
Overview of Glutamatergic Dysregulation in Central Pathologies


Abstract
:1. Introduction
2. Glutamate Receptors and Transporters
2.1. Glutamate Receptors
2.2. Glutamate Transporters
3. Glutamate Dysregulation in Psychiatric Disorders
3.1. Mood and Anxiety Disorders
3.2. Schizophrenia
3.3. Autism Spectrum Disorder
3.4. Attention-Deficit/Hyperactivity Disorder
4. Glutamate Dysregulation in Neurodegenerative Disease
4.1. Epilepsy
4.2. Alzheimer’s Disease
4.3. Huntington’s Disease
4.4. Parkinson’s Disease
4.5. Amyotrophic Lateral Sclerosis (ALS)
4.6. Stroke and Ischemia
5. Glutamate Dysregulation in Gliomas
6. Glutamate Dysregulation in Chronic Pain
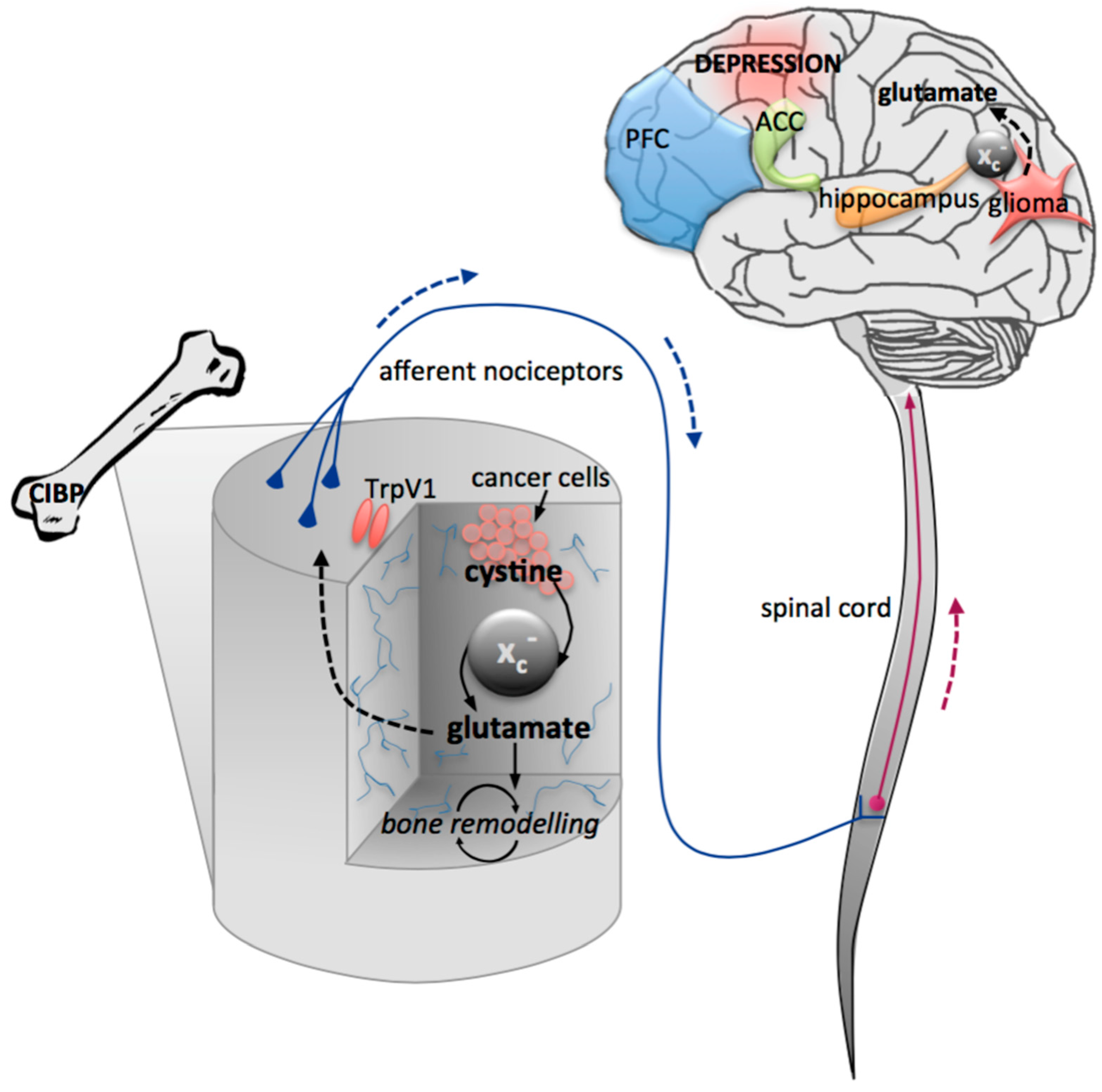
7. Oncodynamic Effects on Central Pathologies
7.1. Cancer-Induced Depression
7.2. Cancer-Induced Bone Pain
8. Conclusions

{kind=link}
| Pathology | Experimental Model/Intervention | Glutamatergic Change | Phenotype | References | ||
|---|---|---|---|---|---|---|
| Psychiatric/Neurodevelopmental | Major depressive disorder | NMDAR antagonist (human and murine) | Activated mTOR pathway, increased BDNF | Reduced depressive symptoms | [41,42,43,44,57] | |
| MRS in patients | Decreased glutamate in PFC and ACC | [47,48] | ||||
| Post-mortem patients | Increased AMPAR expression in hippocampus | [49] | ||||
| Bipolar disorder | NMDAR antagonist in patients | Reduced bipolar depression | [67,68] | |||
| MRS in patients | Decreased glutamate in hippocampus; increased glutamate in ACC, frontal regions, whole brain | [65,69,70] | ||||
| Post-mortem patient hippocampus | Reduced expression of NR1 and NR2A | [72,73] | ||||
| Anxiety | MRS in adolescent GAD patients | Increased Glu/Cr ratio in ACC | [76] | |||
| NMDAR antagonist in PTSD patients | Reduced anxiety symptoms | [77] | ||||
| Obsessive compulsive disorder | MRS in patients | Decreased glutamate in ACC and OFC | [80,81] | |||
| Serology in patients | Increased glutamate in CSF | [82,83] | ||||
| NMDAR antagonist in patients | Reduced OCD symptoms | [84] | ||||
| Riluzole in patients | Increased glutamate uptake by astrocytes and inhibited presynaptic glutamate release | Reduced OCD symptoms | [85] | |||
| Schizophrenia | NMDAR antagonist in healthy subjects | Induced psychotomimetic symptoms | [90,91,92,93] | |||
| MRS in patients | Increased glutamate in PFC, basal ganglia, hippocampus | [94] | ||||
| Post-mortem patients | Decreased dendritic length, number, spine density, synaptophysin protein expression | [98] | ||||
| Autism spectrum disorder | GRM5 knockout | No expression of mGluR5 | Induced ASD behaviours | [104] | ||
| MRS in patients | Increased glutamate in ACC; decreased glutamate in frontal and occipital lobes | [81] | ||||
| Attention-deficit/hyperactivity disorder | MRS in patients | Increased glutamate in PFC, ACC, striatum | [81] | |||
| Genome-wide association | GRM7, GRIN2A, GRIN2B, GRID2, EAAT1 polymorphisms | Association with ADHD symptoms | [107] | |||
| GRM5 knockout/inhibitor | No expression/inhibition of mGluR5 | Locomotor hyperactivity, impaired learning | [107] | |||
| Neurodegenerative/Pain | Epilepsy | EAAT2 knockout | Reduced EAAT2 protein | Seizure and death at 6 weeks | [126] | |
| EAAT3 antisense knockdown | Reduced EAAT3 and GABA | Behavioural abnormalities | [128] | |||
| Alzheimer’s disease | Mutant APP overexpression in transgenic mice | Reduced EAAT1 and EAAT2 protein | Behavioural abnormalities, plaque formation | [109,138] | ||
| Huntington’s disease | Expression of mutant huntington (R6/2) | Reduced EAAT2 protein and mRNA | [111] | |||
| Parkinson’s disease | Disruption of cerebral cortex corpus callosum pathway | Reduced EAAT1 and EAAT2 | [112,113] | |||
| Amyotrophic lateral sclerosis | Mutant SOD1 gene | Reduced EAAT2 protein | Paralysis and spinal neuron degeneration | [114,115] | ||
| EAAT2 antisense knockdown | Reduced EAAT2 protein | Paralysis and spinal neuron degeneration | [123] | |||
| Stroke/Ischemia | Hypoxic neonatal pig | Reduced EAAT2 and EAAT3 | [120] | |||
| Cortical and hippocampal hypoxia-ischemia | Reduced EAAT1, EAAT2, and EAAT3 | [118] | ||||
| MCA occlusion | Reduced EAAT1 and EAAT2 | Contralateral hemiparesis | [122] | |||
| Chronic pain | NMDAR antagonist | Reduced nociception | [178,179] | |||
| Cancer-induced bone pain | System xc− inhibitor | Inhibition of glutamate released by peripheral tumours | Inhibit pain behaviours | [11,171,172] |
Acknowledgments
Conflicts of Interest
References
- Kew, J.N.; Kemp, J.A. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology 2005, 179, 4–29. [Google Scholar] [CrossRef] [PubMed]
- Eulenburg, V.; Gomeza, J. Neurotransmitter transporters expressed in glial cells as regulators of synapse function. Brain Res. Rev. 2010, 63, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [PubMed]
- Hinoi, E.; Takarada, T.; Ueshima, T.; Tsuchihashi, Y.; Yoneda, Y. Glutamate signaling in peripheral tissues. Eur. J. Biochem. FEBS 2004, 271, 1–13. [Google Scholar] [CrossRef]
- Jang, J.H.; Kim, D.W.; Sang Nam, T.; Se Paik, K.; Leem, J.W. Peripheral glutamate receptors contribute to mechanical hyperalgesia in a neuropathic pain model of the rat. Neuroscience 2004, 128, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000, 130, 1007s–1015s. [Google Scholar] [PubMed]
- Javitt, D.C. Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry 2004, 9, 984–997. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.R.; Bray, S.M.; Warren, S.T. Molecular mechanisms of fragile X syndrome: A twenty-year perspective. Annu. Rev. Pathol. 2012, 7, 219–245. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Sontheimer, H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999, 59, 4383–4391. [Google Scholar] [PubMed]
