Decoding F508del Misfolding in Cystic Fibrosis

{kind=link}
Abstract
:1. Introduction
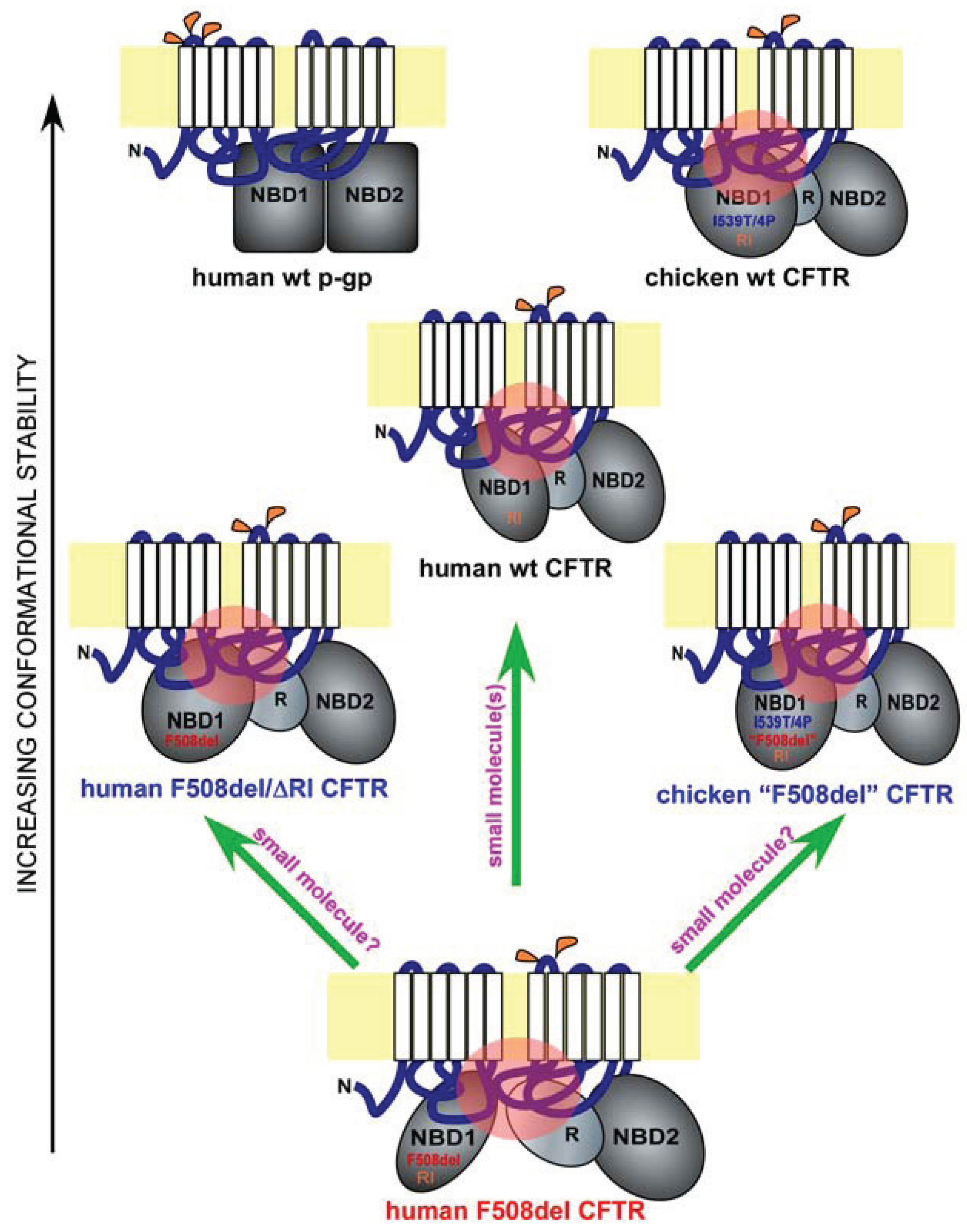
2. NBD1 Misfolding to Domain-Domain Miscontact
3. NBD1 Revisited
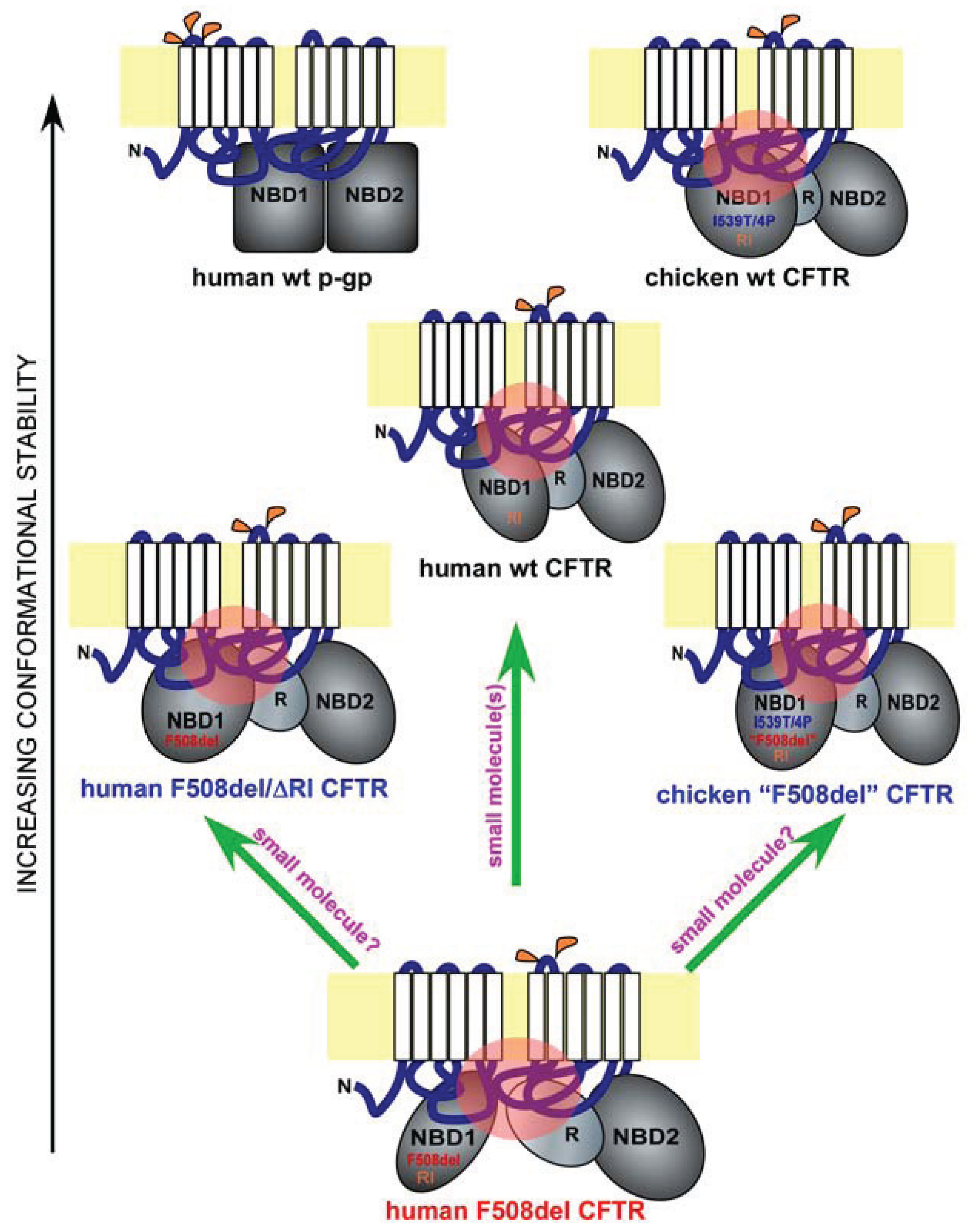
4. Regulatory Insertion
5. A Lesson from Avians
6. Implications for Drug Discovery
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar]
- Jensen, T.J.; Loo, M.A.; Pind, S.; Williams, D.B.; Goldberg, A.L.; Riordan, J.R. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 1995, 83, 129–135. [Google Scholar] [CrossRef]
- Ward, C.L.; Omura, S.; Kopito, R.R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 1995, 83, 121–127. [Google Scholar] [CrossRef]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar]
- Lukacs, G.L.; Chang, X.B.; Bear, C.; Kartner, N.; Mohamed, A.; Riordan, J.R.; Grinstein, S. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem. 1993, 268, 21592–21598. [Google Scholar]
- Okiyoneda, T.; Barriere, H.; Bagdany, M.; Rabeh, W.M.; Du, K.; Hohfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef]
- Roy, G.; Chalfin, E.M.; Saxena, A.; Wang, X. Interplay between ER exit code and domain conformation in CFTR misprocessing and rescue. Mol. Biol. Cell 2010, 21, 597–609. [Google Scholar] [CrossRef]
- Castellani, S.; Guerra, L.; Favia, M.; di Gioia, S.; Casavola, V.; Conese, M. NHERF1 and CFTR restore tight junction organisation and function in cystic fibrosis airway epithelial cells: Role of ezrin and the RhoA/ROCK pathway. Lab. Invest. 2012, 92, 1527–1540. [Google Scholar] [CrossRef]
- Guerra, L.; Fanelli, T.; Favia, M.; Riccardi, S.M.; Busco, G.; Cardone, R.A.; Carrabino, S.; Weinman, E.J.; Reshkin, S.J.; Conese, M.; et al. Na+/H+ exchanger regulatory factor isoform 1 overexpression modulates cystic fibrosis transmembrane conductance regulator (CFTR) expressionand activity in human airway 16HBE14o-cells and rescues DeltaF508 CFTR functional expression in cystic fibrosis cells. J. Biol. Chem. 2005, 280, 40925–40933. [Google Scholar] [CrossRef]
- Moniz, S.; Sousa, M.; Moraes, B.J.; Mendes, A.I.; Palma, M.; Barreto, C.; Fragata, J.I.; Amaral, M.D.; Matos, P. HGF stimulation of Rac1 signaling enhances pharmacological correction of the most prevalent cystic fibrosis mutant F508del-CFTR. ACS Chem. Biol. 2013, 8, 432–442. [Google Scholar]
