
Advances and New Concepts in Alcohol-Induced Organelle Stress, Unfolded Protein Responses and Organ Damage

Abstract
:1. Proteostasis and Alcohol Metabolites: An Overview
2. Alcohol and UPR in the ER
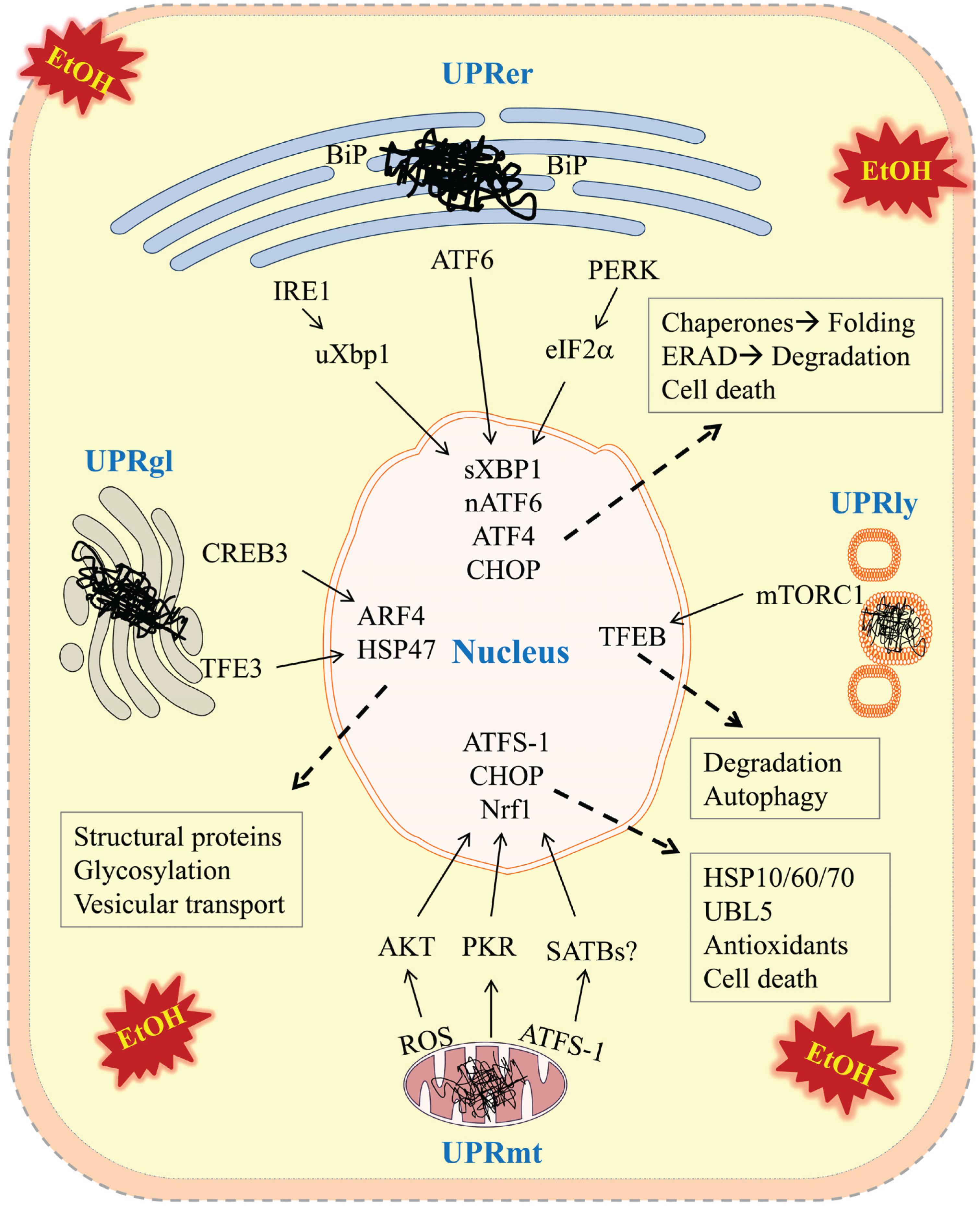
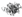 ) in the four types of cellular organelles, which create a stress condition in each of the organelles. The stress activates specialized transcriptional programs (solid arrows) mediated by distinct transducer(s) in each organelle leading to either restoration of organellar protein homeostasis or contribution to cell death (dashed arrows). EtOH, alcohol and/or alcohol metabolites; UPRer or UPR, unfolded protein response in the endoplasmic reticulum (ER); BiP/GRP78, binding immunoglobulin protein also known as 78 kDa glucose-regulated protein; IRE1α, inositol requiring enzyme 1α; ATF 4 or 6, activating transcription factor 4 or 6; nATF6, nuclear ATF6; PERK, PKR-like ER-localized eIF2α kinase; eIF2α, eukaryotic translation initiation factor 2α; uXBP1, un-spliced X-box binding protein 1; sXBP1, spliced XBP1; ERAD, ER associated degradation; CHOP, C/EBP homology protein 10. UPRgl, unfolded protein response in the Golgi apparatus; CREB3; a basic leucine zipper-containing transcription factor; TFE3, a basic-helix-loop-helix type transcription factor; ARF4, a member of the small GTPase family that regulates Golgi-to-ER vesicular transport; HSP47, a 47 kDa collagen-binding glycoprotein. UPRmt, unfolded protein response in the mitochondria; ROS, reactive oxygen species; ATFS-1, activating transcription factor associated with stress-1; AKT, also known as protein kinase B (PKB), is a serine/threonine-specific protein kinase; PKR, protein kinase RNA-activated also known as protein kinase R; Nrf1, the nuclear factor erythroid 2-related factor 1; SATBs, special AT-rich sequence-binding proteins; UBL5, ubiquitin-like 5; HSP10, 60 or 70, heat shock protein 10, 60 or 70 acting as mitochondrial chaperones. UPRly, unfolded protein response in the lysosomes; mTORC1, mammalian target of rapamycin complex 1; TFEB, transcription factor EB.
