Re-Configuration of Sphingolipid Metabolism by Oncogenic Transformation

Abstract
:1. Introduction
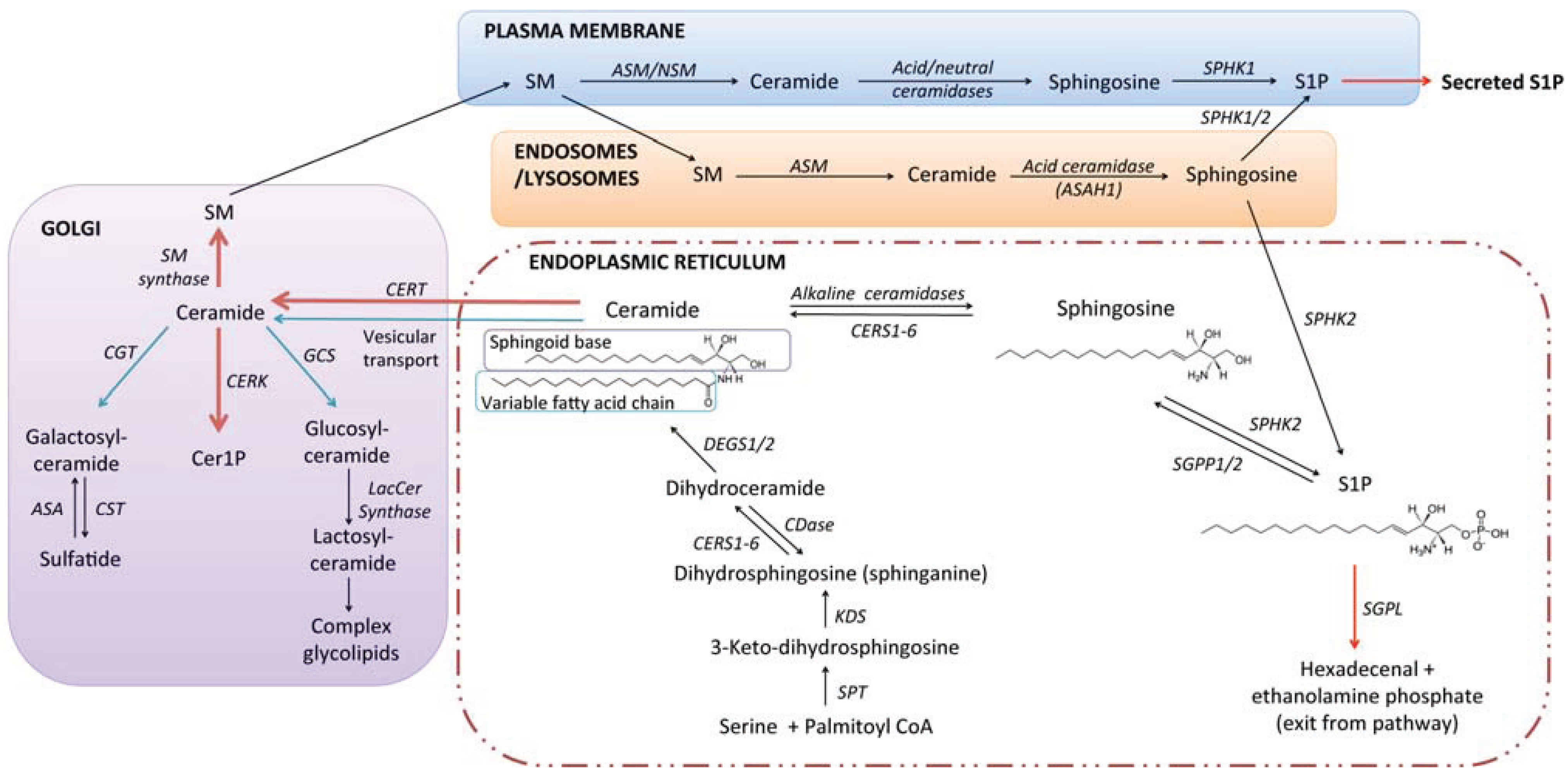
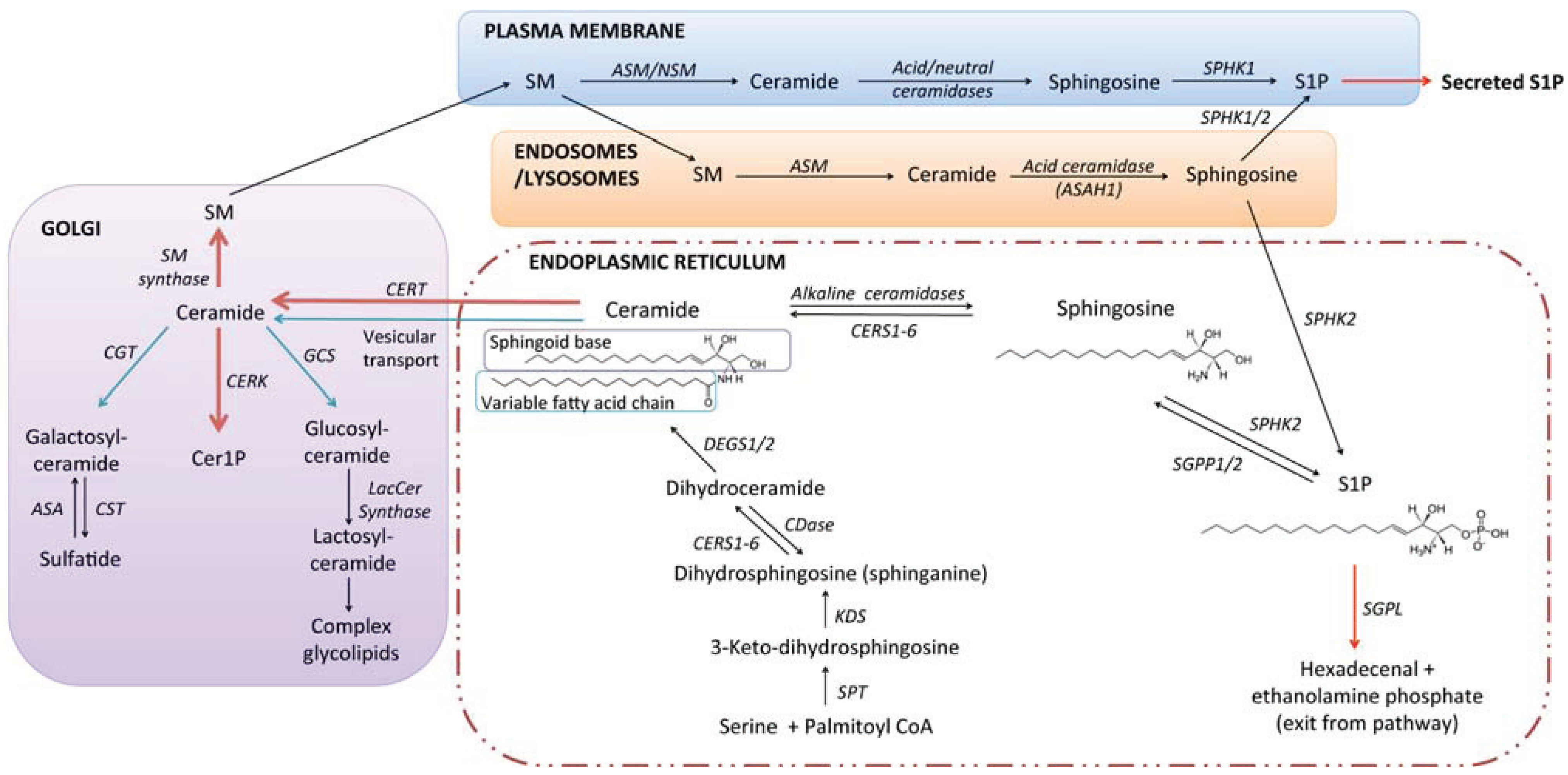
2. Sphingolipid Biosynthesis and Catabolism
3. Signalling Roles of Ceramide, Sphingosine, and S1P
3.1. Ceramide
3.2. Sphingosine and S1P
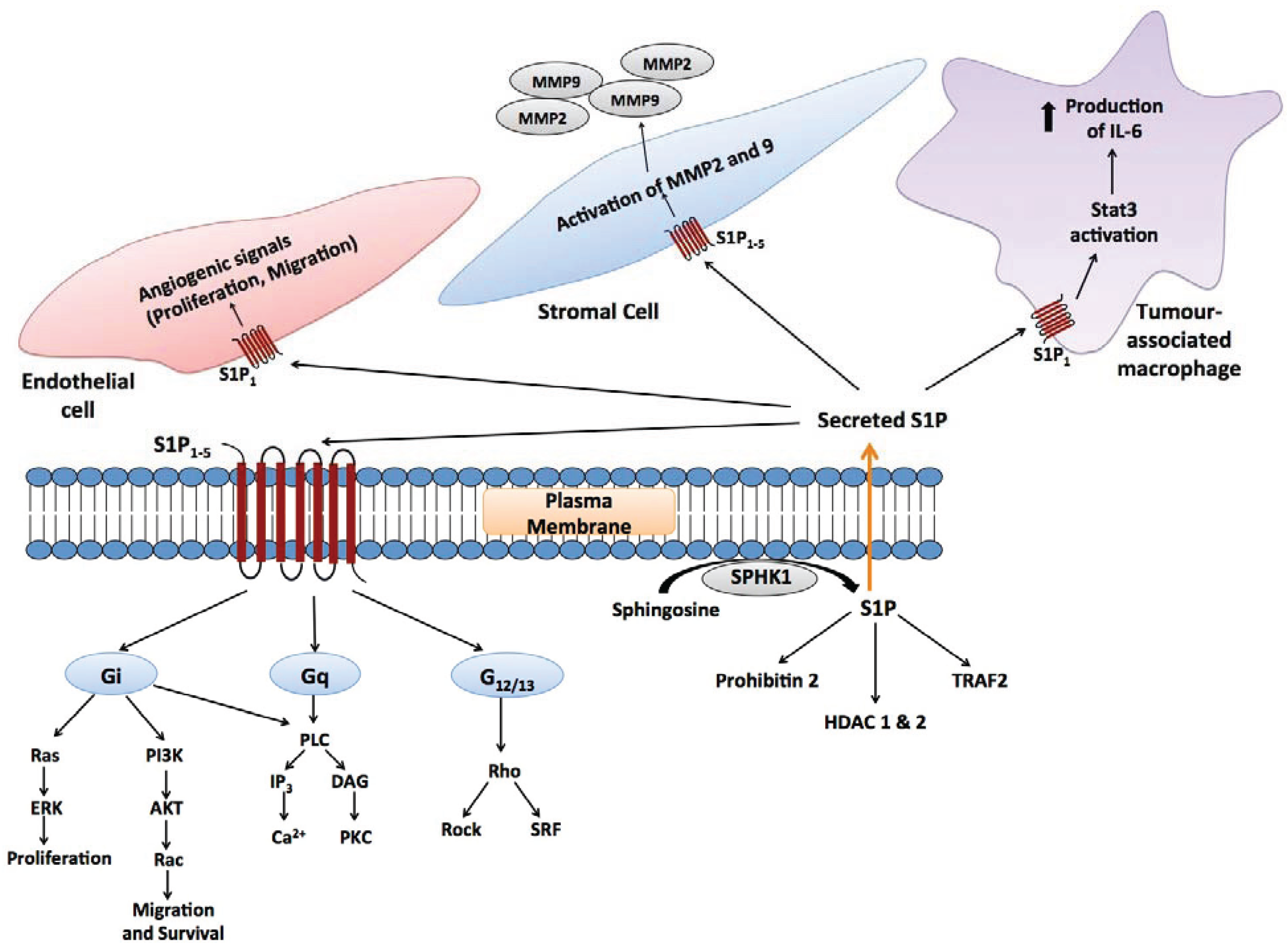