- Monterisi, S.; Favia, M.; Guerra, L.; Cardone, R.A.; Marzulli, D.; Reshkin, S.J.; Casavola, V.; Zaccolo, M. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J. Cell Sci. 2012, 125, 1106–1117. [Google Scholar] [CrossRef]
- Valentine, C.D.; Lukacs, G.L.; Verkman, A.S.; Haggie, P.M. Reduced PDZ interactions of rescued DeltaF508CFTR increases its cell surface mobility. J. Biol. Chem. 2012, 287, 43630–43638. [Google Scholar]
- Thomas, P.J.; Pedersen, P.L. Effects of the delta F508 mutation on the structure, function, and folding of the first nucleotide-binding domain of CFTR. J. Bioenerg. Biomembr. 1993, 25, 11–19. [Google Scholar] [CrossRef]
- Lewis, H.A.; Zhao, X.; Wang, C.; Sauder, J.M.; Rooney, I.; Noland, B.W.; Lorimer, D.; Kearins, M.C.; Conners, K.; Condon, B.; et al. Impact of the deltaF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J. Biol. Chem. 2005, 280, 1346–1353. [Google Scholar] [CrossRef]
- Callebaut, I.; Eudes, R.; Mornon, J.P.; Lehn, P. Nucleotide-binding domains of human cystic fibrosis transmembrane conductance regulator: Detailed sequence analysis and three-dimensional modeling of the heterodimer. Cell Mol. Life Sci. 2004, 61, 230–242. [Google Scholar] [CrossRef]
- Eudes, R.; Lehn, P.; Ferec, C.; Mornon, J.P.; Callebaut, I. Nucleotide binding domains of human CFTR: A structural classification of critical residues and disease-causing mutations. Cell Mol. Life Sci. 2005, 62, 2112–2123. [Google Scholar] [CrossRef]
- Du, K.; Sharma, M.; Lukacs, G.L. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2005, 12, 17–25. [Google Scholar] [CrossRef]
- Thibodeau, P.H.; Brautigam, C.A.; Machius, M.; Thomas, P.J. Side chain and backbone contributions of Phe508 to CFTR folding. Nat. Struct. Mol. Biol. 2005, 12, 10–16. [Google Scholar] [CrossRef]
- Mornon, J.P.; Lehn, P.; Callebaut, I. Atomic model of human cystic fibrosis transmembrane conductance regulator: Membrane-spanning domains and coupling interfaces. Cell Mol. Life Sci. 2008, 65, 2594–2612. [Google Scholar] [CrossRef]
- Mornon, J.P.; Lehn, P.; Callebaut, I. Molecular models of the open and closed states of the whole human CFTR protein. Cell Mol. Life Sci. 2009, 66, 3469–3486. [Google Scholar]
- Serohijos, A.W.; Hegedus, T.; Aleksandrov, A.A.; He, L.; Cui, L.; Dokholyan, N.V.; Riordan, J.R. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. USA 2008, 105, 3256–3261. [Google Scholar]
- Pissarra, L.S.; Farinha, C.M.; Xu, Z.; Schmidt, A.; Thibodeau, P.H.; Cai, Z.; Thomas, P.J.; Sheppard, D.N.; Amaral, M.D. Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chem. Biol. 2008, 15, 62–69. [Google Scholar] [CrossRef]