- Nashed, M.G.; Seidlitz, E.P.; Frey, B.N.; Singh, G. Depressive-like behaviours and decreased dendritic branching in the medial prefrontal cortex of mice with tumors: A novel validated model of cancer-induced depression. Behav. Brain Res. 2015, 294, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Ungard, R.G.; Seidlitz, E.P.; Singh, G. Inhibition of breast cancer-cell glutamate release with sulfasalazine limits cancer-induced bone pain. Pain 2014, 155, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Fonnum, F. Glutamate: A neurotransmitter in mammalian brain. J. Neurochem. 1984, 42, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A. Transport and metabolism of glutamate and gaba in neuron and glial cells. Int. Rev. Neurobiol. 1981, 22, 1–45. [Google Scholar] [PubMed]
- Nicholls, D.; Attwell, D. The release and uptake of excitatory amino acids. Trends Pharmacol. Sci. 1990, 11, 462–468. [Google Scholar] [CrossRef]
- Daikhin, Y.; Yudkoff, M. Compartmentation of brain glutamate metabolism in neurons and glia. J. Nutr. 2000, 130, 1026S–1031S. [Google Scholar] [PubMed]
- Reissner, K.J.; Kalivas, P.W. Using glutamate homeostasis as a target for treating addictive disorders. Behav. Pharmacol. 2010, 21, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 514–522. [Google Scholar] [CrossRef]
- Grewer, C.; Rauen, T. Electrogenic glutamate transporters in the CNS: Molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J. Membr. Biol. 2005, 203, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Tzingounis, A.V.; Wadiche, J.I. Glutamate transporters: Confining runaway excitation by shaping synaptic transmission. Nat. Rev. Neurosci. 2007, 8, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, R.J.; Ryan, R.M. Mechanisms of glutamate transport. Physiol. Rev. 2013, 93, 1621–1657. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate receptors and the induction of excitotoxic neuronal death. Prog. Brain Res. 1994, 100, 47–51. [Google Scholar] [PubMed]
- Choi, D.W.; Maulucci-Gedde, M.; Kriegstein, A.R. Glutamate neurotoxicity in cortical cell culture. J. Neurosci. 1987, 7, 357–368. [Google Scholar] [PubMed]
- Hollmann, M.; Heinemann, S. Cloned glutamate receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef] [PubMed]
- Schoepfer, R.; Monyer, H.; Sommer, B.; Wisden, W.; Sprengel, R.; Kuner, T.; Lomeli, H.; Herb, A.; Köhler, M.; Burnashev, N. Molecular biology of glutamate receptors. Prog. Neurobiol. 1994, 42, 353–357. [Google Scholar] [CrossRef]
- Borges, K.; Dingledine, R. AMPA receptors: Molecular and functional diversity. Prog. Brain Res. 1998, 116, 153–170. [Google Scholar] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar] [PubMed]
- Kanai, Y.; Hediger, M.A. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 1992, 360, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Fairman, W.A.; Vandenberg, R.J.; Arriza, J.L.; Kavanaugh, M.P.; Amara, S.G. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 1995, 375, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Pines, G.; Danbolt, N.C.; Bjørås, M.; Zhang, Y.; Bendahan, A.; Eide, L.; Koepsell, H.; Storm-Mathisen, J.; Seeberg, E.; Kanner, B.I. Cloning and expression of a rat brain l-glutamate transporter. Nature 1992, 360, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Storck, T.; Schulte, S.; Hofmann, K.; Stoffel, W. Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc. Natl. Acad. Sci. USA 1992, 89, 10955–10959. [Google Scholar] [CrossRef] [PubMed]
- Arriza, J.L.; Eliasof, S.; Kavanaugh, M.P.; Amara, S.G. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. USA 1997, 94, 4155–4160. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, O.; Ben-Hur, H.; Biegon, A.; Groner, Y.; Dewhurst, S.; Furuta, A.; Rothstein, J.D. Distribution of glutamate transporter subtypes during human brain development. J. Neurochem. 1997, 69, 2571–2580. [Google Scholar] [CrossRef] [PubMed]
- Furuta, A.; Martin, L.J.; Lin, C.L.; Dykes-Hoberg, M.; Rothstein, J.D. Cellular and synaptic localization of the neuronal glutamate transporters excitatory amino acid transporter 3 and 4. Neuroscience 1997, 81, 1031–1042. [Google Scholar] [CrossRef]
- Baker, D.A.; Xi, Z.-X.; Shen, H.; Swanson, C.J.; Kalivas, P.W. The origin and neuronal function of in vivo nonsynaptic glutamate. J. Neurosci. 2002, 22, 9134–9141. [Google Scholar] [PubMed]
- McBean, G.J. Cerebral cystine uptake: A tale of two transporters. Trends Pharmacol. Sci. 2002, 23, 299–302. [Google Scholar] [CrossRef]
- Sagara, J.I.; Miura, K.; Bannai, S. Maintenance of neuronal glutathione by glial cells. J. Neurochem. 1993, 61, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The x(c)- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell. Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Merikangas, K.R.; Low, N.C. The epidemiology of mood disorders. Curr. Psychiatry Rep. 2004, 6, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Andrade, L.; Caraveo-Anduaga, J.J.; Berglund, P.; Bijl, R.V.; de Graaf, R.; Vollebergh, W.; Dragomirecka, E.; Kohn, R.; Keller, M.; Kessler, R.C.; et al. The epidemiology of major depressive episodes: Results from the international consortium of psychiatric epidemiology (ICPE) surveys. Int. J. Methods Psychiatr. Res. 2003, 12, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Ibrahim, L.; Diazgranados, N.; Franco-Chaves, J.; Brutsche, N.; Henter, I.D.; Kronstein, P.; Moaddel, R.; Wainer, I.; Luckenbaugh, D.A.; Manji, H.K.; et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: Results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology 2012, 37, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Lally, N.; Nugent, A.C.; Luckenbaugh, D.A.; Niciu, M.J.; Roiser, J.P.; Zarate, C.A., Jr. Neural correlates of change in major depressive disorder anhedonia following open-label ketamine. J. Psychopharmacol. 2015, 29, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A., Jr.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.; Panchalingam, K.; Rapoport, A.; Gershon, S.; McClure, R.J.; Pettegrew, J.W. Increased cerebrospinal fluid glutamine levels in depressed patients. Biol. Psychiatry 2000, 47, 586–593. [Google Scholar] [CrossRef]