) in the four types of cellular organelles, which create a stress condition in each of the organelles. The stress activates specialized transcriptional programs (solid arrows) mediated by distinct transducer(s) in each organelle leading to either restoration of organellar protein homeostasis or contribution to cell death (dashed arrows). EtOH, alcohol and/or alcohol metabolites; UPRer or UPR, unfolded protein response in the endoplasmic reticulum (ER); BiP/GRP78, binding immunoglobulin protein also known as 78 kDa glucose-regulated protein; IRE1α, inositol requiring enzyme 1α; ATF 4 or 6, activating transcription factor 4 or 6; nATF6, nuclear ATF6; PERK, PKR-like ER-localized eIF2α kinase; eIF2α, eukaryotic translation initiation factor 2α; uXBP1, un-spliced X-box binding protein 1; sXBP1, spliced XBP1; ERAD, ER associated degradation; CHOP, C/EBP homology protein 10. UPRgl, unfolded protein response in the Golgi apparatus; CREB3; a basic leucine zipper-containing transcription factor; TFE3, a basic-helix-loop-helix type transcription factor; ARF4, a member of the small GTPase family that regulates Golgi-to-ER vesicular transport; HSP47, a 47 kDa collagen-binding glycoprotein. UPRmt, unfolded protein response in the mitochondria; ROS, reactive oxygen species; ATFS-1, activating transcription factor associated with stress-1; AKT, also known as protein kinase B (PKB), is a serine/threonine-specific protein kinase; PKR, protein kinase RNA-activated also known as protein kinase R; Nrf1, the nuclear factor erythroid 2-related factor 1; SATBs, special AT-rich sequence-binding proteins; UBL5, ubiquitin-like 5; HSP10, 60 or 70, heat shock protein 10, 60 or 70 acting as mitochondrial chaperones. UPRly, unfolded protein response in the lysosomes; mTORC1, mammalian target of rapamycin complex 1; TFEB, transcription factor EB.
) in the four types of cellular organelles, which create a stress condition in each of the organelles. The stress activates specialized transcriptional programs (solid arrows) mediated by distinct transducer(s) in each organelle leading to either restoration of organellar protein homeostasis or contribution to cell death (dashed arrows). EtOH, alcohol and/or alcohol metabolites; UPRer or UPR, unfolded protein response in the endoplasmic reticulum (ER); BiP/GRP78, binding immunoglobulin protein also known as 78 kDa glucose-regulated protein; IRE1α, inositol requiring enzyme 1α; ATF 4 or 6, activating transcription factor 4 or 6; nATF6, nuclear ATF6; PERK, PKR-like ER-localized eIF2α kinase; eIF2α, eukaryotic translation initiation factor 2α; uXBP1, un-spliced X-box binding protein 1; sXBP1, spliced XBP1; ERAD, ER associated degradation; CHOP, C/EBP homology protein 10. UPRgl, unfolded protein response in the Golgi apparatus; CREB3; a basic leucine zipper-containing transcription factor; TFE3, a basic-helix-loop-helix type transcription factor; ARF4, a member of the small GTPase family that regulates Golgi-to-ER vesicular transport; HSP47, a 47 kDa collagen-binding glycoprotein. UPRmt, unfolded protein response in the mitochondria; ROS, reactive oxygen species; ATFS-1, activating transcription factor associated with stress-1; AKT, also known as protein kinase B (PKB), is a serine/threonine-specific protein kinase; PKR, protein kinase RNA-activated also known as protein kinase R; Nrf1, the nuclear factor erythroid 2-related factor 1; SATBs, special AT-rich sequence-binding proteins; UBL5, ubiquitin-like 5; HSP10, 60 or 70, heat shock protein 10, 60 or 70 acting as mitochondrial chaperones. UPRly, unfolded protein response in the lysosomes; mTORC1, mammalian target of rapamycin complex 1; TFEB, transcription factor EB.