- Teem, J.L.; Carson, M.R.; Welsh, M.J. Mutation of R555 in CFTR-delta F508 enhances function and partially corrects defective processing. Recept. Channels 1996, 4, 63–72. [Google Scholar]
- Atwell, S.; Brouillette, C.G.; Conners, K.; Emtage, S.; Gheyi, T.; Guggino, W.B.; Hendle, J.; Hunt, J.F.; Lewis, H.A.; Lu, F.; et al. Structures of a minimal human CFTR first nucleotide-binding domain as a monomer, head-to-tail homodimer, and pathogenic mutant. Protein Eng. Des. Sel. 2010, 23, 375–384. [Google Scholar]
- Lewis, H.A.; Buchanan, S.G.; Burley, S.K.; Conners, K.; Dickey, M.; Dorwart, M.; Fowler, R.; Gao, X.; Guggino, W.B.; Hendrickson, W.A.; et al. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004, 23, 282–293. [Google Scholar] [CrossRef]
- Csanady, L.; Chan, K.W.; Nairn, A.C.; Gadsby, D.C. Functional roles of nonconserved structural segments in CFTR’s NH2-terminal nucleotide binding domain. J. Gen. Physiol. 2005, 125, 43–55. [Google Scholar]
- Lewis, H.A.; Wang, C.; Zhao, X.; Hamuro, Y.; Conners, K.; Kearins, M.C.; Lu, F.; Sauder, J.M.; Molnar, K.S.; Coales, S.J.; et al. Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J. Mol. Biol. 2010, 396, 406–430. [Google Scholar] [CrossRef]
- Protasevich, I.; Yang, Z.; Wang, C.; Atwell, S.; Zhao, X.; Emtage, S.; Wetmore, D.; Hunt, J.F.; Brouillette, C.G. Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 2010, 19, 1917–1931. [Google Scholar] [CrossRef]
- Wang, C.; Protasevich, I.; Yang, Z.; Seehausen, D.; Skalak, T.; Zhao, X.; Atwell, S.; Spencer Emtage, J.; Wetmore, D.R.; Brouillette, C.G.; et al. Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci. 2010, 19, 1932–1947. [Google Scholar] [CrossRef]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef]
- Wieczorek, G.; Zielenkiewicz, P. DeltaF508 mutation increases conformational flexibility of CFTR protein. J. Cyst. Fibros 2008, 7, 295–300. [Google Scholar] [CrossRef]
- Park, I.; Fan, Y.; Bhattacharya, S.; Chettiar, S.; Regan, N.; Bhasin, D.; Giovannucci, D.R.; Li, P.; Frizzell, R.A.; Wang, X.; et al. Development of ΔF508 correctors by NBD1 conformational rescue. Pediatr. Pulmonol. Suppl. 2012, 47, 232. [Google Scholar]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar]