- Mitani, H.; Shirayama, Y.; Yamada, T.; Maeda, K.; Ashby, C.R., Jr.; Kawahara, R. Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2006, 30, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Auer, D.P.; Putz, B.; Kraft, E.; Lipinski, B.; Schill, J.; Holsboer, F. Reduced glutamate in the anterior cingulate cortex in depression: An in vivo proton magnetic resonance spectroscopy study. Biol. Psychiatry 2000, 47, 305–313. [Google Scholar] [CrossRef]
- Hasler, G.; van der Veen, J.W.; Tumonis, T.; Meyers, N.; Shen, J.; Drevets, W.C. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 2007, 64, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Duric, V.; Banasr, M.; Stockmeier, C.A.; Simen, A.A.; Newton, S.S.; Overholser, J.C.; Jurjus, G.J.; Dieter, L.; Duman, R.S. Altered expression of synapse and glutamate related genes in post-mortem hippocampus of depressed subjects. Int. J. Neuropsychopharmacol. 2013, 16, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; Juhasz, G.; Kekesi, K.A.; Kovacs, K.J. Corticosterone peak is responsible for stress-induced elevation of glutamate in the hippocampus. Stress 1998, 2, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Stein-Behrens, B.A.; Lin, W.J.; Sapolsky, R.M. Physiological elevations of glucocorticoids potentiate glutamate accumulation in the hippocampus. J. Neurochem. 1994, 63, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Lowy, M.T.; Gault, L.; Yamamoto, B.K. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J. Neurochem. 1993, 61, 1957–1960. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.; Mackay, D.; Chana, G.; Beasley, C.; Landau, S.; Everall, I.P. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 2002, 12, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.; Mackay, D.; Landau, S.; Kerwin, R.; Everall, I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch. Gen. Psychiatry 2001, 58, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Drevets, W.C.; Price, J.L. Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol. Psychiatry 2004, 55, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Voleti, B. Signaling pathways underlying the pathophysiology and treatment of depression: Novel mechanisms for rapid-acting agents. Trends Neurosci. 2012, 35, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Geddes, J.R.; Miklowitz, D.J. Treatment of bipolar disorder. Lancet 2013, 381, 1672–1682. [Google Scholar] [CrossRef]
- Cade, J.F. Lithium salts in the treatment of psychotic excitement. 1949. Bull. World Health Organ. 2000, 78, 518–520. [Google Scholar] [PubMed]
- Geddes, J.R.; Burgess, S.; Hawton, K.; Jamison, K.; Goodwin, G.M. Long-term lithium therapy for bipolar disorder: Systematic review and meta-analysis of randomized controlled trials. Am. J. Psychiatry 2004, 161, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Barbui, C.; Salanti, G.; Rendell, J.; Brown, R.; Stockton, S.; Purgato, M.; Spineli, L.M.; Goodwin, G.M.; Geddes, J.R. Comparative efficacy and acceptability of antimanic drugs in acute mania: A multiple-treatments meta-analysis. Lancet 2011, 378, 1306–1315. [Google Scholar] [CrossRef]
- Frye, M.A.; Ha, K.; Kanba, S.; Kato, T.; McElroy, S.L.; Ozerdem, A.; Vazquez, G.; Vieta, E. International consensus group on depression prevention in bipolar disorder. J. Clin. Psychiatry 2011, 72, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Gijsman, H.J.; Geddes, J.R.; Rendell, J.M.; Nolen, W.A.; Goodwin, G.M. Antidepressants for bipolar depression: A systematic review of randomized, controlled trials. Am. J. Psychiatry 2004, 161, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Sachs, G.S.; Nierenberg, A.A.; Calabrese, J.R.; Marangell, L.B.; Wisniewski, S.R.; Gyulai, L.; Friedman, E.S.; Bowden, C.L.; Fossey, M.D.; Ostacher, M.J.; et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N. Engl. J. Med. 2007, 356, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Gigante, A.D.; Bond, D.J.; Lafer, B.; Lam, R.W.; Young, L.T.; Yatham, L.N. Brain glutamate levels measured by magnetic resonance spectroscopy in patients with bipolar disorder: A meta-analysis. Bipolar Disord. 2012, 14, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Scarr, E.; Pavey, G.; Sundram, S.; MacKinnon, A.; Dean, B. Decreased hippocampal NMDA, but not kainate or AMPA receptors in bipolar disorder. Bipolar Disord. 2003, 5, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Diazgranados, N.; Ibrahim, L.; Brutsche, N.E.; Newberg, A.; Kronstein, P.; Khalife, S.; Kammerer, W.A.; Quezado, Z.; Luckenbaugh, D.A.; Salvadore, G.; et al. A randomized add-on trial of an N-methyl-d-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiatry 2010, 67, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A., Jr.; Brutsche, N.E.; Ibrahim, L.; Franco-Chaves, J.; Diazgranados, N.; Cravchik, A.; Selter, J.; Marquardt, C.A.; Liberty, V.; Luckenbaugh, D.A. Replication of ketamine’s antidepressant efficacy in bipolar depression: A randomized controlled add-on trial. Biol. Psychiatry 2012, 71, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Chitty, K.M.; Lagopoulos, J.; Hickie, I.B.; Hermens, D.F. Hippocampal glutamatergic/nmda receptor functioning in bipolar disorder: A study combining mismatch negativity and proton magnetic resonance spectroscopy. Psychiatry Res. 2015, 233, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, A.; Schubert, F.; Pehrs, C.; Gallinat, J. Alterations of cerebral glutamate in the euthymic state of patients with bipolar disorder. Psychiatry Res. 2015, 233, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, R.; Fekkes, D.; Loonen, A.J.; Pepplinkhuizen, L.; Tuinier, S.; Verhoeven, W.M. Bipolar mania and plasma amino acids: Increased levels of glycine. Eur. Neuropsychopharmacol. 2006, 16, 71–77. [Google Scholar] [CrossRef] [PubMed]