) in the four types of cellular organelles, which create a stress condition in each of the organelles. The stress activates specialized transcriptional programs (solid arrows) mediated by distinct transducer(s) in each organelle leading to either restoration of organellar protein homeostasis or contribution to cell death (dashed arrows). EtOH, alcohol and/or alcohol metabolites; UPRer or UPR, unfolded protein response in the endoplasmic reticulum (ER); BiP/GRP78, binding immunoglobulin protein also known as 78 kDa glucose-regulated protein; IRE1α, inositol requiring enzyme 1α; ATF 4 or 6, activating transcription factor 4 or 6; nATF6, nuclear ATF6; PERK, PKR-like ER-localized eIF2α kinase; eIF2α, eukaryotic translation initiation factor 2α; uXBP1, un-spliced X-box binding protein 1; sXBP1, spliced XBP1; ERAD, ER associated degradation; CHOP, C/EBP homology protein 10. UPRgl, unfolded protein response in the Golgi apparatus; CREB3; a basic leucine zipper-containing transcription factor; TFE3, a basic-helix-loop-helix type transcription factor; ARF4, a member of the small GTPase family that regulates Golgi-to-ER vesicular transport; HSP47, a 47 kDa collagen-binding glycoprotein. UPRmt, unfolded protein response in the mitochondria; ROS, reactive oxygen species; ATFS-1, activating transcription factor associated with stress-1; AKT, also known as protein kinase B (PKB), is a serine/threonine-specific protein kinase; PKR, protein kinase RNA-activated also known as protein kinase R; Nrf1, the nuclear factor erythroid 2-related factor 1; SATBs, special AT-rich sequence-binding proteins; UBL5, ubiquitin-like 5; HSP10, 60 or 70, heat shock protein 10, 60 or 70 acting as mitochondrial chaperones. UPRly, unfolded protein response in the lysosomes; mTORC1, mammalian target of rapamycin complex 1; TFEB, transcription factor EB.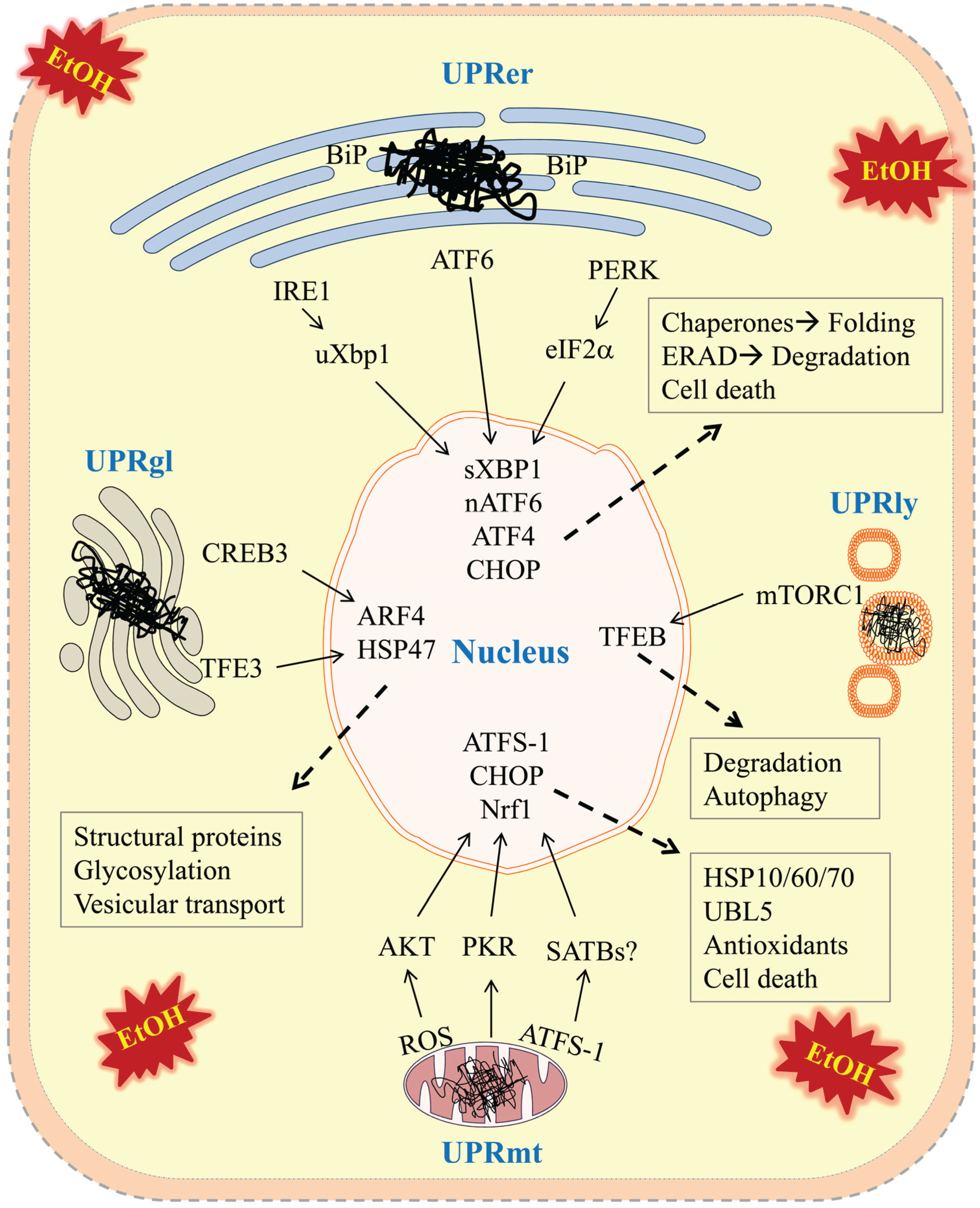
3. Alcohol and UPR in the Mitochondria

{kind=link}
| Organelle | Molecular Factors | References |
|---|---|---|
| Mitochondria | Activating transcription factor associated with stress (ATFS) | [23,24,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75] |
| Special AT-rich sequence-binding proteins (SATBs) | ||
| Protein kinase RNA-activated/protein kinase R (PKR) | ||
| Serine/threonine-specific protein kinase (AKT) | ||
| C/EBP homology protein 10 (CHOP) | ||
| Estrogen receptor a (ERa) | ||
| The nuclear factor erythroid 2-related factor 1 (Nrf1) | ||
| Ubiquitin-like 5 (UBL5) | ||
| The mitochondrial specific deacetylase (SIRT3) | ||
| Heat shock protein 10, 60 or 70 (HSP10, 60, or 70) | ||
| Golgi | The basic-helix-loop-helix type transcription factor (TFE3) | [76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105] |
| The basic leucine zipper-containing transcription factor (CREB3) | ||
| Member of the small GTPase family that regulates Golgi-to-ER vesicular transport (ARF4) | ||
| The 47 kDa collagen-binding glycoprotein (HSP47) | ||
| Glycosyl transferase | ||
| Lysosomes | Mammalian target of rapamycin complex 1 (mTORC1) | [106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127] |
| Transcription factor EB (TFEB) | ||
| Eukaryotic initiation factor 4E-binding protein 1 (4E1-EBP1) | ||
| Unc-51 like autophagy activating kinase 1 (ULK1) | ||
| Eukaryotic initiation factor 4G/4E (eIF4G/4E) | ||
| The Bcl-2e associated athanogen 3 (BAG3) | ||
| Ribosomal protein S6 kinase (S6K) |
4. Alcohol and UPR in the Golgi
5. Alcohol and UPR in the Lysosomes
6. Alcohol and Inter-Organellar Crosstalk
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Altelaar, A.F.; Munoz, J.; Heck, A.J. Next-generation proteomics: Towards an integrative view of proteome dynamics. Nat. Rev. Genet. 2013, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Fedyukina, D.V.; Cavagnero, S. Protein folding at the exit tunnel. Annu. Rev. Biophys. 2011, 40, 337–359. [Google Scholar] [CrossRef] [PubMed]
- Lemus, L.; Goder, V. Regulation of endoplasmic reticulum-associated protein degradation (ERAD) by ubiquitin. Cells 2014, 3, 824–847. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, S.; Mariño, G.; Fernández, A.F.; López-Otín, C. Autophagy, proteases and the sense of balance. Autophagy 2010, 6, 961–963. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic liver disease: The gut microbiome and liver cross talk. Alcohol. Clin. Exp. Res. 2015, 39, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F. Focus on: Neuroscience and treatment: The potential of neuroscience to inform treatment. Alcohol. Res. Health 2010, 33, 144–151. [Google Scholar] [PubMed]
- González-Reimers, E.; Santolaria-Fernández, F.; Martín-González, M.C.; Fernández-Rodríguez, C.M.; Quintero-Platt, G. Alcoholism: A systemic proinflammatory condition. World J. Gastroenterol. 2014, 20, 14660–14671. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, M.S.; Kraemer, K.L. Focus on the heart: Alcohol consumption, HIV infection, and cardiovascular disease. Alcohol. Res. Health 2010, 33, 237–246. [Google Scholar] [PubMed]