- Hyde, S.C.; Emsley, P.; Hartshorn, M.J.; Mimmack, M.M.; Gileadi, U.; Pearce, S.R.; Gallagher, M.P.; Gill, D.R.; Hubbard, R.E.; Higgins, C.F. Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature 1990, 346, 362–365. [Google Scholar] [CrossRef]
- Loo, M.A.; Jensen, T.J.; Cui, L.; Hou, Y.; Chang, X.B.; Riordan, J.R. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998, 17, 6879–6887. [Google Scholar] [CrossRef]
- Aleksandrov, A.A.; Kota, P.; Aleksandrov, L.A.; He, L.; Jensen, T.; Cui, L.; Gentzsch, M.; Dokholyan, N.V.; Riordan, J.R. Regulatory insertion removal restores maturation, stability and function of DeltaF508 CFTR. J. Mol. Biol. 2010, 401, 194–210. [Google Scholar] [CrossRef]
- Kanelis, V.; Hudson, R.P.; Thibodeau, P.H.; Thomas, P.J.; Forman-Kay, J.D. NMR evidence for differential phosphorylation-dependent interactions in WT and DeltaF508 CFTR. EMBO J. 2010, 29, 263–277. [Google Scholar] [CrossRef]
- Ostedgaard, L.S.; Rogers, C.S.; Dong, Q.; Randak, C.O.; Vermeer, D.W.; Rokhlina, T.; Karp, P.H.; Welsh, M.J. Processing and function of CFTR-DeltaF508 are species-dependent. Proc. Natl. Acad. Sci. USA 2007, 104, 15370–15375. [Google Scholar] [CrossRef]
- Aleksandrov, A.A.; Kota, P.; Cui, L.; Jensen, T.; Alekseev, A.E.; Reyes, S.; He, L.; Gentzsch, M.; Aleksandrov, L.A.; Dokholyan, N.V.; et al. Allosteric modulation balances thermodynamic stability and restores function of DeltaF508 CFTR. J. Mol. Biol. 2012, 419, 41–60. [Google Scholar] [CrossRef]
- McPhail, G.L.; Clancy, J.P. Ivacaftor: The first therapy acting on the primary cause of cystic fibrosis. Drugs Today Barc. 2013, 49, 253–260. [Google Scholar]
- Yu, H.; Burton, B.; Huang, C.J.; Worley, J.; Cao, D.; Johnson, J.P., Jr.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros 2012, 11, 237–245. [Google Scholar] [CrossRef]
- Pedemonte, N.; Lukacs, G.L.; Du, K.; Caci, E.; Zegarra-Moran, O.; Galietta, L.J.; Verkman, A.S. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 2005, 115, 2564–2571. [Google Scholar] [CrossRef]
- Van Goor, F.; Straley, K.S.; Cao, D.; Gonzalez, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of {Delta}F508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef]
- Dalemans, W.; Barbry, P.; Champigny, G.; Jallat, S.; Dott, K.; Dreyer, D.; Crystal, R.G.; Pavirani, A.; Lecocq, J.P.; Lazdunski, M. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 1991, 354, 526–528. [Google Scholar] [CrossRef]
- DeCarvalho, A.C.; Gansheroff, L.J.; Teem, J.L. Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator delta F508. J. Biol. Chem. 2002, 277, 35896–35905. [Google Scholar] [CrossRef]