- McCullumsmith, R.E.; Kristiansen, L.V.; Beneyto, M.; Scarr, E.; Dean, B.; Meador-Woodruff, J.H. Decreased NR1, NR2A, and SAP102 transcript expression in the hippocampus in bipolar disorder. Brain Res. 2007, 1127, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R.; Ibrahim, L.; Henter, I.D.; Zarate, C.A., Jr. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol. Biochem. Behav. 2012, 100, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.; Charney, D. Comorbidity of mood and anxiety disorders. Depression Anxiety 2000, 12, S69–S76. [Google Scholar] [CrossRef]
- Dunlop, B.W.; Davis, P.G. Combination treatment with benzodiazepines and ssris for comorbid anxiety and depression: A review. Prim. Care Companion J. Clin. Psychiatry 2008, 10, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Strawn, J.R.; Chu, W.J.; Whitsel, R.M.; Weber, W.A.; Norris, M.M.; Adler, C.M.; Eliassen, J.C.; Phan, K.L.; Strakowski, S.M.; DelBello, M.P. A pilot study of anterior cingulate cortex neurochemistry in adolescents with generalized anxiety disorder. Neuropsychobiology 2013, 67, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.M.; Zhou, W.W.; Ji, Y.J.; Li, Y.; Zhao, N.; Chen, H.X.; Xue, R.; Mei, X.G.; Zhang, Y.Z.; Wang, H.L.; et al. Anxiolytic effects of ketamine in animal models of posttraumatic stress disorder. Psychopharmacology 2015, 232, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Silvestre, J.S.; Nadal, R.; Pallares, M.; Ferre, N. Acute effects of ketamine in the holeboard, the elevated-plus maze, and the social interaction test in wistar rats. Depression Anxiety 1997, 5, 29–33. [Google Scholar] [CrossRef]
- Zimmer, E.R.; Torrez, V.R.; Kalinine, E.; Augustin, M.C.; Zenki, K.C.; Almeida, R.F.; Hansel, G.; Muller, A.P.; Souza, D.O.; Machado-Vieira, R.; et al. Long-term NMDAR antagonism correlates reduced astrocytic glutamate uptake with anxiety-like phenotype. Front. Cell. Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Brennan, B.P.; Rauch, S.L.; Jensen, J.E.; Pope, H.G., Jr. A critical review of magnetic resonance spectroscopy studies of obsessive-compulsive disorder. Biol. Psychiatry 2013, 73, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.E.; Uchida, M.; Kenworthy, T.; Keary, C.J.; Biederman, J. Glutamatergic dysregulation in pediatric psychiatric disorders: A systematic review of the magnetic resonance spectroscopy literature. J. Clin. Psychiatry 2014, 75, 1226–1241. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, K.; Bhattacharyya, S.; Christopher, R.; Khanna, S. Glutamatergic dysfunction in OCD. Neuropsychopharmacology 2005, 30, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Khanna, S.; Chakrabarty, K.; Mahadevan, A.; Christopher, R.; Shankar, S.K. Anti-brain autoantibodies and altered excitatory neurotransmitters in obsessive-compulsive disorder. Neuropsychopharmacology 2009, 34, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, C. Glutamate modulators in the treatment of obsessive-compulsive disorder. Psychiatr. Ann. 2015, 45, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Kariuki-Nyuthe, C.; Gomez-Mancilla, B.; Stein, D.J. Obsessive compulsive disorder and the glutamatergic system. Curr. Opin. Psychiatry 2014, 27, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Van Os, J.; Kapur, S. Schizophrenia. Lancet 2009, 374, 635–645. [Google Scholar] [CrossRef]
- Tandon, R.; Gaebel, W.; Barch, D.M.; Bustillo, J.; Gur, R.E.; Heckers, S.; Malaspina, D.; Owen, M.J.; Schultz, S.; Tsuang, M.; et al. Definition and description of schizophrenia in the DSM-5. Schizophr. Res. 2013, 150, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, D.; Owen, M.J.; Heckers, S.; Tandon, R.; Bustillo, J.; Schultz, S.; Barch, D.M.; Gaebel, W.; Gur, R.E.; Tsuang, M.; et al. Schizoaffective disorder in the DSM-5. Schizophr. Res. 2013, 150, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kornhuber, H.H.; Holzmuller, B.; Schmid-Burgk, W.; Mergner, T.; Krzepinski, G. Reduction of cerebrospinal fluid glutamic acid in huntington’s chorea and in schizophrenic patients. Arch. Psychiatr. Nervenkr. 1980, 228, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T. The glutamatergic dysfunction hypothesis for schizophrenia. Harv. Rev. Psychiatry 1996, 3, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C. Negative schizophrenic symptomatology and the PCP (phencyclidine) model of schizophrenia. Hillside J. Clin. Psychiatry 1987, 9, 12–35. [Google Scholar] [PubMed]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B., Jr.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Lipska, B.K.; Weinberger, D.R. To model a psychiatric disorder in animals: Schizophrenia as a reality test. Neuropsychopharmacology 2000, 23, 223–239. [Google Scholar] [CrossRef]
- Poels, E.M.; Kegeles, L.S.; Kantrowitz, J.T.; Javitt, D.C.; Lieberman, J.A.; Abi-Dargham, A.; Girgis, R.R. Glutamatergic abnormalities in schizophrenia: A review of proton MRS findings. Schizophr. Res. 2014, 152, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Plitman, E.; Nakajima, S.; de la Fuente-Sandoval, C.; Gerretsen, P.; Chakravarty, M.M.; Kobylianskii, J.; Chung, J.K.; Caravaggio, F.; Iwata, Y.; Remington, G.; et al. Glutamate-mediated excitotoxicity in schizophrenia: A review. Eur. Neuropsychopharmacol. 2014, 24, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Zsiros, V.; Jiang, Z.; Nakao, K.; Kolata, S.; Zhang, S.; Belforte, J.E. Gabaergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 2012, 62, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.M.; Morrison, P.D.; Pilowsky, L.S. Glutamate and dopamine dysregulation in schizophrenia—A synthesis and selective review. J. Psychopharmacol. 2007, 21, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; MacDonald, M.L.; Elswick, D.E.; Sweet, R.A. The glutamate hypothesis of schizophrenia: Evidence from human brain tissue studies. Ann. N. Y. Acad. Sci. 2015, 1338, 38–57. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, M.C.; Craddock, N.J.; Owen, M.J. Genetics of psychosis; insights from views across the genome. Hum. Genet. 2009, 126, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Koga, M.; Ishiguro, H.; Horiuchi, Y.; Arai, M.; Niizato, K.; Itokawa, M.; Inada, T.; Iwata, N.; Iritani, S.; et al. A polymorphism of the metabotropic glutamate receptor mGluR7 (GRM7) gene is associated with schizophrenia. Schizophr. Res. 2008, 101, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, H.; Tani, A.; Chikuhara, T.; Kikuta, R.; Sakai, M.; Ninomiya, H.; Tashiro, N.; Iwata, N.; Ozaki, N.; Fukumaki, Y. Association study of polymorphisms in the group iii metabotropic glutamate receptor genes, GRM4 and GRM7, with schizophrenia. Psychiatry Res. 2009, 167, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2015, 386, 743–800. [Google Scholar]