- Turner, R.T. Skeletal response to alcohol. Alcohol. Clin. Exp. Res. 2000, 24, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Gao, B.; Zakhari, S.; Nagy, L.E. Inflammation in alcoholic liver disease. Annu. Rev. Nutr. 2012, 32, 343–368. [Google Scholar] [CrossRef] [PubMed]
- Stevens, V.J.; Fantl, W.J.; Newman, C.B.; Sims, R.V.; Cerami, A.; Peterson, C.M. Acetaldehyde adducts with hemoglobin. J. Clin. Invest. 1981, 67, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Behrens, U.J.; Hoerner, M.; Lasker, J.M.; Lieber, C.S. Formation of acetaldehyde adducts with ethanol-inducible P450IIE1 in vivo. Biochem. Biophys. Res. Commun. 1988, 154, 584–590. [Google Scholar] [CrossRef]
- Israel, Y.; Hurwitz, E.; Niemelä, O.; Arnon, R. Monoclonal and polyclonal antibodies against acetaldehyde-containing epitopes in acetaldehyde-protein adducts. Proc. Natl. Acad. Sci. USA 1986, 83, 7923–7927. [Google Scholar] [CrossRef] [PubMed]
- Thiele, G.M.; Tuma, D.J.; Willis, M.S.; Miller, J.A.; McDonald, T.L.; Sorrell, M.F.; Klassen, L.W. Soluble proteins modified with acetaldehyde and malondialdehyde are immunogenic in the absence of adjuvant. Alcohol. Clin. Exp. Res. 1998, 22, 1731–1739. [Google Scholar] [CrossRef] [PubMed]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Molecular basis of homocysteine toxicity in humans. Cell. Mol. Life Sci. 2004, 61, 470–487. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.G.; Ji, S.; Lemasters, J.J. Alcohol-induced liver injury. The role of oxygen. Recent Dev. Alcohol. 1984, 2, 103–117. [Google Scholar] [PubMed]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Gunzerath, L.; Hewitt, B.G.; Li, T.K.; Warren, K.R. Alcohol research: Past, present, and future. Ann. NY Acad. Sci. 2011, 1216, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Yoshida, H. Organelle autoregulation-stress responses in the ER, Golgi, mitochondria and lysosome. J. Biochem. 2015, 157, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Ji, C. New insights into the pathogenesis of alcohol-induced ER stress and liver diseases. Int. J. Hepatol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ji, C. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem. Res. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef]
- Longato, L.; Ripp, K.; Setshedi, M.; Dostalek, M.; Akhlaghi, F.; Branda, M.; Wands, J.R.; de la Monte, S.M. Insulin resistance, ceramide accumulation, and endoplasmic reticulum stress in human chronic alcohol-related liver disease. Oxid. Med. Cell. Longev. 2012. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.; Derdak, Z.; Wands, J.R. Alcohol, insulin resistance and the liver-brain axis. J. Gastroenterol. Hepatol. 2012, 27, S33–S41. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, F.; Villanueva, J.A.; Wong, D.H.; French, S.W.; Halsted, C.H. Chronic ethanol feeding and folate deficiency activate hepatic endoplasmic reticulum stress pathway in micropigs. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G54–G63. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, F.; You, M.; Villanueva, J.A.; Wong, D.H.; French, S.W.; Halsted, C.H. S-adenosylmethionine attenuates hepatic lipid synthesis in micropigs fed ethanol with a folate-deficient diet. Alcohol. Clin. Exp. Res. 2007, 31, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Seth, D.; Leo, M.A.; McGuinness, P.H.; Lieber, C.S.; Brennan, Y.; Williams, R.; Wang, X.M.; McCaughan, G.W.; Gorrell, M.D.; Haber, P.S. Gene expression profiling of alcoholic liver disease in the baboon (Papio hamadryas) and human liver. Am. J. Pathol. 2003, 163, 2303–2317. [Google Scholar] [CrossRef]
- Tazi, K.A.; Bièche, I.; Paradis, V.; Guichard, C.; Laurendeau, I.; Dargère, D.; Legrand, A.; Fay, M.; Pedruzzi, E.; Robin, M.A.; et al. In vivo altered unfolded protein response and apoptosis in livers from lipopolysaccharide-challenged cirrhotic rats. J. Hepatol. 2007, 46, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Kubisch, C.H.; Sans, M.D.; Arumugam, T.; Ernst, S.A.; Williams, J.A.; Logsdon, C.D. Early activation of endoplasmic reticulum stress is associated with arginine-induced acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G238–G245. [Google Scholar] [CrossRef] [PubMed]
- Lugea, A.; Tischler, D.; Nguyen, J.; Gong, J.; Gukovsky, I.; French, S.W.; Gorelick, F.S.; Pandol, S.J. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 2011, 140, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Lugea, A.; Waldron, R.T.; Pandol, S.J. Pancreatic adaptive responses in alcohol abuse: Role of the unfolded protein response. Pancreatology 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Ren, J. Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: Role of insulin signaling and ER stress. J. Mol. Cell. Cardiol. 2008, 44, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Ceylan-Isik, A.F.; Zhao, P.; Zhang, B.; Xiao, X.; Su, G.; Ren, J. Cardiac overexpression of metallothionein rescues cardiac contractile dysfunction and endoplasmic reticulum stress but not autophagy in sepsis. J. Mol. Cell. Cardiol. 2010, 48, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Paschen, W. Endoplasmic reticulum: A primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium. 2003, 34, 365–383. [Google Scholar] [CrossRef]