4. Studies on Individual Enzymes and Sphingolipid Metabolites in Cancer
4.1. Sphingosine Kinase 1 (SPHK1)
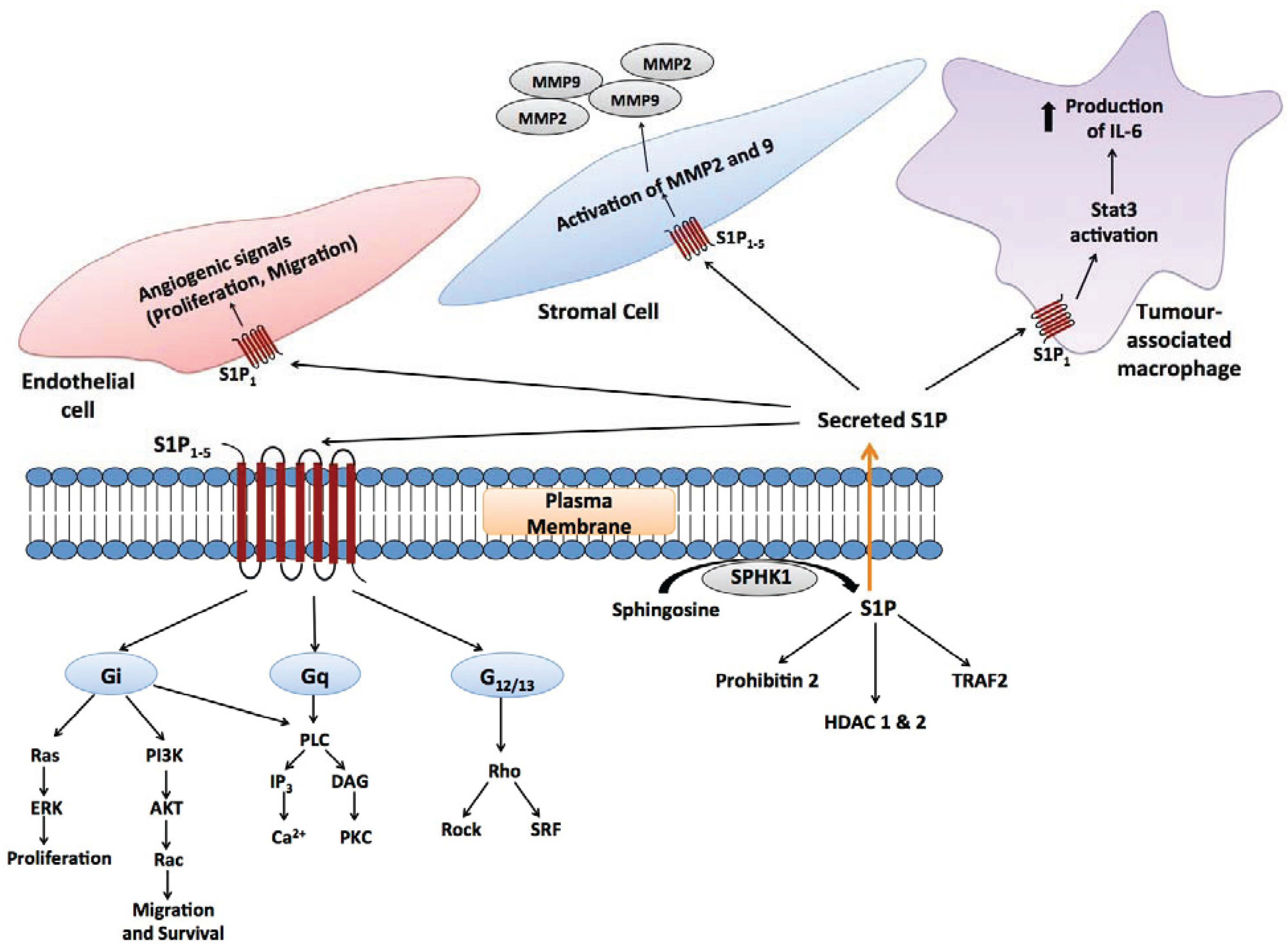
4.2. S1P Signalling in the Cancer Microenvironment
4.3. Sphingosine Kinase 2 (SPHK2)
4.4. Sphingosine 1-Phosphate Lyase (SGPL)
4.5. Sphingosine 1-Phosphate Phosphatases
4.6. Ceramide and Ceramide Synthases
4.7. Acid Ceramidase