- Dolen, G.; Osterweil, E.; Rao, B.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of fragile X syndrome in mice. Neuron 2007, 56, 955–962. [Google Scholar] [PubMed]
- Zantomio, D.; Chana, G.; Laskaris, L.; Testa, R.; Everall, I.; Pantelis, C.; Skafidas, E. Convergent evidence for mGluR5 in synaptic and neuroinflammatory pathways implicated in ASD. Neurosci. Biobehav. Rev. 2015, 52, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Betancur, C.; Quach, H.; Philippe, A.; Fellous, M.; Giros, B.; Gillberg, C.; Leboyer, M.; Bourgeron, T. Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry 2002, 7, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Shinohe, A.; Hashimoto, K.; Nakamura, K.; Tsujii, M.; Iwata, Y.; Tsuchiya, K.J.; Sekine, Y.; Suda, S.; Suzuki, K.; Sugihara, G.; et al. Increased serum levels of glutamate in adult patients with autism. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2006, 30, 1472–1477. [Google Scholar]
- Lesch, K.P.; Merker, S.; Reif, A.; Novak, M. Dances with black widow spiders: Dysregulation of glutamate signalling enters centre stage in ADHD. Eur. Neuropsychopharmacol. 2013, 23, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mallory, M.; Alford, M.; Tanaka, S.; Masliah, E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J. Neuropathol. Exp. Neurol. 1997, 56, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Alford, M.; Mallory, M.; Rockenstein, E.; Moechars, D.; van Leuven, F. Abnormal glutamate transport function in mutant amyloid precursor protein transgenic mice. Exp. Neurol. 2000, 163, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Liévens, J.C.; Woodman, B.; Mahal, A.; Spasic-Boscovic, O.; Samuel, D.; Kerkerian-Le Goff, L.; Bates, G.P. Impaired glutamate uptake in the R6 huntington’s disease transgenic mice. Neurobiol. Disease 2001, 8, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Behrens, P.F.; Franz, P.; Woodman, B.; Lindenberg, K.S.; Landwehrmeyer, G.B. Impaired glutamate transport and glutamate-glutamine cycling: Downstream effects of the huntington mutation. Brain J. Neurol. 2002, 125, 1908–1922. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Martin, L.J.; Rothstein, J.D. Regional deafferentation down-regulates subtypes of glutamate transporter proteins. J. Neurochem. 1995, 65, 2800–2803. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.M.; Lehre, K.P.; Walaas, S.I.; Storm-Mathisen, J.; Danbolt, N.C. Down-regulation of glial glutamate transporters after glutamatergic denervation in the rat brain. Eur. J. Neurosci. 1995, 7, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 2002, 99, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-C.; Hsu-Chou, H.; Lu, J.-L.; Chiang, Y.-C.; Huang, H.-M.; Wang, H.-L.; Wu, T.; Liao, J.-J.; Yeh, T.-S. Down-regulation of the glial glutamate transporter GLT-1 in rat hippocampus and striatum and its modulation by a group III metabotropic glutamate receptor antagonist following transient global forebrain ischemia. Neuropharmacology 2005, 49, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Fukamachi, S.; Furuta, A.; Ikeda, T.; Ikenoue, T.; Kaneoka, T.; Rothstein, J.D.; Iwaki, T. Altered expressions of glutamate transporter subtypes in rat model of neonatal cerebral hypoxia-ischemia. Brain Res. Dev. Brain Res. 2001, 132, 131–139. [Google Scholar] [CrossRef]
- Inage, Y.W.; Itoh, M.; Wada, K.; Takashima, S. Expression of two glutamate transporters, GLAST and EAAT4, in the human cerebellum: Their correlation in development and neonatal hypoxic-ischemic damage. J. Neuropathol. Exp. Neurol. 1998, 57, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Brambrink, A.M.; Lehmann, C.; Portera-Cailliau, C.; Koehler, R.; Rothstein, J.; Traystman, R.J. Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann. Neurol. 1997, 42, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra Rao, V.L.; Rao, A.M.; Dogan, A.; Bowen, K.K.; Hatcher, J.; Rothstein, J.D.; Dempsey, R.J. Glial glutamate transporter glt-1 down-regulation precedes delayed neuronal death in gerbil hippocampus following transient global cerebral ischemia. Neurochem. Int. 2000, 36, 531–537. [Google Scholar] [CrossRef]
- Rao, V.L.; Bowen, K.K.; Dempsey, R.J. Transient focal cerebral ischemia down-regulates glutamate transporters GLT-1 and EAAC1 expression in rat brain. Neurochem. Res. 2001, 26, 497–502. [Google Scholar] [PubMed]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Yeh, T.-H.; Hwang, H.-M.; Chen, J.-J.; Wu, T.; Li, A.H.; Wang, H.-L. Glutamate transporter function of rat hippocampal astrocytes is impaired following the global ischemia. Neurobiol. Disease 2005, 18, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S. The role of glutamate in epilepsy and other CNS disorders. Neurology 1994, 44, S14–S23. [Google Scholar] [PubMed]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Morimoto, K.; Hirao, T.; Suwaki, H.; Watase, K.; Tanaka, K. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter glast-deficient mice. Brain Res. 1999, 845, 92–96. [Google Scholar] [CrossRef]
