégard, S.; Beauvillain, J.-C.; Sergeant, N.; Hamdane, M.; Buée, L. Two-dimensional electrophoresis of tau mutants reveals specific phosphorylation pattern likely linked to early tau conformational changes. PLoS ONE 2009, 4, e4843. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Diana, A.; Hof, P.R. Phosphorylation pattern of tau associated with distinct changes of the growth cone cytoskeleton. Prog. Mol. Subcell. Biol. 2003, 32, 33–48. [Google Scholar] [PubMed]
- Kingwell, K. Neurodegenerative disease: Targeting tau acetylation attenuates neurodegeneration. Nat. Rev. Drug Discov. 2015, 14, 748–749. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, G.V.W. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Y.; Weisgraber, K.H.; Goedert, M.; Saunders, A.M.; Roses, A.D.; Strittmatter, W.J. ApoE3 binding to tau tandem repeat I is abolished by tau serine262 phosphorylation. Neurosci. Lett. 1995, 192, 209–212. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Garwood, C.J.; Wray, S.; Price, C.; Kellie, S.; Perera, T.; Zvelebil, M.; Yang, A.; Sheppard, P.W.; Varndell, I.M.; et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cγ1, Grb2, and Src family kinases. J. Biol. Chem. 2008, 283, 18177–18186. [Google Scholar] [CrossRef] [PubMed]
- Surridge, C.D.; Burns, R.G. The difference in the binding of phosphatidylinositol distinguishes MAP2 from MAP2C and Tau. Biochemistry 1994, 33, 8051–8057. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, L.A.; Cunningham, C.C.; Chen, J.; Prestwich, G.D.; Kosik, K.S.; Janmey, P.A. The structure of divalent cation-induced aggregates of PIP2 and their alteration by gelsolin and tau. Biophys. J. 1997, 73, 1440–1447. [Google Scholar] [CrossRef]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Himmelstein, D.S.; Ward, S.M.; Lancia, J.K.; Patterson, K.R.; Binder, L.I. Tau as a therapeutic target in neurodegenerative disease. Pharmacol. Ther. 2012, 136, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Kramer, E.-M.; Cardine, A.-M.; Schraven, B.; Brandt, R.; Trotter, J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J. Neurosci. 2002, 22, 698–707. [Google Scholar] [PubMed]
- Jadhav, S.; Cubinkova, V.; Zimova, I.; Brezovakova, V.; Madari, A.; Cigankova, V.; Zilka, N. Tau-mediated synaptic damage in Alzheimer’s disease. Transl. Neurosci. 2015, 6, 214–226. [Google Scholar] [CrossRef]
- Mondragón-Rodríguez, S.; Trillaud-Doppia, E.; Dudilot, A.; Bourgeois, C.; Lauzon, M.; Leclerc, N.; Boehm, J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-d-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 2012, 287, 32040–32053. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; van der Jeugd, A.; Blum, D.; Galas, M.-C.; D’Hooge, R.; Buée, L.; Balschun, D. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol. Aging 2014, 35, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- Regan, P.; Piers, T.; Yi, J.-H.; Kim, D.-H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J. Neurosci. 2015, 35, 4804–4812. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Santa-Maria, I.; de Gomez Barreda, E.; Zhu, X.; Cuadros, R.; Cabrero, J.R.; Sanchez-Madrid, F.; Dawson, H.N.; Vitek, M.P.; Perry, G.; et al. Tau-an inhibitor of deacetylase HDAC6 function. J. Neurochem. 2009, 109, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Leyk, J.; Goldbaum, O.; Noack, M.; Richter-Landsberg, C. Inhibition of HDAC6 modifies tau inclusion body formation and impairs autophagic clearance. J. Mol. Neurosci. 2015, 55, 1031–1046. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Wong, C.W.; Quaranta, V.; Glenner, G.G. Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc. Natl. Acad. Sci. USA 1985, 82, 8729–8732. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Blanquet, V.; Goldgaber, D.; Turleau, C.; Créau-Goldberg, N.; Delabar, J.; Sinet, P.M.; Roudier, M.; de Grouchy, J. The beta amyloid protein (AD-AP) cDNA hybridizes in normal and Alzheimer individuals near the interface of 21q21 and q22.1. Ann. Génétique 1987, 30, 68–69. [Google Scholar]
- Robakis, N.; Wisniewski, H.; Jenkins, E.; Devine-Gage, E.; Houck, G.; Yao, X.; Ramakrishna, N.; Wolfe, G.; Silverman, W.; Brown, W. Chromosome 21q21 sublocalisation of gene encoding beta-amyloid peptide in cerebral vessels and neuritic (senile) plaques of people with Alzheimer disease and Down syndrome. Lancet 1987, 1, 384–385. [Google Scholar] [CrossRef]