- He, L.; Aleksandrov, L.A.; Cui, L.; Jensen, T.J.; Nesbitt, K.L.; Riordan, J.R. Restoration of domain folding and interdomain assembly by second-site suppressors of the DeltaF508 mutation in CFTR. FASEB J. 2010, 24, 3103–3112. [Google Scholar] [CrossRef]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Processing mutations disrupt interactions between the nucleotide binding and transmembrane domains of P-glycoprotein and the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 2008, 283, 28190–28197. [Google Scholar] [CrossRef]
- Serohijos, A.W.; Hegedus, T.; Riordan, J.R.; Dokholyan, N.V. Diminished self-chaperoning activity of the DeltaF508 mutant of CFTR results in protein misfolding. PLoS Comput. Biol. 2008, 4, e1000008. [Google Scholar] [CrossRef]
- Teem, J.L.; Berger, H.A.; Ostedgaard, L.S.; Rich, D.P.; Tsui, L.C.; Welsh, M.J. Identification of revertants for the cystic fibrosis delta F508 mutation using STE6-CFTR chimeras in yeast. Cell 1993, 73, 335–346. [Google Scholar] [CrossRef]
- Thibodeau, P.H.; Richardson, J.M., 3rd; Wang, W.; Millen, L.; Watson, J.; Mendoza, J.L.; Du, K.; Fischman, S.; Senderowitz, H.; Lukacs, G.L.; et al. The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 2010, 285, 35825–35835. [Google Scholar] [CrossRef]
- Rabeh, W.M.; Bossard, F.; Xu, H.; Okiyoneda, T.; Bagdany, M.; Mulvihill, C.M.; Du, K.; di Bernardo, S.; Liu, Y.; Konermann, L.; et al. Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell 2012, 148, 150–163. [Google Scholar] [CrossRef]
- Mendoza, J.L.; Schmidt, A.; Li, Q.; Nuvaga, E.; Barrett, T.; Bridges, R.J.; Feranchak, A.P.; Brautigam, C.A.; Thomas, P.J. Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell 2012, 148, 164–174. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, X.R.; Li, C. Decoding F508del Misfolding in Cystic Fibrosis. Biomolecules 2014, 4, 498-509. https://doi.org/10.3390/biom4020498
Wang XR, Li C. Decoding F508del Misfolding in Cystic Fibrosis. Biomolecules. 2014; 4(2):498-509. https://doi.org/10.3390/biom4020498
Chicago/Turabian StyleWang, Xiaodong Robert, and Chenglong Li. 2014. "Decoding F508del Misfolding in Cystic Fibrosis" Biomolecules 4, no. 2: 498-509. https://doi.org/10.3390/biom4020498
APA StyleWang, X. R., & Li, C. (2014). Decoding F508del Misfolding in Cystic Fibrosis. Biomolecules, 4(2), 498-509. https://doi.org/10.3390/biom4020498