- Sepkuty, J.P.; Cohen, A.S.; Eccles, C.; Rafiq, A.; Behar, K.; Ganel, R.; Coulter, D.A.; Rothstein, J.D. A neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J. Neurosci. 2002, 22, 6372–6379. [Google Scholar] [PubMed]
- Simantov, R.; Liu, W.; Broutman, G.; Baudry, M. Antisense knockdown of glutamate transporters alters the subfield selectivity of kainate-induced cell death in rat hippocampal slice cultures. J. Neurochem. 1999, 73, 1828–1835. [Google Scholar] [PubMed]
- Furuta, A.; Noda, M.; Suzuki, S.O.; Goto, Y.; Kanahori, Y.; Rothstein, J.D.; Iwaki, T. Translocation of glutamate transporter subtype excitatory amino acid carrier 1 protein in kainic acid-induced rat epilepsy. Am. J. Pathol. 2003, 163, 779–787. [Google Scholar] [CrossRef]
- Miller, H.P.; Levey, A.I.; Rothstein, J.D.; Tzingounis, A.V.; Conn, P.J. Alterations in glutamate transporter protein levels in kindling-induced epilepsy. J. Neurochem. 1997, 68, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B.; Jin, H.; Shumate, M.D.; Robinson, M.B.; Coulter, D.A.; Brooks-Kayal, A.R. Increased expression of the neuronal glutamate transporter (EAAT3/EAAC1) in hippocampal and neocortical epilepsy. Epilepsia 2002, 43, 211–218. [Google Scholar] [CrossRef] [PubMed]
- During, M.J.; Spencer, D.D. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993, 341, 1607–1610. [Google Scholar] [CrossRef]
- Tessler, S.; Danbolt, N.C.; Faull, R.L.; Storm-Mathisen, J.; Emson, P.C. Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience 1999, 88, 1083–1091. [Google Scholar] [CrossRef]
- Masliah, E.; Alford, M.; deTeresa, R.; Mallory, M.; Hansen, L. Deficient glutamate transport is associated with neurodegeneration in alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Raber, J.; Alford, M.; Mallory, M.; Mattson, M.P.; Yang, D.; Wong, D.; Mucke, L. Amyloid protein precursor stimulates excitatory amino acid transport. Implications for roles in neuroprotection and pathogenesis. J. Biol. Chem. 1998, 273, 12548–12554. [Google Scholar] [CrossRef] [PubMed]
- Ikegaya, Y.; Matsuura, S.; Ueno, S.; Baba, A.; Yamada, M.K.; Nishiyama, N.; Matsuki, N. Beta-amyloid enhances glial glutamate uptake activity and attenuates synaptic efficacy. J. Biol. Chem. 2002, 277, 32180–32186. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Nakanishi, H.; Akaike, N. Glutamate release from microglia via glutamate transporter is enhanced by amyloid-beta peptide. Neuroscience 1999, 92, 1465–1474. [Google Scholar] [CrossRef]
- Scott, H.L.; Pow, D.V.; Tannenberg, A.E.G.; Dodd, P.R. Aberrant expression of the glutamate transporter excitatory amino acid transporter 1 (EAAT1) in alzheimer’s disease. J. Neurosci. 2002, 22, RC206. [Google Scholar] [PubMed]
- Thai, D.R. Excitatory amino acid transporter EAAT-2 in tangle-bearing neurons in Alzheimer’s disease. Brain Pathol. 2002, 12, 405–411. [Google Scholar] [PubMed]
- Parsons, C.G.; Danysz, W.; Dekundy, A.; Pulte, I. Memantine and cholinesterase inhibitors: Complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox. Res. 2013, 24, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Mangano, R.M.; Schwarcz, R. Platelet glutamate and aspartate uptake in Huntington’s disease. J. Neurochem. 1981, 37, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Hazell, A.S.; Itzhak, Y.; Liu, H.; Norenberg, M.D. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) decreases glutamate uptake in cultured astrocytes. J. Neurochem. 1997, 68, 2216–2219. [Google Scholar] [CrossRef] [PubMed]
- Liévens, J.C.; Salin, P.; Had-Aissouni, L.; Mahy, N.; Kerkerian-Le Goff, L. Differential effects of corticostriatal and thalamostriatal deafferentation on expression of the glutamate transporter GLT1 in the rat striatum. J. Neurochem. 2000, 74, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Spencer, P.S.; Nunn, P.B.; Hugon, J.; Ludolph, A.C.; Ross, S.M.; Roy, D.N.; Robertson, R.C. Guam amyotrophic lateral sclerosis-parkinsonism-dementia linked to a plant excitant neurotoxin. Science 1987, 237, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Siddique, T.; Figlewicz, D.A.; Pericak-Vance, M.A.; Haines, J.L.; Rouleau, G.; Jeffers, A.J.; Sapp, P.; Hung, W.Y.; Bebout, J.; McKenna-Yasek, D. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N. Engl. J. Med. 1991, 324, 1381–1384. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Tsai, G.; Kuncl, R.W.; Clawson, L.; Cornblath, D.R.; Drachman, D.B.; Pestronk, A.; Stauch, B.L.; Coyle, J.T. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 1990, 28, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321. [Google Scholar] [PubMed]
- Torp, R.; Lekieffre, D.; Levy, L.M.; Haug, F.M.; Danbolt, N.C.; Meldrum, B.S.; Ottersen, O.P. Reduced postischemic expression of a glial glutamate transporter, glt1, in the rat hippocampus. Exp. Brain Res. 1995, 103, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, M.; Domercq, M.; Matute, C. Altered expression of the glutamate transporter eaac1 in neurons and immature oligodendrocytes after transient forebrain ischemia. J. Cereb. Blood Flow Metab. 2000, 20, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Ikematsu, K.; Tsuda, R.; Orihara, Y.; Nakasono, I. The expression of excitatory amino acid transporter 2 (EAAT2) in forensic autopsy cases. Forensic Sci. Int. 2001, 118, 49–55. [Google Scholar] [CrossRef]