- St George-Hyslop, P.H.; Tanzi, R.E.; Polinsky, R.J.; Haines, J.L.; Nee, L.; Watkins, P.C.; Myers, R.H.; Feldman, R.G.; Pollen, D.; Drachman, D.; et al. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science 1987, 235, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Goldgaber, D.; Lerman, M.; McBride, W.; Saffiotti, U.; Gajdusek, D. Isolation, characterization, and chromosomal localization of human brain cDNA clones coding for the precursor of the amyloid of brain in Alzheimer’s disease, Down’s syndrome and aging. J. Neural Transm. 1987, 24, 23–28. [Google Scholar]
- Van Broeckhoven, C.; Haan, J.; Bakker, E.; Hardy, J.; van Hul, W.; Webnert, A.; der Vegter-Van Vlis, M.; Roos, R. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science 1990, 248, 1120–1122. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Carman, M.; Fernandez-Madrid, I. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Lowenberg, K.; Waggoner, R. Familial organic psychosis (Alzheimer’s type). Arch. Neurol. 1934, 31, 737–754. [Google Scholar] [CrossRef]
- St George-Hyslop, P.; Haines, J.; Rogaev, E.; Mortilla, M.; Vaula, G.; Pericak-Vance, M.; Foncin, J.F.; Montesi, M.; Bruni, A.; Sorbi, S.; et al. Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome 14. Nat. Genet. 1992, 2, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, G.D.; Bird, T.D.; Wijsman, E.M.; Orr, H.T.; Anderson, L.; Nemens, E.; White, J.A.; Bonnycastle, L.; Weber, J.L.; Alonso, M.E. Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science 1992, 258, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Czech, C.; Tremp, G.; Pradier, L. Presenilins and Alzheimer’s disease: Biological functions and pathogenic mechanisms. Prog. Neurobiol. 2000, 60, 363–384. [Google Scholar] [CrossRef]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Levesque, G.; Johnson-Wood, K.; Lee, M.; Seubert, P.; Davis, A.; et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 1997, 3, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Okochi, M.; Steiner, H.; Fukumori, A.; Tanii, H.; Tomita, T.; Tanaka, T.; Iwatsubo, T.; Kudo, T.; Takeda, M.; Haass, C. Presenilins mediate a dual intramembranous γ-secretase cleavage of Notch-1. EMBO J. 2002, 21, 5408–5416. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Bronson, R.T.; Chen, D.F.; Xia, W.; Selkoe, D.J.; Tonegawa, S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 1997, 89, 629–639. [Google Scholar] [CrossRef]
- Van Groen, T. DNA Methylation and Alzheimer’s Disease. In Epigenetics of Aging; Tollefsbol, T., Ed.; Springer: New York, NY, USA, 2010; pp. 315–326. [Google Scholar]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J. Old drug, new hope for Alzheimer’s disease. Science 2012, 335, 1447–1448. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.-X.; Gong, Y.; Moore, C.; Fu, M.; German, D.C.; Chang, L.-Y.; Rosenberg, R.; Diaz-Arrastia, R. Beta-amyloid auto-antibodies are reduced in Alzheimer’s disease. J. Neuroimmunol. 2014, 274, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Pharmacogenetic basis for therapeutic optimization in Alzheimer’s disease. Mol. Diagn. Ther. 2007, 11, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.; Carrasquillo, M.; Abraham, R.; Hamshere, M.; Pahwa, J.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naj, A.; Jun, G.; Beecham, G.; Wang, L.; Vardarajan, B.; Buros, J.; Gallins, P.; Buxbaum, J.; Jarvik, G.; Crane, P.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.M.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L. A.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Cruchaga, C.; Karch, C.M.; Jin, S.C.; Benitez, B.A.; Cai, Y.; Guerreiro, R.; Harari, O.; Norton, J.; Budde, J.; Bertelsen, S.; et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 2014, 505, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S. K.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Bierer, L.M.; Hof, P.R.; Purohit, D.P.; Carlin, L.; Schmeidler, J.; Davis, K.L.; Perl, D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch. Neurol. 1995, 52, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Bancher, C.; Brunner, C.; Lassmann, H.; Budka, H.; Jellinger, K.; Wiche, G.; Seitelberger, F.; Grundke-Iqbal, I.; Iqbal, K.; Wisniewski, H.M. Accumulation of abnormally phosphorylated τ precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 1989, 477, 90–99. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E.; Strothjohann, M. Abnormally phosphorylated tau protein related to the formation of neurofibrillary tangles and neuropil threads in the cerebral cortex of sheep and goat. Neurosci. Lett. 1994, 171, 1–4. [Google Scholar] [CrossRef]
