
Molecular Interactions between NR4A Orphan Nuclear Receptors and NF-κB Are Required for Appropriate Inflammatory Responses and Immune Cell Homeostasis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction: Appropriate Inflammation and NF-κB Activity
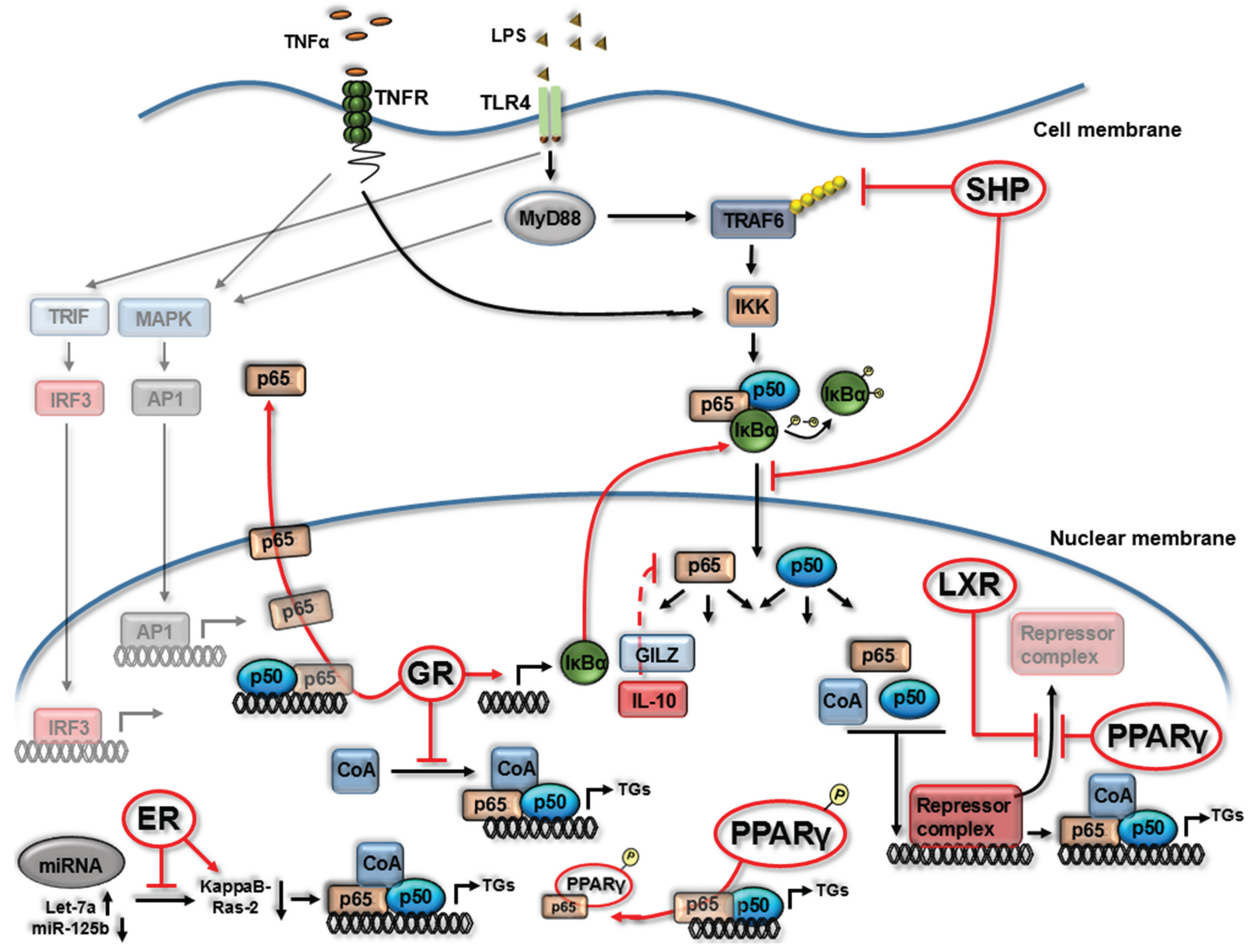
2. Nuclear Receptor (NR) Modulation of NF-κB Signaling
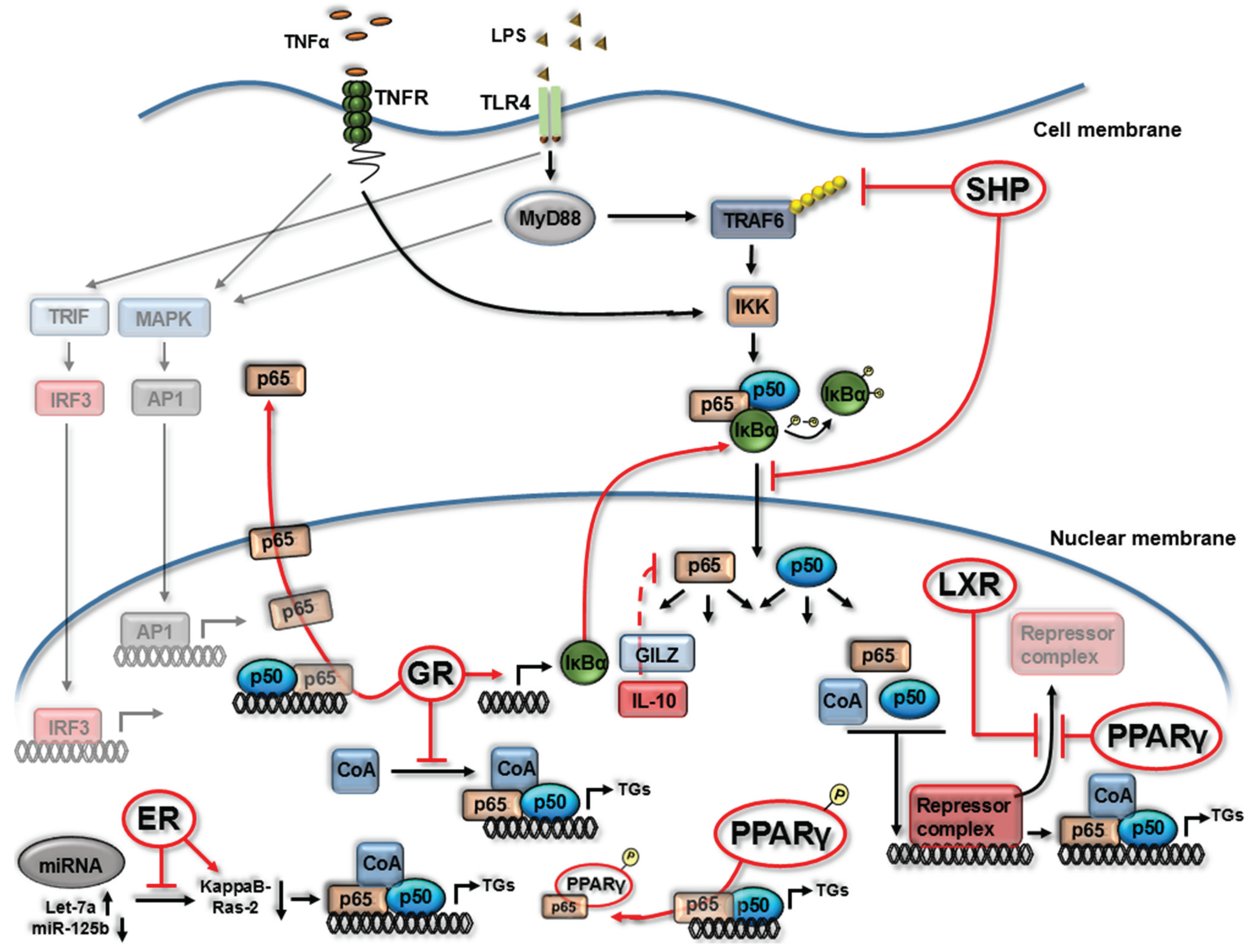
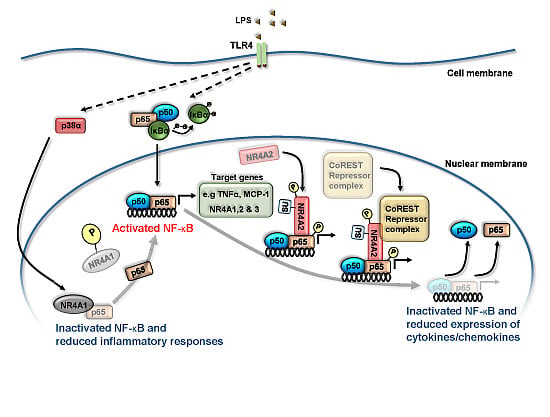
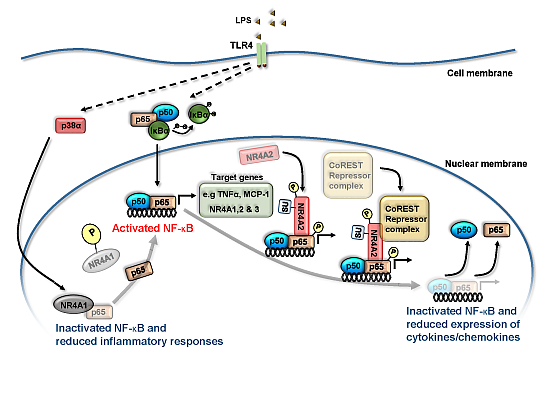
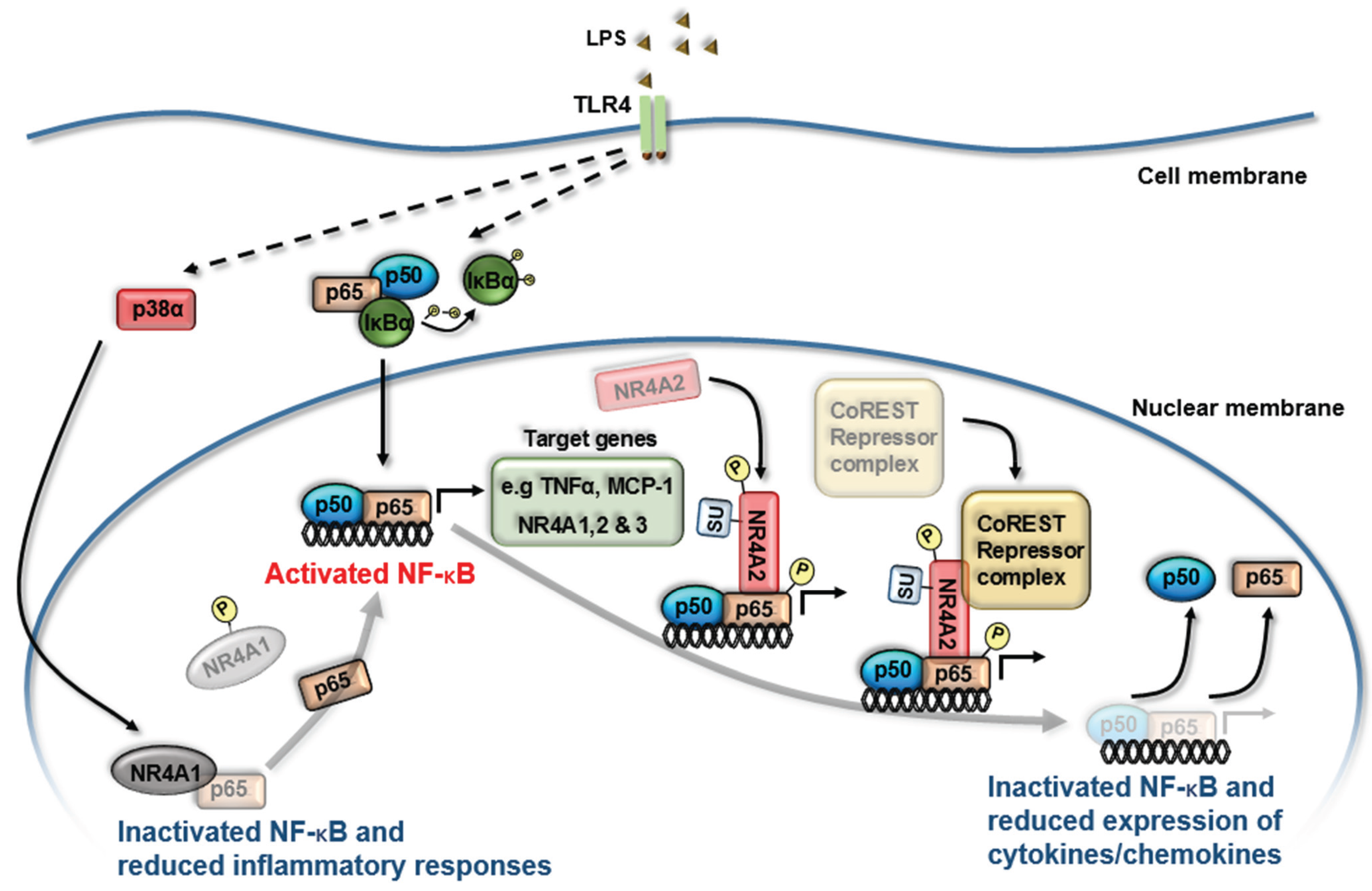
3. NR4A Nuclear Receptors and NF-κB Signaling
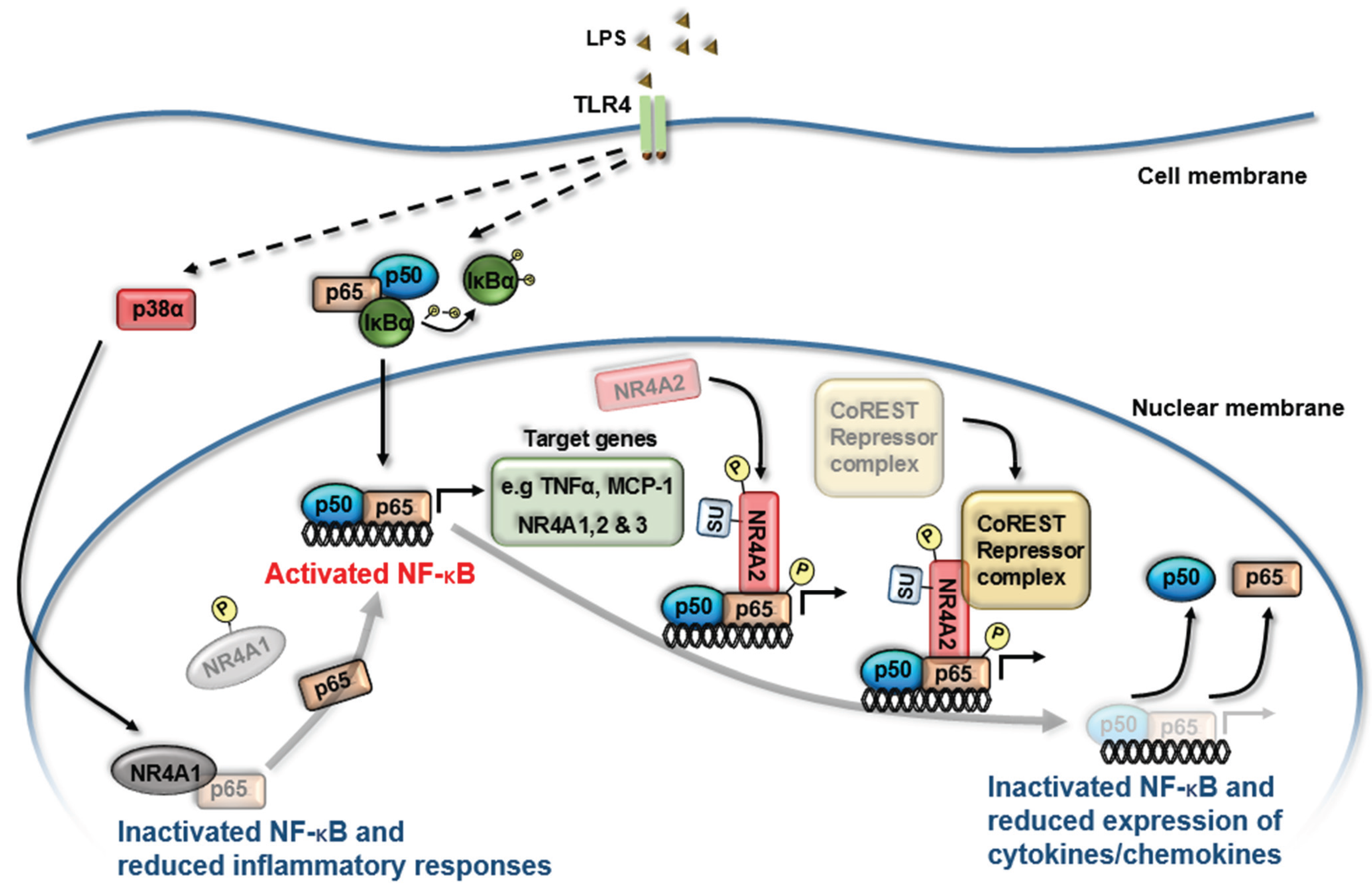
4. NR4A Control of Acute Inflammation, Inflammatory Resolution and Tissue Repair
4.1. NR4A1 is Crucial for the Development of Ly6Clow Reparative Monocytes and Has an Anti-Inflammatory Effect in Monocyte/Macrophage Cells
4.2. Generation and Maintenance of Regulatory T Cells by NR4A Receptors
4.3. Role of NR4A Receptors in Maintaining Endothelial Cell Homeostasis
4.4. NR4A Receptors Mediate Growth Factor and Cytokine Responses in Mesenchymal Cells
5. Therapeutic Potential of Altering NR4A/NF-κB Interactions to Control Inflammation
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Glass, C.K. Nuclear receptors and inflammation control: Molecular mechanisms and pathophysiological relevance. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.; Godson, C. Lipoxins: Regulators of resolution. Curr. Opin. Pharmacol. 2010, 10, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Maderna, P.; Godson, C. Lipoxins: Resolutionary road. Br. J. Pharmacol. 2009, 158, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Bellini, A.; Mattoli, S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab. Invest. 2007, 87, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Gilroy, D.W.; Colville-Nash, P.R.; Willoughby, D.A. Possible new role for NF-kappaB in the resolution of inflammation. Nat. Med. 2001, 7, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- D’Acquisto, F.; May, M.J.; Ghosh, S. Inhibition of nuclear factor kappa B (NF-B): An emerging theme in anti-inflammatory therapies. Mol. Interv. 2002, 2, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Scheinman, R.I.; Cogswell, P.C.; Lofquist, A.K.; Baldwin, A.S., Jr. Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science 1995, 270, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Crinelli, R.; Antonelli, A.; Bianchi, M.; Gentilini, L.; Scaramucci, S.; Magnani, M. Selective inhibition of NF-κB activation and TNF-alpha production in macrophages by red blood cell-mediated delivery of dexamethasone. Blood Cells Mol. Dis. 2000, 26, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Driessler, F.; Venstrom, K.; Sabat, R.; Asadullah, K.; Schottelius, A.J. Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: A role for p50. Clin. Exp. Immunol. 2004, 135, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, P.; Siegel, M.I.; Egan, R.W.; Billah, M.M. Interleukin (IL)10 inhibits nuclear factor kappa B (NF kappa B) activation in human monocytes. IL-10 and IL-4suppress cytokine synthesis by different mechanisms. J. Biol. Chem. 1995, 270, 9558–9563. [Google Scholar] [PubMed]
- Berrebi, D.; Bruscoli, S.; Cohen, N.; Foussat, A.; Migliorati, G.; Bouchet-Delbos, L.; Maillot, M.C.; Portier, A.; Couderc, J.; Galanaud, P.; et al. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: An anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood 2003, 101, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Wilde, G.J.; Spiller, D.G.; Kennedy, S.M.; Ray, D.W.; Sullivan, E.; Unitt, J.F.; White, M.R. NF-kappaB signalling is inhibited by glucocorticoid receptor and STAT6 via distinct mechanisms. J. Cell Sci. 2003, 116, 2495–2503. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Tontonoz, P. Liver X receptor signaling pathways and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Ghisletti, S.; Huang, W.; Jepsen, K.; Benner, C.; Hardiman, G.; Rosenfeld, M.G.; Glass, C.K. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 2009, 23, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Hiroshima, A.; Ariga, A.; Honzumi, S.; Koieyama, T.; Inaba, T.; Fujiwara, T. Liver X receptor agonists inhibit tissue factor expression in macrophages. FEBS J. 2005, 272, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, A.; Joseph, S.B.; Marathe, C.; Mangelsdorf, D.J.; Tontonoz, P. Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J. Biol. Chem. 2003, 278, 10443–10449. [Google Scholar] [CrossRef] [PubMed]