- Lopez-Valdes, H.E.; Clarkson, A.N.; Ao, Y.; Charles, A.C.; Carmichael, S.T.; Sofroniew, M.V.; Brennan, K.C. Memantine enhances recovery from stroke. Stroke 2014, 45, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Mohammad-Gharibani, P.; Modi, J.; Menzie, J.; Genova, R.; Ma, Z.; Tao, R.; Prentice, H.; Wu, J.Y. Mode of action of S-methyl-N,N-diethylthiocarbamate sulfoxide (DETC-MeSO) as a novel therapy for stroke in a rat model. Mol. Neurobiol. 2014, 50, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncology 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Roesler, R.; Brunetto, A.T.; Abujamra, A.L.; de Farias, C.B.; Brunetto, A.L.; Schwartsmann, G. Current and emerging molecular targets in glioma. Expert Rev. Anticancer Ther. 2010, 10, 1735–1751. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 who classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.; Sontheimer, H. Glutamate and the biology of gliomas. Glia 2011, 59, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Gochenauer, G.E.; Robinson, M.B. Dibutyryl-cAMP (dbcAMP) up-regulates astrocytic chloride-dependent l-[3h]glutamate transport and expression of both system xc(−) subunits. J. Neurochem. 2001, 78, 276–286. [Google Scholar] [CrossRef] [PubMed]
- McBean, G.J.; Flynn, J. Molecular mechanisms of cystine transport. Biochem. Soc. Trans. 2001, 29, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.M.; Sontheimer, H. Glutamate transporters in the biology of malignant gliomas. Cell. Mol. Life Sci. 2014, 71, 1839–1854. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, H. Ion channels and amino acid transporters support the growth and invasion of primary brain tumors. Mol. Neurobiol. 2004, 29, 61–71. [Google Scholar] [CrossRef]
- Sontheimer, H. A role for glutamate in growth and invasion of primary brain tumors. J. Neurochem. 2008, 105, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Willard, S.S.; Koochekpour, S. Glutamate signaling in benign and malignant disorders: Current status, future perspectives, and therapeutic implications. Int. J. Biol. Sci. 2013, 9, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Negishi, K.; Mawatari, K.; Kuo, C.H. A mechanism for glutamate toxicity in the C6 glioma cells involving inhibition of cystine uptake leading to glutathione depletion. Neuroscience 1992, 48, 903–914. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kanai, Y.; Chairoungdua, A.; Cha, S.H.; Matsuo, H.; Kim, D.K.; Inatomi, J.; Sawa, H.; Ida, Y.; Endou, H. Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochim. Biophys. Acta 2001, 1512, 335–344. [Google Scholar] [CrossRef]
- Sontheimer, H. Malignant gliomas: Perverting glutamate and ion homeostasis for selective advantage. Trends Neurosci. 2003, 26, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.K.; Seidlitz, E.P.; Singh, G. Cancer cells release glutamate via the cystine/glutamate antiporter. Biochem. Biophys. Res. Commun. 2010, 391, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Seidlitz, E.P.; Sharma, M.K.; Saikali, Z.; Ghert, M.; Singh, G. Cancer cell lines release glutamate into the extracellular environment. Clin. Exp. Metastasis 2009, 26, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.J.; Lyons, S.A.; Nelson, G.M.; Hamza, H.; Gladson, C.L.; Gillespie, G.Y.; Sontheimer, H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 2005, 25, 7101–7110. [Google Scholar] [CrossRef] [PubMed]
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007, 67, 9463–9471. [Google Scholar] [CrossRef] [PubMed]
- Liubinas, S.V.; O’Brien, T.J.; Moffat, B.M.; Drummond, K.J.; Morokoff, A.P.; Kaye, A.H. Tumour associated epilepsy and glutamate excitotoxicity in patients with gliomas. J. Clin. Neurosci. 2014, 21, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Chizh, B.A. Novel approaches to targeting glutamate receptors for the treatment of chronic pain: Review article. Amino Acids 2002, 23, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Sandkühler, J. Models and mechanisms of hyperalgesia and allodynia. Physiol. Rev. 2009, 89, 707–758. [Google Scholar] [CrossRef] [PubMed]
- Chizh, B.A.; Schlütz, H.; Scheede, M.; Englberger, W. The N-methyl-d-aspartate antagonistic and opioid components of d-methadone antinociception in the rat spinal cord. Neurosci. Lett. 2000, 296, 117–120. [Google Scholar] [CrossRef]
- Davidson, E.M.; Coggeshall, R.E.; Carlton, S.M. Peripheral NMDA and non-NMDA glutamate receptors contribute to nociceptive behaviors in the rat formalin test. Neuroreport 1997, 8, 941–946. [Google Scholar] [CrossRef] [PubMed]
- McRoberts, J.A.; Coutinho, S.V.; Marvizón, J.C.; Grady, E.F.; Tognetto, M.; Sengupta, J.N.; Ennes, H.S.; Chaban, V.V.; Amadesi, S.; Creminon, C.; et al. Role of peripheral N-methyl-d-aspartate (NMDA) receptors in visceral nociception in rats. Gastroenterology 2001, 120, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Wallace, M.S.; Ridgeway, B.; Yaksh, T. Concentration-effect relationship of intravenous alfentanil and ketamine on peripheral neurosensory thresholds, allodynia and hyperalgesia of neuropathic pain. Pain 2001, 91, 177–187. [Google Scholar] [CrossRef]
- Caplette-Gingras, A.; Savard, J. Depression in women with metastatic breast cancer: A review of the literature. Palliat. Support. Care 2008, 6, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Litofsky, N.S.; Farace, E.; Anderson, F., Jr.; Meyers, C.A.; Huang, W.; Laws, E.R., Jr. Depression in patients with high-grade glioma: Results of the glioma outcomes project. Neurosurgery 2004, 54, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Green, A.I.; Austin, C.P. Psychopathology of pancreatic cancer. A psychobiologic probe. Psychosomatics 1993, 34, 208–221. [Google Scholar] [CrossRef]
- Jacobsson, L.; Ottosson, J.O. Initial mental disorders in carcinoma of pancreas and stomach. Acta Psychiatr. Scand. Suppl. 1971, 221, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Passik, S.D.; Roth, A.J. Anxiety symptoms and panic attacks preceding pancreatic cancer diagnosis. Psycho-Oncology 1999, 8, 268–272. [Google Scholar] [CrossRef]