- Joshi, Y.B.; Praticò, D. Neuroinflammation and Alzheimer’s disease: Lessons learned from 5-lypoxigenase. Transl. Neurosci. 2014, 5, 197–202. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Stucky, A.; Hahn, C.-G.; Wilson, R.; Bennett, D.; Arnold, S. BDNF-trkB signaling in late life cognitive decline and Alzheimer’s disease. Transl. Neurosci. 2011, 2, 91–100. [Google Scholar] [CrossRef]
- Pollock, N.; Mirra, S.; Binder, L.; Hansen, L.; Wood, J. Filamentous aggregates in Pick’s disease, progressive supranuclear palsy, and Alzheimer’s disease share antigenic determinants with microtubule-associated protein, tau. Lancet 1986, 204–205. [Google Scholar] [CrossRef]
- Sergeant, N.; Delacourte, A.; Buée, L. Tau protein as a differential biomarker of tauopathies. Biochim. Biophys. Acta 2005, 1739, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Flament, S.; Delacourte, A.; Verny, M.; Hauw, J.J.; Javoy-Agid, F. Abnormal Tau proteins in progressive supranuclear palsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type. Acta Neuropathol. 1991, 81, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Ksiezak-Reding, H.; Morgan, K.; Mattiace, L.A.; Davies, P.; Liu, W.K.; Yen, S.H.; Weidenheim, K.; Dickson, D.W. Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am. J. Pathol. 1994, 145, 1496–1508. [Google Scholar] [PubMed]
- Šimić, G. Pathological tau proteins in argyrophilic grain disease. Lancet Neurol. 2002. [Google Scholar] [CrossRef]
- Delacourte, A.; Robitaille, Y.; Sergeant, N.; Buée, L.; Hof, P.R.; Wattez, A.; Laroche-Cholette, A.; Mathieu, J.; Chagnon, P.; Gauvreau, D. Specific pathological tau protein variants characterize Pick’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998, 21, 428–433. [Google Scholar] [CrossRef]
- Wilhelmsen, K.C.; Lynch, T.; Pavlou, E.; Higgins, M.; Nygaard, T.G. Localization of disinhibition-dementia-parkinsonism-amyotrophy complex to 17q21–22. Am. J. Hum. Genet. 1994, 55, 1159–1165. [Google Scholar] [PubMed]
- Spillantini, M.G.; Goedert, M.; Crowther, R.A.; Murrell, J.R.; Farlow, M.R.; Ghetti, B. Familial multiple system tauopathy with presenile dementia: A disease with abundant neuronal and glial tau filaments. Proc. Natl. Acad. Sci. USA 1997, 94, 4113–4118. [Google Scholar] [CrossRef] [PubMed]
- Murrell, J.R.; Koller, D.; Foroud, T.; Goedert, M.; Spillantini, M.G.; Edenberg, H.J.; Farlow, M.R.; Ghetti, B. Familial multiple-system tauopathy with presenile dementia is localized to chromosome 17. Am. J. Hum. Genet. 1997, 61, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.L.; Wilhelmsen, K.; Sima, A.A.; Jones, M.Z.; D’Amato, C.J.; Gilman, S. Frontotemporal dementia and parkinsonism linked to chromosome 17: A consensus conference. Conference Participants. Ann. Neurol. 1997, 41, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef] [PubMed]
- Poorkaj, P.; Bird, T.D.; Wijsman, E.; Nemens, E.; Garruto, R.M.; Anderson, L.; Andreadis, A.; Wiederholt, W.C.; Raskind, M.; Schellenberg, G.D. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 1998, 43, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; Hackett, J.; et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Smith, M.J.; Goedert, M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998, 437, 207–210. [Google Scholar] [CrossRef]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Gnjidić, M.; Kostović, I. Cytoskeletal changes as an alternative view on pathogenesis of Alzheimer’s disease. Period. Biol. 1998, 100, 165–173. [Google Scholar]
- Bhatia, N.; Hall, G.F. Untangling the role of tau in Alzheimer’s disease: A unifying hypothesis. Transl. Neurosci. 2013, 4, 115–133. [Google Scholar] [CrossRef]