- Töröcsik, D.; Baráth, M.; Benko, S.; Széles, L.; Dezso, B.; Póliska, S.; Hegyi, Z.; Homolya, L.; Szatmári, I.; Lányi, A.; et al. Activation of liver X receptor sensitizes human dendritic cells to inflammatory stimuli. J. Immunol. 2010, 184, 5456–5465. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Kim, J.J.; Kim, S.W.; Jin, H.S.; Yang, C.S.; Park, K.A.; Chanda, D.; Kim, D.K.; et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 2011, 12, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Guyre, P.M.; Pioli, P.A. Estradiol suppresses NF-kappa B activation through coordinated regulation of let-7a and miR-125b in primary human macrophages. J. Immunol. 2010, 184, 5029–5037. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bruemmer, D. NR4A orphan nuclear receptors: Transcriptional regulators of gene expression in metabolism and vascular biology. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- McMorrow, J.P.; Murphy, E.P. Inflammation: A role for NR4A orphan nuclear receptors? Biochem. Soc. Trans. 2011, 39, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; Liu, Y.; Zhao, Y.; Aono, J.; Jones, K.L.; Heywood, E.B.; Howatt, D.; Binkley, C.M.; Daugherty, A.; Liang, Y.; et al. Deficiency of the NR4A orphan nuclear receptor NOR1 in hematopoietic stem cells accelerates atherosclerosis. Stem Cells 2014, 32, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Jin, U.H.; Hedrick, E.; Reeder, A.; Lee, S.O. Minireview: Role of orphan nuclear receptors in cancer and potential as drug targets. Mol. Endocrinol. 2014, 28, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Pearen, M.A.; Muscat, G.E. Minireview: Nuclear hormone receptor 4A signaling: Implications for metabolic disease. Mol. Endocrinol. 2010, 24, 1891–1903. [Google Scholar] [CrossRef] [PubMed]
- Mohan, H.M.; Aherne, C.M.; Rogers, A.C.; Baird, A.W.; Winter, D.C.; Murphy, E.P. Molecular pathways: The role of NR4A orphan nuclear receptors in cancer. Clin. Cancer Res. 2012, 18, 3223–3228. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Castrillo, A.; Chen, M.; Hoffmann, A.; Tontonoz, P. Induction of NR4A orphan nuclear receptor expression in macrophages in response to inflammatory stimuli. J. Biol. Chem. 2005, 280, 29256–29262. [Google Scholar] [CrossRef] [PubMed]
- Bonta, P.I.; van Tiel, C.M.; Vos, M.; Pols, T.W.; van Thienen, J.V.; Ferreira, V.; Arkenbout, E.K.; Seppen, J.; Spek, C.A.; van der Poll, T.; et al. Nuclear receptors Nur77, Nurr1, and NOR-1 expressed in atherosclerotic lesion macrophages reduce lipid loading and inflammatory responses. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2288–2294. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.N.; Carlin, L.M.; Hubbeling, H.G.; Nackiewicz, D.; Green, A.M.; Punt, J.A.; Geissmann, F.; Hedrick, C.C. The transcriptional factor NR4A1 controls bone marrow differentiation and the survival of Ly6C-monocytes. Nat. Immunol. 2011, 12, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.N.; Shaked, I.; Hubbeling, H.G.; Punt, J.A.; Wu, R.; Herrley, E.; Zaugg, C.; Pei, H.; Geissmann, F.; Ley, K.; et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ. Res. 2012, 110, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Hamers, A.A.; Vos, M.; Rassam, F.; Marinković, G.; Kurakula, K.; van Gorp, P.J.; de Winther, M.P.; Gijbels, M.J.; de Waard, V.; de Vries, C.J. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ. Res. 2012, 110, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Carlin, L.M.; Stamatiades, E.G.; Auffray, C.; Hanna, R.N.; Glover, L.; Vizcay-Barrena, G.; Hedrick, C.C.; Cook, H.T.; Diebold, S.; Geissmann, F. Nr4a1-dependent Ly6Clow monocytes monitor endothelial cells and orchestrate their disposal. Cell 2013, 153, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Winner, B.; Carson, C.T.; Collier, J.G.; Boyer, L.; Rosenfeld, M.G.; Gage, F.H.; Glass, C.K. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009, 137, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Saini, A.; Chandra, V.; Nanduri, R.; Kalra, R.; Bhagyaraj, E.; Khatri, N.; Gupta, P. Nuclear receptor Nr4a2 promotes alternative polarization of macrophages and confer protection in sepsis. J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- De Paoli, F.; Eeckhoute, J.; Copin, C.; Vanhoutte, J.; Duhem, C.; Derudas, B.; Dubois-Chevalier, J.; Colin, S.; Zawadzki, C.; Jude, B.; et al. The neuron-derived orphan receptor 1 (NOR1) is induced upon human alternative macrophage polarization and stimulates the expression of markers of the M2 phenotype. Atherosclerosis 2015, 241, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Crean, D.; Cummins, E.P.; Bahar, B.; Mohan, H.M.; McMorrow, J.P.; Murphy, E.P. Adenosine modulates NR4A orphan nuclear receptors to attenuate hyper-inflammatory responses in monocytic cells. J. Immunol. 2015, in press. [Google Scholar]