- Van Esch, L.; Roukema, J.A.; Ernst, M.F.; Nieuwenhuijzen, G.A.; de Vries, J. Combined anxiety and depressive symptoms before diagnosis of breast cancer. J. Affect. Disord. 2012, 136, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Jehn, C.F.; Kuehnhardt, D.; Bartholomae, A.; Pfeiffer, S.; Krebs, M.; Regierer, A.C.; Schmid, P.; Possinger, K.; Flath, B.C. Biomarkers of depression in cancer patients. Cancer 2006, 107, 2723–2729. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Lamkin, D.M.; Lutgendorf, S.K.; Lubaroff, D.; Sood, A.K.; Beltz, T.G.; Johnson, A.K. Cancer induces inflammation and depressive-like behavior in the mouse: Modulation by social housing. Brain Behav. Immun. 2011, 25, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, S.; Furst, C.J. Symptoms in advanced cancer: Relationship to endogenous cortisol levels. Palliat. Med. 2003, 17, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Pariante, C.M.; Lightman, S.L. The HPA axis in major depression: Classical theories and new developments. Trends Neurosci. 2008, 31, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Bentea, E.; Demuyser, T.; van Liefferinge, J.; Albertini, G.; Deneyer, L.; Nys, J.; Merckx, E.; Michotte, Y.; Sato, H.; Arckens, L.; et al. Absence of system xc− in mice decreases anxiety and depressive-like behavior without affecting sensorimotor function or spatial vision. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 59C, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.A. The blood-brain barrier and glutamate. Am. J. Clin. Nutr. 2009, 90, 867S–874S. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.Y.; Greco, S.J.; Patel, P.S.; Trzaska, K.A.; Rameshwar, P. Re-1-silencing transcription factor shows tumor-suppressor functions and negatively regulates the oncogenic TAC1 in breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 4408–4413. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.L.; Jiang, S.; Fu, Y.; Avraham, S.; Avraham, H.K. The proinflammatory peptide substance p promotes blood-brain barrier breaching by breast cancer cells through changes in microvascular endothelial cell tight junctions. Int. J. Cancer 2014, 134, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Cochaud, S.; Giustiniani, J.; Thomas, C.; Laprevotte, E.; Garbar, C.; Savoye, A.M.; Cure, H.; Mascaux, C.; Alberici, G.; Bonnefoy, N.; et al. IL-17A is produced by breast cancer TILs and promotes chemoresistance and proliferation through ERK1/2. Sci. Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.D.; Witt, K.A.; Hom, S.; Egleton, R.D.; Mark, K.S.; Davis, T.P. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1241–H1248. [Google Scholar] [PubMed]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Guise, T. Examining the metastatic Niche: Targeting the microenvironment. Semin. Oncol. 2010, 37, S2–S14. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Seidlitz, E.P.; Sharma, M.K.; Singh, G. A by-product of glutathione production in cancer cells may cause disruption in bone metabolic processes. Can. J. Physiol. Pharmacol. 2010, 88, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Skerry, T.M. The role of glutamate in the regulation of bone mass and architecture. J. Musculoskelet. Neuronal Interact. 2008, 8, 166–173. [Google Scholar] [PubMed]
- Seidlitz, E.P.; Sharma, M.K.; Singh, G. Extracellular glutamate alters mature osteoclast and osteoblast functions. Can. J. Physiol. Pharmacol. 2010, 88, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Cowan, R.W.; Seidlitz, E.P.; Singh, G. Glutamate signaling in healthy and diseased bone. Front. Endocrinol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Takarada, T.; Yoneda, Y. Pharmacological topics of bone metabolism: Glutamate as a signal mediator in bone. J. Pharmacol. Sci. 2008, 106, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.A.N.; Siegel, P.M. Breast cancer-derived factors facilitate osteolytic bone metastasis. Bull. Cancer 2006, 93, 931–943. [Google Scholar] [PubMed]
- Coleman, R.E.; Lipton, A.; Roodman, G.D.; Guise, T.A.; Boyce, B.F.; Brufsky, A.M.; Clézardin, P.; Croucher, P.I.; Gralow, J.R.; Hadji, P.; et al. Metastasis and bone loss: Advancing treatment and prevention. Cancer Treat. Rev. 2010, 36, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Chaisson, M. The RANK/RANKL/OPG triad in cancer-induced bone diseases. Cancer Metastasis Rev. 2006, 25, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Serre, C.M.; Farlay, D.; Delmas, P.D.; Chenu, C. Evidence for a dense and intimate innervation of the bone tissue, including glutamate-containing fibers. Bone 1999, 25, 623–629. [Google Scholar] [CrossRef]
- Cairns, B.E.; Svensson, P.; Wang, K.; Castrillon, E.; Hupfeld, S.; Sessle, B.J.; Arendt-Nielsen, L. Ketamine attenuates glutamate-induced mechanical sensitization of the masseter muscle in human males. Exp. Brain Res. 2006, 169, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Meotti, F.C.; Coelho Idos, S.; Santos, A.R. The nociception induced by glutamate in mice is potentiated by protons released into the solution. J. Pain 2010, 11, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.X.; Gu, X.P.; Zheng, Y.G.; Liu, C.L.; Wang, D.; Sun, Y.E.; Ma, Z.L. Intrathecal injection of metabotropic glutamate receptor subtype 3 and 5 agonist/antagonist attenuates bone cancer pain by inhibition of spinal astrocyte activation in a mouse model. Anesthesiology 2012, 116, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhang, J.; Ma, Z.; Wang, J.; Zhou, X.; Jin, Y.; Xia, X.; Gao, Q.; Mei, F. The role of N-methyl-d-aspartate receptor subunit NR2B in spinal cord in cancer pain. Eur. J. Pain 2010, 14, 496–502. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miladinovic, T.; Nashed, M.G.; Singh, G. Overview of Glutamatergic Dysregulation in Central Pathologies. Biomolecules 2015, 5, 3112-3141. https://doi.org/10.3390/biom5043112
Miladinovic T, Nashed MG, Singh G. Overview of Glutamatergic Dysregulation in Central Pathologies. Biomolecules. 2015; 5(4):3112-3141. https://doi.org/10.3390/biom5043112
Chicago/Turabian StyleMiladinovic, Tanya, Mina G. Nashed, and Gurmit Singh. 2015. "Overview of Glutamatergic Dysregulation in Central Pathologies" Biomolecules 5, no. 4: 3112-3141. https://doi.org/10.3390/biom5043112