- Dermaut, B.; Kumar-Singh, S.; Engelborghs, S.; Theuns, J.; Rademakers, R.; Saerens, J.; Pickut, B.A.; Peeters, K.; van den Broeck, M.; Vennekens, K.; et al. A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann. Neurol. 2004, 55, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Vandrovcova, J.; Pittman, A.M.; Malzer, E.; Abou-Sleiman, P.M.; Lees, A.J.; Wood, N.W.; de Silva, R. Association of MAPT haplotype-tagging SNPs with sporadic Parkinson’s disease. Neurobiol. Aging 2009, 30, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Dawson, H.N.; Ferreira, A.; Eyster, M.V.; Ghoshal, N.; Binder, L.I.; Vitek, M.P. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 2001, 114, 1179–1187. [Google Scholar] [PubMed]
- Terry, R.D. The pathogenesis of Alzheimer’s disease: An alternative to the amyloid hypothesis. J. Neuropathol. Exp. Neurol. 1996, 55, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Kopeikina, K.; Hyman, B.; Spires-Jones, T. Soluble forms of tau are toxic in Alzheimer’s disease. Transl. Neurosci. 2012, 3, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.R.; Mukaetova-Ladinska, E.B.; Hills, R.; Edwards, P.C.; de Montejo Garcini, E.; Novak, M.; Wischik, C.M. Measurement of distinct immunochemical presentations of tau protein in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 5842–5846. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Kabat, J.; Wischik, C.M. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993, 12, 365–370. [Google Scholar] [PubMed]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.R.; Storey, J.M.D.; Clunas, S.; Harrington, K.A.; Horsley, D.; Ishaq, A.; Kemp, S.J.; Larch, C.P.; Marshall, C.; Nicoll, S.L.; et al. Cellular models of aggregation-dependent template-directed proteolysis to characterize tau aggregation inhibitors for treatment of Alzheimer’s Disease. J. Biol. Chem. 2015, 290, 10862–10875. [Google Scholar] [CrossRef] [PubMed]
- Hrnkova, M.; Zilka, N.; Minichova, Z.; Koson, P.; Novak, M. Neurodegeneration caused by expression of human truncated tau leads to progressive neurobehavioural impairment in transgenic rats. Brain Res. 2007, 1130, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Zilka, N.; Filipcik, P.; Koson, P.; Fialova, L.; Skrabana, R.; Zilkova, M.; Rolkova, G.; Kontsekova, E.; Novak, M. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006, 580, 3582–3588. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Harrington, C.R.; Storey, J.M.D. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Mukaetova-Ladinska, E.B.; Garcia-Siera, F.; Hurt, J.; Gertz, H.J.; Xuereb, J.H.; Hills, R.; Brayne, C.; Huppert, F.A.; Paykel, E.S.; McGee, M.; et al. Staging of cytoskeletal and beta-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s disease. Am. J. Pathol. 2000, 157, 623–636. [Google Scholar] [CrossRef]
- Callahan, L.M.; Vaules, W.A.; Coleman, P.D. Progressive reduction of synaptophysin message in single neurons in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2002, 61, 384–395. [Google Scholar] [PubMed]
- Liu, X.; Brun, A. Regional and laminar synaptic pathology in frontal lobe degeneration of non-Alzheimer type. Int. J. Geriatr. Psychiatry 1996, 11, 47–55. [Google Scholar] [CrossRef]
- Suzuki, K.; Parker, C.C.; Pentchev, P.G.; Katz, D.; Ghetti, B.; D’Agostino, A.N.; Carstea, E.D. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol. 1995, 89, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Lécolle, K.; Caillierez, R.; Bégard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Aurégan, G.; Winderickx, J.; et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.F.; Patuto, B.A. Is tau ready for admission to the prion club? Prion 2012, 6, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanamandra, K.; Kfoury, N.; Jiang, H.; Mahan, T.E.; Ma, S.; Maloney, S.E.; Wozniak, D.F.; Diamond, M.I.; Holtzman, D.M. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 2013, 80, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Gómez-Ramos, A.; Bolós, M. AD genetic risk factors and tau spreading. Front. Aging Neurosci. 2015, 7, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Le Corre, S.; Klafki, H.W.; Plesnila, N.; Hübinger, G.; Obermeier, A.; Sahagún, H.; Monse, B.; Seneci, P.; Lewis, J.; Eriksen, J.; et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9673–9678. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.; Ho, L.; Wang, J.; Zhao, W.; Levine, S.; Ono, K.; Mannino, S.; Pasinetti, G. Green coffee as a novel agent for Alzheimer’s disease prevention by attenuating diabetes. Transl. Neurosci. 2014, 5, 111–116. [Google Scholar] [CrossRef]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Troquier, L.; Caillierez, R.; Burnouf, S.; Fernandez-Gomez, F.J.; Grosjean, M.-E.; Zommer, N.; Sergeant, N.; Schraen-Maschke, S.; Blum, D.; Buée, L. Targeting phospho-Ser422 by active Tau Immunotherapy in the THYTau22 mouse model: A suitable therapeutic approach. Curr. Alzheimer Res. 2012, 9, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Dawidowitz, E.; Moe, J. Targeting tau for Alzheimer’s disease and related neurodegenerative disorders. Drug Discov. 2012, 3, 16–21. [Google Scholar]
- Del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andrés, M.V.; Gómez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; León, T. Treatment of Alzheimer’s Disease with the GSK-3 Inhibitor Tideglusib: A Pilot Study. J. Alzheimers. Dis. 2013, 33, 205–215. [Google Scholar] [PubMed]
- Baddeley, T.C.; McCaffrey, J.; Storey, J.M.D.; Cheung, J.K.S.; Melis, V.; Horsley, D.; Harrington, C.R.; Wischik, C.M. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2015, 352, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Staff, R.T.; Wischik, D.J.; Bentham, P.; Murray, A.D.; Storey, J.M.D.; Kook, K.A.; Harrington, C.R. Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimers. Dis. 2015, 44, 705–720. [Google Scholar] [PubMed]
- Grinberg, L.T.; Wang, X.; Wang, C.; Sohn, P.D.; Theofilas, P.; Sidhu, M.; Arevalo, J.B.; Heinsen, H.; Huang, E.J.; Rosen, H.; et al. Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation. Acta Neuropathol. 2013, 125, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Mohideen, S.; Yamasaki, Y.; Omata, Y.; Tsuda, L.; Yoshiike, Y. Nontoxic singlet oxygen generator as a therapeutic candidate for treating tauopathies. Sci. Rep. 2015. [Google Scholar] [CrossRef]
- Melis, V.; Magbagbeolu, M.; Rickard, J.E.; Horsley, D.; Davidson, K.; Harrington, K.A.; Goatman, K.; Goatman, E.A.; Deiana, S.; Close, S.P.; et al. Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav. Pharmacol. 2015, 26, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Stack, C.; Jainuddin, S.; Elipenahli, C.; Gerges, M.; Starkova, N.; Starkov, A.; Jové, M.; Portero-Otin, M.; Launay, N.; Pujol, A.; et al. Methylene blue upregulates Nrf2/ARE genes and prevents tau-relatd neurotoxicity. Hum. Mol. Genet. 2014, 23, 3716–3732. [Google Scholar] [CrossRef] [PubMed]
- Pakavathkumar, P.; Sharma, G.; Kaushal, V.; Foveau, B.; LeBlanc, A. Methylene blue inhibits caspases by oxidation of the catalytic cysteine. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hochgräfe, K.; Sydow, A.; Matenia, D.; Cadinu, D.; Könen, S.; Petrova, O.; Pickhardt, M.; Goll, P.; Morellini, F.; Mandelkow, E.; et al. Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta Neuropathol. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bulic, B.; Pickhardt, M.; Mandelkow, E.-M.; Mandelkow, E. Tau protein and tau aggregation inhibitors. Neuropharmacology 2010, 59, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Jun, G.; Ibrahim-Verbaas, C.A.; Vronskaya, M.; Lambert, J.-C.; Chung, J.; Naj, A.C.; Kunkle, B.W.; Wang, L.-S.; Bis, J.C.; Bellenguez, C.; et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol. Psychiatry 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šimić, G.; Stanić, G.; Mladinov, M.; Jovanov-Milošević, N.; Kostović, I.; Hof, P.R. Does Alzheimer’s disease begin in the brainstem? Neuropathol. Appl. Neurobiol. 2009, 35, 532–554. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; De Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. https://doi.org/10.3390/biom6010006
Šimić G, Babić Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milošević N, Bažadona D, Buée L, De Silva R, Di Giovanni G, et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules. 2016; 6(1):6. https://doi.org/10.3390/biom6010006
Chicago/Turabian StyleŠimić, Goran, Mirjana Babić Leko, Selina Wray, Charles Harrington, Ivana Delalle, Nataša Jovanov-Milošević, Danira Bažadona, Luc Buée, Rohan De Silva, Giuseppe Di Giovanni, and et al. 2016. "Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies" Biomolecules 6, no. 1: 6. https://doi.org/10.3390/biom6010006