- Li, L.; Liu, Y.; Chen, H.Z.; Li, F.W.; Wu, J.F.; Zhang, H.K.; He, J.P.; Xing, Y.Z.; Chen, Y.; Wang, W.J.; et al. Impeding the interaction between Nur77 and p38 reduces LPS-induced inflammation. Nat. Chem. Biol. 2015, 11, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Castrillo, A.; Tontonoz, P. Regulation of macrophage inflammatory gene expression by the orphan nuclear receptor Nur77. Mol. Endocrinol. 2006, 20, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Arkenbout, E.K.; de Waard, V.; van Bragt, M.; van Achterberg, T.A.; Grimbergen, J.M.; Pichon, B.; Pannekoek, H.; de Vries, C.J. Protective function of transcription factor TR3 orphan receptor in atherogenesis: Decreased lesion formation in carotid artery ligation model in TR3 transgenic mice. Circulation 2002, 106, 1530–1535. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Calvo, R.; Guadall, A.; Calvayrac, O.; Navarro, M.A.; Alonso, J.; Ferrán, B.; de Diego, A.; Muniesa, P.; Osada, J.; Rodríguez, C.; et al. Over-expression of neuron-derived orphan receptor-1 (NOR-1) exacerbates neointimal hyperplasia after vascular injury. Hum. Mol. Genet. 2013, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
- Calvayrac, O.; Rodríguez-Calvo, R.; Martí-Pamies, I.; Alonso, J.; Ferrán, B.; Aguiló, S.; Crespo, J.; Rodríguez-Sinovas, A.; Rodríguez, C.; Martínez-González, J. NOR-1 modulates the inflammatory response of vascular smooth muscle cells by preventing NFκB activation. J. Mol. Cell. Cardiol. 2015, 80, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Calvo, R.; Ferrán, B.; Alonso, J.; Martí-Pàmies, I.; Aguiló, S.; Calvayrac, O.; Rodríguez, C.; Martínez-González, J. NR4A receptors up-regulate the antiproteinase alpha-2 macroglobulin (A2M) and modulate MMP-2 and MMP-9 in vascular smooth muscle cells. Thromb. Haemost. 2015, 113, 1323–1324. [Google Scholar] [CrossRef] [PubMed]
- Nomiyama, T.; Zhao, Y.; Gizard, F.; Findeisen, H.M.; Heywood, E.B.; Jones, K.L.; Conneely, O.M.; Bruemmer, D. Deficiency of the NR4A neuron-derived orphan receptor-1 attenuates neointima formation after vascular injury. Circulation 2009, 119, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, T.; Schulz, C. Myocardial infarction: A critical role of macrophages in cardiac remodeling. Front. Physiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Dewald, O.; Frangogiannis, N.G. Inflammatory mechanisms in myocardial infarction. Curr. Drug Targets Inflamm. Allergy 2003, 2, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorf, I.; Gerhardt, L.M.; Tan, T.C.; Winter, C.; Holderried, T.A.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; Hedrick, C.C.; et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-κB in the heart: To be or not to NF-κB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.L.; Leung, K.K.; Bennett, M.J.; Winoto1, A. T cell-specific inhibition of multiple apoptotic pathways blocks negative selection and causes autoimmunity. eLife 2014, 3, e03468. [Google Scholar] [CrossRef] [PubMed]