- Paschen, W. Endoplasmic reticulum dysfunction in brain pathology: Critical role of protein synthesis. Curr. Neurovasc. Res. 2004, 1, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K.; Ojala, J. ER stress in Alzheimer’s disease: A novel neuronal trigger for inflammation and Alzheimer’s pathology. J. Neuroinflammation 2009. [Google Scholar] [CrossRef] [PubMed]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013, 12, 105–118. [Google Scholar] [CrossRef]
- Ke, Z.; Wang, X.; Liu, Y.; Fan, Z.; Chen, G.; Xu, M.; Bower, K.A.; Frank, J.A.; Li, M.; Fang, S.; et al. Ethanol induces endoplasmic reticulum stress in the developing brain. Alcohol. Clin. Exp. Res. 2011, 35, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Alimov, A.; Wang, H.; Liu, M.; Frank, J.A.; Xu, M.; Ou, X.; Luo, J. Expression of autophagy and UPR genes in the developing brain during ethanol-sensitive and resistant periods. Metab. Brain Dis. 2013, 28, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Dlugos, C.A. ATF6 and caspase 12 expression in Purkinje neurons in acute slices from adult, ethanol-fed rats. Brain Res. 2014, 1577, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, F.; Medici, V.; Wong, D.H.; Jose, S.; Dolatshahi, M.; Quinlivan, E.; Dayal, S.; Lentz, S.R.; Tsukamoto, H.; Zhang, Y.H.; et al. Epigenetic regulation of hepatic endoplasmic reticulum stress pathways in the ethanol-fed cystathionine beta synthase-deficient mouse. Hepatology 2010, 51, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Hu, J.; Lau, M.Y.; Feng, M.; Petrovic, L.M.; Ji, C. Altered methylation and expression of ER-associated degradation factors in long-term alcohol and constitutive ER stress-induced murine hepatic tumors. Front Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.C.; Crawford, D.H.; Jaskowski, L.A.; Subramaniam, V.N.; Clouston, A.D.; Crane, D.I.; Bridle, K.R.; Anderson, G.J.; Fletcher, L.M. Excess iron modulates endoplasmic reticulum stress-associated pathways in a mouse model of alcohol and high-fat diet-induced liver injury. Lab Invest. 2013, 93, 1295–1312. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhong, W.; Zhang, W.; Li, Q.; Sun, X.; Tan, X.; Sun, X.; Dong, D.; Zhou, Z. Zinc deficiency mediates alcohol-induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hiroyuki, T.; Yuxia, Z.; Sangmin, L.; Rana, S.; Li, W. Circadian clock mediates ER stress signaling in alcoholic fatty liver (1116.12). FASEB J. 2014. [Google Scholar] [CrossRef]
- Nikolova-Karakashian, M.N.; Rozenova, K.A. Ceramide in stress response. Adv. Exp. Med. Biol. 2010, 688, 86–108. [Google Scholar] [PubMed]
- Zeidan, Y.H.; Hannun, Y.A. The acid sphingomyelinase/ceramide pathway: Biomedical significance and mechanisms of regulation. Curr. Mol. Med. 2010, 10, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Tirodkar, T.S.; Voelkel-Johnson, C. Sphingolipids in apoptosis. Exp. Oncol. 2012, 34, 231–242. [Google Scholar] [PubMed]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial cholesterol accumulation in alcoholic liver disease: Role of ASMase and endoplasmic reticulum stress. Redox Biol. 2014, 3, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Matias, N.; Fucho, R.; Ribas, V.; von Montfort, C.; Nuño, N.; Baulies, A.; Martinez, L.; Tarrats, N.; Mari, M.; et al. ASMase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. J. Hepatol. 2013, 59, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef] [PubMed]
- Kao, E.; Shinohara, M.; Feng, M.; Lau, M.Y.; Ji, C. Human immunodeficiency virus protease inhibitors modulate Ca2+ homeostasis and potentiate alcoholic stress and injury in mice and primary mouse and human hepatocytes. Hepatology 2012, 56, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Ron, D. The mitochondrial UPR-protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Benedetti, C.; Urano, F.; Clark, S.G.; Harding, H.P.; Ron, D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 2004, 117, 4055–4066. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Poplawski, M.; Yen, K.; Cheng, H.; Bloss, E.; Zhu, X.; Patel, H.; Mobbs, C.V. Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biol. 2009, 7, e1000245. [Google Scholar] [CrossRef] [PubMed]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Høj, P.B.; Hoogenraad, N.J. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Horibe, T.; Hoogenraad, N.J. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2007, 2, e835. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Radke, S.; Chander, H.; Schäfer, P.; Meiss, G.; Krüger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [PubMed]
- Alli, A.A.; Brewer, E.M.; Montgomery, D.S.; Ghant, M.S.; Eaton, D.C.; Brown, L.A.; Helms, M.N. Chronic ethanol exposure alters the lung proteome and leads to mitochondrial dysfunction in alveolar type 2 cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L1026–L1035. [Google Scholar] [CrossRef] [PubMed]