- Harant, H.; Lindley, I.J. Negative cross-talk between the human orphan nuclear receptor Nur77/NAK-1/TR3 and nuclear factor-kappaB. Nucleic Acids Res. 2004, 32, 5280–5290. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, T.; Kashiwagi, I.; Yoshida, R.; Fukaya, T.; Morita, R.; Kimura, A.; Ichinose, H.; Metzger, D.; Chambon, P.; Yoshimura, A. Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat. Immunol. 2013, 14, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, T.; Kashiwagi, I.; Inoue, N.; Morita, R.; Hori, S.; Waldmann, H.; Rudensky, A.Y.; Ichinose, H.; Metzger, D.; Chambon, P.; et al. The nuclear orphan receptor Nr4a2 induces Foxp3 and regulates differentiation of CD4+ T cells. Nat. Commun. 2011. [Google Scholar] [CrossRef] [PubMed]
- Nowyhed, H.N.; Huynh, T.R.; Blatchley, A.; Wu, R.; Thomas, G.D.; Hedrick, C.C. The nuclear receptor Nr4a1 controls CD8 T cell development through transcriptional suppression of Runx3. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Muto, G.; Kotani, H.; Kondo, T.; Morita, R.; Tsuruta, S.; Kobayashi, T.; Luche, H.; Fehling, H.J.; Walsh, M.; Choi, Y.; Yoshimura, A. TRAF6 is essential for maintenance of regulatory T cells that suppress Th2 type autoimmunity. PLoS ONE 2013, 8, e74639. [Google Scholar] [CrossRef] [PubMed]
- You, B.; Jiang, Y.Y.; Chen, S.; Yan, G.; Sun, J. The orphan nuclear receptor Nur77 suppresses endothelial cell activation through induction of IkappaB alpha expression. Circ. Res. 2009, 104, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Hamers, A.A.; Uleman, S.; van Tiel, C.M.; Kruijswijk, D.; van Stalborch, A.M.; Huveneers, S.; de Vries, C.J.; van’t Veer, C. Limited role of nuclear receptor Nur77 in Escherichia coli-induced peritonitis. Infect. Immun. 2014, 82, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Qin, L.; Bourbon, P.M.; James, L.; Dvorak, H.F.; Zeng, H. Orphan nuclear transcription factor TR3/Nur77 regulates microvessel permeability by targeting endothelial nitricoxide synthase and destabilizing endothelial junctions. Proc. Natl. Acad. Sci. USA 2011, 108, 12066–12071. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Qin, L.; Zhao, D.; Tan, X.; Manseau, E.J.; van Hoang, M.; Senger, D.R.; Brown, L.F.; Nagy, J.A.; Dvorak, H.F. Orphan nuclear receptor TR3/Nur77 regulates VEGF-A-induced angiogenesis through its transcriptional activity. J. Exp. Med. 2006, 203, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Desai, S.; Zeng, H. VEGF stimulates PKD-mediated CREB-dependent orphan nuclear receptor Nurr1 expression: Role in VEGF-induced angiogenesis. Int. J. Cancer 2011, 128, 2602–2612. [Google Scholar] [CrossRef] [PubMed]
- McMorrow, J.P.; Crean, D.; Gogarty, M.; Smyth, A.; Connolly, M.; Cummins, E.; Veale, D.; Fearon, U.; Tak, P.P.; Fitzgerald, O.; et al. Tumor necrosis factor inhibition modulates thrombospondin-1 expression in human inflammatory joint disease through altered NR4A2 activity. Am. J. Pathol. 2013, 183, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Kaipainen, A.; Kieran, M.W.; Huang, S.; Butterfield, C.; Bielenberg, D.; Mostoslavsky, G.; Mulligan, R.; Folkman, J.; Panigrahy, D. PPARalpha deficiency in inflammatory cells suppresses tumor growth. PLoS ONE 2007, 2, e260. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Campbell, S.C.; Bedford, D.F.; Nelius, T.; Veliceasa, D.; Shroff, E.H.; Henkin, J.; Schneider, A.; Bouck, N.; Volpert, O.V. Peroxisome proliferator-activated receptor gamma ligands improve the antitumor efficacy of thrombospondin peptide ABT510. Mol. Cancer Res. 2004, 2, 541–550. [Google Scholar] [PubMed]
- Palumbo-Zerr, K.; Zerr, P.; Distler, A.; Fliehr, J.; Mancuso, R.; Huang, J.; Mielenz, D.; Tomcik, M.; Fürnrohr, B.G.; Scholtysek, C.; et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-β signaling and fibrosis. Nat. Med. 2015, 21, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Ye, T.; Qin, L.; Bourbon, P.M.; Chang, C.; Zhao, S.; Li, Y.; Zhou, L.; Cui, P.; Rabinovitz, I.; et al. Orphan nuclear receptor TR3/Nur77 improves wound healing by upregulating the expression of integrin β4. FASEB J. 2015, 29, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Mix, K.S.; McMahon, K.; McMorrow, J.P.; Walkenhorst, D.E.; Smyth, A.M.; Petrella, B.L.; Gogarty, M.; Fearon, U.; Veale, D.; Attur, M.G.; et al. Orphan nuclear receptor NR4A2 induces synoviocyte proliferation, invasion, and matrix metalloproteinase 13 transcription. Arthritis Rheum. 2012, 64, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Aherne, C.M.; McMorrow, J.; Kane, D.; FitzGerald, O.; Mix, K.S.; Murphy, E.P. Identification of NR4A2 as a transcriptional activator of IL-8 expression in human inflammatory arthritis. Mol. Immunol. 2009, 46, 3345–3357. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, Y.; Sawada, T.; Nagatani, K.; Komagata, Y.; Inoue, T.; Muto, S.; Itai, A.; Yamamoto, K. Effect of nuclear factor-kappaB inhibition on rheumatoid fibroblast-like synoviocytes and collagen induced arthritis. J. Rheumatol. 2005, 32, 1440–1447. [Google Scholar] [PubMed]
- Maijenburg, M.W.; Gilissen, C.; Melief, S.M.; Kleijer, M.; Weijer, K.; Ten Brinke, A.; Roelofs, H.; van Tiel, C.M.; Veltman, J.A.; de Vries, C.J.; et al. Nuclear receptors Nur77 and Nurr1 modulate mesenchymal stromal cell migration. Stem Cells Dev. 2012, 21, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Drabsch, Y.; Dekker, T.J.; de Vinuesa, A.G.; Li, Y.; Hawinkels, L.J.; Sheppard, K.A.; Goumans, M.J.; Luwor, R.B.; de Vries, C.J.; et al. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-β signalling. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, K.; Koenis, D.S.; van Tiel, C.M.; de Vries, C.J. NR4A nuclear receptors are orphans but not lonesome. Biochim Biophys Acta 2014, 1843, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, B.R.; Popichak, K.A.; Hammond, S.L.; Jorgensen, B.A.; Phillips, A.T.; Safe, S.; Tjalkens, R.B. The Nurr1 Activator 1,1-bis(3'-indolyl)-1-(p-chlorophenyl)methane Blocks Inflammatory Gene Expression in BV-2 Microglial Cells by Inhibiting NF-κB. Mol. Pharmacol. 2015, 87, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, E.P.; Crean, D. Molecular Interactions between NR4A Orphan Nuclear Receptors and NF-κB Are Required for Appropriate Inflammatory Responses and Immune Cell Homeostasis. Biomolecules 2015, 5, 1302-1318. https://doi.org/10.3390/biom5031302
Murphy EP, Crean D. Molecular Interactions between NR4A Orphan Nuclear Receptors and NF-κB Are Required for Appropriate Inflammatory Responses and Immune Cell Homeostasis. Biomolecules. 2015; 5(3):1302-1318. https://doi.org/10.3390/biom5031302
Chicago/Turabian StyleMurphy, Evelyn P., and Daniel Crean. 2015. "Molecular Interactions between NR4A Orphan Nuclear Receptors and NF-κB Are Required for Appropriate Inflammatory Responses and Immune Cell Homeostasis" Biomolecules 5, no. 3: 1302-1318. https://doi.org/10.3390/biom5031302