- Andringa, K.K.; Udoh, U.S.; Landar, A.; Bailey, S.M. Proteomic analysis of 4 hydroxynonenal (4-HNE) modified proteins in liver mitochondria from chronic ethanol-fed rats. Redox Biol. 2014, 2, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Fritz, K.S.; Galligan, J.J.; Hirschey, M.D.; Verdin, E.; Petersen, D.R. Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J. Proteome Res. 2012, 11, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Mehrian-Shai, R.; Chan, C.; Hsu, Y.H.; Kaplowitz, N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol. Clin. Exp. Res. 2005, 29, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.E.; Horibe, T.; Hoogenraad, N.J. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2007, 2, e874. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.Y.; Han, H.; Hu, J.; Ji, C. Association of cyclin D and estrogen receptor α36 with hepatocellular adenomas of female mice under chronic endoplasmic reticulum stress. J. Gastroenterol. Hepatol. 2013, 28, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Han, H.; Lau, M.Y.; Lee, H.; MacVeigh-Aloni, M.; Ji, C. Effects of combined alcohol and anti-HIV drugs on cellular stress responses in primary hepatocytes and hepatic stellate and kupffer cells. Alcohol. Clin. Exp. Res. 2015, 39, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, W.G.; Fries, E.; Urbani, L.J.; Rothman, J.E. Early and late functions associated with the Golgi apparatus reside in distinct compartments. Proc. Natl. Acad. Sci. USA 1981, 78, 7453–7457. [Google Scholar] [CrossRef] [PubMed]
- Bierring, F. Electron microscopic observations on the mucus production in human and rat intestinal goblet cells. Acta Pathol. Microbiol. Scand. 1962, 54, 241–52. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.A. Fine structure of the goblet cell mucous secretory process. Anat. Rec. 1962, 144, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Clermont, Y.; Xia, L.; Rambourg, A.; Turner, J.D.; Hermo, L. Structure of the Golgi apparatus in stimulated and nonstimulated acinar cells of mammary glands of the rat. Anat. Rec. 1993, 237, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Rambourg, A.; Clermont, Y.; Chrétien, M.; Olivier, L. Modulation of the Golgi apparatus in stimulated and nonstimulated prolactin cells of female rats. Anat. Rec. 1993, 235, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, E.; di Gaeta, S.; Torrisi, M.R.; Ward, P.L.; Roizman, B.; Campadelli-Fiume, G. Redistribution of microtubules and Golgi apparatus in herpes simplex virus-infected cells and their role in viral exocytosis. J. Virol. 1995, 69, 7472–7482. [Google Scholar] [PubMed]
- Oku, M.; Tanakura, S.; Uemura, A.; Sohda, M.; Misumi, Y.; Taniguchi, M.; Wakabayashi, S.; Yoshida, H. Novel cis-acting element GASE regulates transcriptional induction by the Golgi stress response. Cell Struct. Funct. 2011, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Nadanaka, S.; Tanakura, S.; Sawaguchi, S.; Midori, S.; Kawai, Y.; Yamaguchi, S.; Shimada, Y.; Nakamura, Y.; Matsumura, Y.; et al. TFE3 is a bHLH-ZIP-type transcription factor that regulates the mammalian Golgi stress response. Cell Struct. Funct. 2015, 40, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; You, H.; Mo, X.; He, W.; Tang, X.; Jiang, Z.; Chen, S.; Chen, Y.; Zhang, J.; Hu, Z. GOLPH3 mediated Golgi stress response in modulating N2A cell death upon oxygen-glucose deprivation and reoxygenation Injury. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Reiling, J.H.; Olive, A.J.; Sanyal, S.; Carette, J.E.; Brummelkamp, T.R.; Ploegh, H.L.; Starnbach, M.N.; Sabatini, D.M. A CREB3-ARF4 signalling pathway mediates the response to Golgi stress and susceptibility to pathogens. Nat. Cell Biol. 2013, 15, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Raggo, C.; Rapin, N.; Stirling, J.; Gobeil, P.; Smith-Windsor, E.; O’Hare, P.; Misra, V. Luman, the cellular counterpart of herpes simplex virus VP16, is processed by regulated intramembrane proteolysis. Mol. Cell. Biol. 2002, 22, 5639–5649. [Google Scholar] [CrossRef] [PubMed]
- DenBoer, L.M.; Hardy-Smith, P.W.; Hogan, M.R.; Cockram, G.P.; Audas, T.E.; Lu, R. Luman iscapable of binding and activating transcription from the unfolded protein response element. Biochem. Biophys. Res. Commun. 2005, 331, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Audas, T.E.; Li, Y.; Cockram, G.P.; Dean, J.D.; Martyn, A.C.; Kokame, K.; Lu, R. Luman/CREB3 induces transcription of the endoplasmic reticulum (ER) stress response protein Herp through an ER stress response element. Mol. Cell. Biol. 2006, 26, 7999–8010. [Google Scholar] [CrossRef] [PubMed]
- Nakai, W.; Kondo, Y.; Saitoh, A.; Naito, T.; Nakayama, K.; Shin, H.W. ARF1 and ARF4 regulate recycling endosomal morphology and retrograde transport from endosomes to the Golgi apparatus. Mol. Biol. Cell. 2013, 24, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, S.; Mizuno, T.; Koyama, Y.; Katayama, T.; Tohyama, M. The endoplasmic reticulum-resident chaperone heat shock protein 47 protects the Golgi apparatus from the effects of O-glycosylation inhibition. PLoS ONE 2013, 8, e69732. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.; Rybak, B.J.; Lindenbaum, J.; Gerson, C.D.; Walker, G.; Lieber, C.S. Ultrastructural changes in the small intestine induced by ethanol. Gastroenterology 1972, 63, 801–814. [Google Scholar] [PubMed]
- Vilaró, S.; Viñas, O.; Remesar, X. Altered ultrastructure of lactating rat mammary epithelial cells induced by chronic ethanol ingestion. Alcohol. Clin. Exp. Res. 1989, 13, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Takada, A.; Takase, S.; Yasuhara, M. Effects of ethanol on the secretion of hepatic secretory protein in rat alcoholic liver injury. Alcohol 1991, 8, 433–437. [Google Scholar] [CrossRef]
- Madeira, M.D.; Sousa, N.; Lieberman, A.R.; Paula-Barbosa, M.M. Effects of chronic alcohol consumption and of dehydration on the supraoptic nucleus of adult male and female rats. Neuroscience 1993, 56, 657–672. [Google Scholar] [CrossRef]
- Ruela, C.; Sousa, N.; Madeira, M.D.; Paula-Barbosa, M.M. Stereological study of the ultrastructural changes induced by chronic alcohol consumption and dehydration in the supraoptic nucleus of the rat hypothalamus. J. Neurocytol. 1994, 23, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Sousa, N.; Madeira, M.D.; Ruela, C.; Paula-Barbosa, M.M. Structural reorganization in the supraoptic nucleus of withdrawn rats following long-term alcohol consumption. Alcohol. Clin. Exp. Res. 1995, 19, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Cagnon, V.H.; Tomazini, F.M.; Garcia, P.J.; Martinez, M.; Padovani, C.R.; Martinez, F.E. Structure and ultrastructure of the ventral prostate of isogenic mice (C57B1/6J) submitted to chronic alcohol ingestion. Tissue Cell. 2001, 33, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.M.; Renau-Piqueras, J.; Marín, M.P.; Esteban-Pretel, G. Chronic alcohol exposure affects the cell components involved in membrane traffic in neuronal dendrites. Neurotox Res. 2015, 27, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Gang, H.; Lieber, C.S.; Rubin, E. Ethanol increases glycosyl transferase activity in the hepatic Golgi apparatus. Nat. New Biol. 1973, 243, 123–125. [Google Scholar] [PubMed]
- Cottalasso, D.; Gazzo, P.; Dapino, D.; Domenicotti, C.; Pronzato, M.A.; Traverso, N.; Bellocchio, A.; Nanni, G.; Marinari, U.M. Effect of chronic ethanol consumption on glycosylation processes in rat liver microsomes and Golgi apparatus. Alcohol Alcohol. 1996, 31, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Mozo, L.; Simó, A.; Suárez, A.; Rodrigo, L.; Gutiérrez, C. Autoantibodies to Golgi proteins in hepatocellular carcinoma: Case report and literature review. Eur. J. Gastroenterol. Hepatol. 2002, 14, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Hale, E.A.; Raza, S.K.; Ciecierski, R.G.; Ghosh, P. Deleterious actions of chronic ethanol treatment on the glycosylation of rat brain clusterin. Brain Res. 1998, 785, 158–166. [Google Scholar] [CrossRef]
- Han, B.H.; DeMattos, R.B.; Dugan, L.L.; Kim-Han, J.S.; Brendza, R.P.; Fryer, J.D.; Kierson, M.; Cirrito, J.; Quick, K.; Harmony, J.A.; et al. Clusterin contributes to caspase-3-independent brain injury following neonatal hypoxia-ischemia. Nat. Med. 2001, 7, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Garige, M.; Hirsch, K.; Lakshman, M.R. Liver Galbeta1,4GlcNAc alpha2,6-sialyltransferase is down-regulated in human alcoholics: Possible cause for the appearance of asialoconjugates. Metabolism 2007, 56, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.M.; Ding, W.X.; Gao, W. Autophagy in the liver. Hepatology 2008, 47, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Mizushima, N. At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 2014, 39, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Brüning, A.; Jückstock, J. Misfolded proteins: From little villains to little helpers in the fight against cancer. Front Oncol. 2015, 5. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Carra, S.; Behl, C. Emerging roles of molecular chaperones and co-chaperones in selective autophagy: Focus on BAG proteins. J. Mol. Med. 2011, 89, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Gamerdinger, M.; Kaya, A.M.; Wolfrum, U.; Clement, A.M.; Behl, C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011, 12, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Peña-Llopis, S.; Vega-Rubin-de-Celis, S.; Schwartz, J.C.; Wolff, N.C.; Tran, T.A.; Zou, L.; Xie, X.J.; Corey, D.R.; Brugarolas, J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011, 30, 3242–3258. [Google Scholar] [CrossRef] [PubMed]
- Panchaud, N.; Péli-Gulli, M.P.; de Virgilio, C. Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci Signal. 2013. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012. [Google Scholar] [CrossRef] [PubMed]
- Paula-Barbosa, M.M.; Borges, M.M.; Cadete-Leite, A.; Tavares, M.A. Giant multivesicular bodies in the rat hippocampal pyramidal cells after chronic alcohol consumption. Neurosci. Lett. 1986, 64, 345–349. [Google Scholar] [CrossRef]
- Cadete-Leite, A.; Alves, M.C.; Tavares, M.A. Lysosomal abnormalities in the pyramidal cells of the rat medial prefrontal cortex after chronic alcohol consumption and withdrawal. J. Submicrosc. Cytol. Pathol. 1988, 20, 115–122. [Google Scholar] [PubMed]
- Rinderknecht, H.; Renner, I.G.; Koyama, H.H. Lysosomal enzymes in pure pancreatic juice from normal healthy volunteers and chronic alcoholics. Dig. Dis. Sci. 1979, 24, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Manabe, T. Effect of ethanol on pancreatic lysosomes in rats: A possible mechanism for alcoholic pancreatitis. Nihon Geka Hokan 1993, 62, 16–23. [Google Scholar] [PubMed]
- Baraona, E.; Lieber, C.S. Effects of alcohol on hepatic transport of proteins. Annu. Rev. Med. 1982, 33, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; McVicker, D.L.; Zetterman, R.K.; Donohue, T.M., Jr. Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes. Biochim. Biophys. Acta 1995, 1245, 421–429. [Google Scholar] [CrossRef]
- Donohue, T.M., Jr. Autophagy and ethanol-induced liver injury. World J. Gastroenterol. 2009, 15, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Vary, T.C.; Lynch, C.J.; Lang, C.H. Effects of chronic alcohol consumption on regulation of myocardial protein synthesis. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1242–H12451. [Google Scholar] [PubMed]
- McClintick, J.N.; Xuei, X.; Tischfield, J.A.; Goate, A.; Foroud, T.; Wetherill, L.; Ehringer, M.A.; Edenberg, H.J. Stress-response pathways are altered in the hippocampus of chronic alcoholics. Alcohol 2013, 47, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniele, T.; Schiaffino, M.V. Organelle biogenesis and interorganellar connections: Better in contact than in isolation. Commun. Integr. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Dlugos, C.A. Ethanol-induced alterations in Purkinje neuron dendrites in adult and aging rats: A review. Cerebellum 2015. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.; Schwindling, C.; Wenning, A.S.; Becherer, U.; Rettig, J.; Schwarz, E.C.; Hoth, M. T cell activation requires mitochondrial translocation to the immunological synapse. Proc. Natl. Acad. Sci. USA 2007, 104, 14418–14423. [Google Scholar] [CrossRef] [PubMed]
- Holownia, A.; Ledig, M.; Copin, J.C.; Tholey, G. The effect of ethanol on HSP70 in cultured rat glial cells and in brain areas of rat pups exposed to ethanol in utero. Neurochem. Res. 1995, 20, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, Q.; Mao, Z. Chaperone-mediated autophagy: Machinery, regulation and biological consequences. Cell. Mol. Life Sci. 2011, 68, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Rovetta, F.; Stacchiotti, A.; Consiglio, A.; Cadei, M.; Grigolato, P.G.; Lavazza, A.; Rezzani, R.; Aleo, M.F. ER signaling regulation drives the switch between autophagy and apoptosis in NRK-52E cells exposed to cisplatin. Exp. Cell Res. 2012, 318, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Benbrook, D.M.; Long, A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp. Oncol. 2012, 34, 286–297. [Google Scholar] [PubMed]
- Schreiber, A.; Peter, M. Substrate recognition in selective autophagy and the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, W.; Sun, D.; Wang, J.; Li, C.; Zhang, R.; Babcock, S.A.; Li, Y.; Liu, M.; Ma, M.; et al. A novel protective mechanism for mitochondrial aldehyde dehydrogenase (ALDH2) in type I diabetes-induced cardiac dysfunction: Role of AMPK-regulated autophagy. Biochim. Biophys. Acta 2015, 1852, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, S.; Silvers, J.M.; Matthews, D.B. Chronic intermittent ethanol exposure during adolescence blocks ethanol-induced inhibition of spontaneously active hippocampal pyramidal neurons. Alcohol. Clin. Exp. Res. 2006, 30, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kojima, E.; Takeuchi, A.; Haneda, M.; Yagi, A.; Hasegawa, T.; Yamaki, K.; Takeda, K.; Akira, S.; Shimokata, K.; Isobe, K. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: Elucidation by GADD34-deficient mice. FASEB J. 2003, 17, 1573–1575. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell. Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, N.; Sugiyama, Y.; Miyazaki, S.; Nakagawa, H.; Nishimura, K.; Matsuo, S. An ATF4-signal-modulating machine other than GADD34 acts in ATF4-to-CHOP signaling to block CHOP expression in ER-stress-related autophagy. J. Cell. Biochem. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, C. Advances and New Concepts in Alcohol-Induced Organelle Stress, Unfolded Protein Responses and Organ Damage. Biomolecules 2015, 5, 1099-1121. https://doi.org/10.3390/biom5021099
Ji C. Advances and New Concepts in Alcohol-Induced Organelle Stress, Unfolded Protein Responses and Organ Damage. Biomolecules. 2015; 5(2):1099-1121. https://doi.org/10.3390/biom5021099
Chicago/Turabian StyleJi, Cheng. 2015. "Advances and New Concepts in Alcohol-Induced Organelle Stress, Unfolded Protein Responses and Organ Damage" Biomolecules 5, no. 2: 1099-1121. https://doi.org/10.3390/biom5021099