4.8. Ceramide Kinase (CERK)
4.9. Sphingomyelin (SM) and Sphingomyelinases
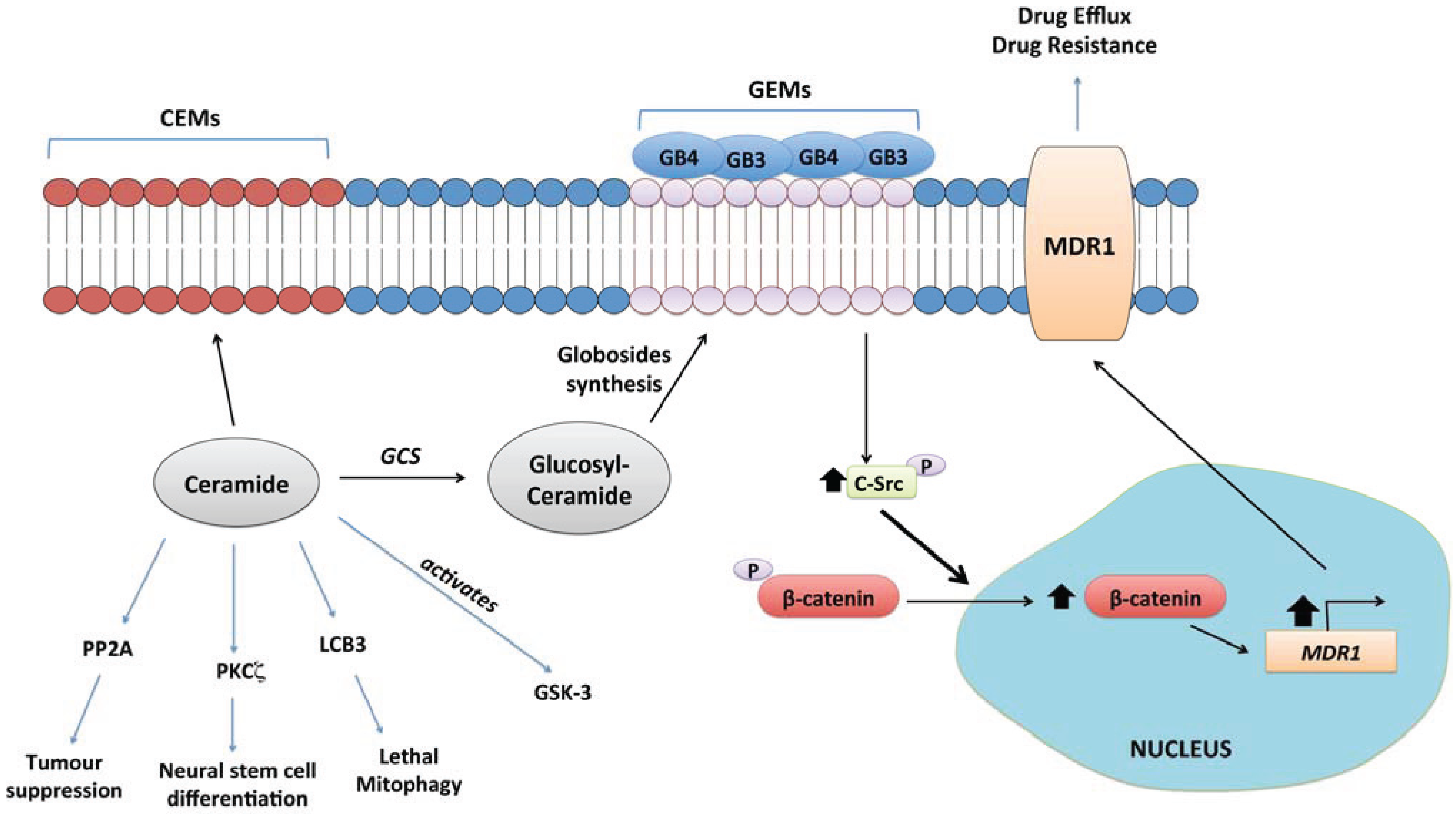
4.10. Glucosylceramide Synthase (GCS)
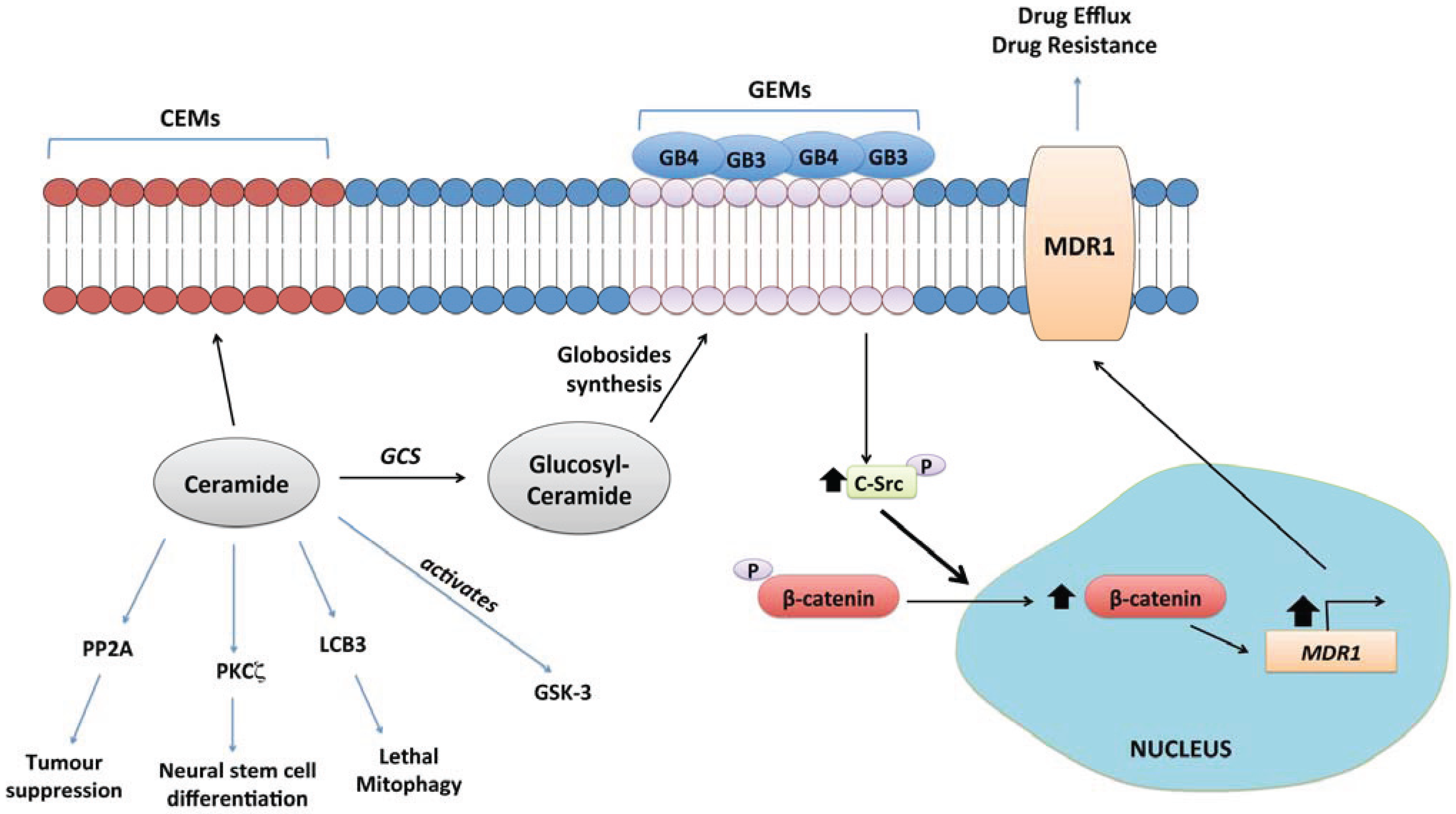
4.11. Lactosylceramide and Complex Glycosphingolipids

{kind=link}
{kind=link}
{kind=link}
| Cancer type | Differential expression/functional significance of glycolipids | Reference |
|---|---|---|
| Breast Cancer | Elevated ganglioside GB5 and globohexaosylceramide (Globo H) | [220] |
| in breast cancer cells with a stem cell phenotype | ||
| Colon Cancer | Increased levels of lactosylceramide in association with up-regulation of human plasma membrane-associated sialidase (Neu3) | [221,222] |
| Addition of lactosylceramide or transfection with Neu3 inhibits apoptosis, associated with increased Bcl-2 expression, in cultured colon cancer cells | ||
| Elevated expression of ganglioside GB3, which converts noninvasive epithelial cells into cells with an invasive and migratory phenotype | [223,224] | |
| Glioblastoma Multiforme | Increased levels of simple ganglioside GM3, GD3; | [225,226] |
| Decreased levels of complex gangliosides GT1b, GQ1b and GD1b | ||
| GD1b expression is inversely proportional to astrocytoma grade | ||
| Glioma | Increased levels of ganglioside GD3 and lacto-series ganglioside 3'-isoLM1 | [227,228] |
| Lung Cancer | Ganglioside GM2 important in maintaining growth of lung cancer cells in the presence of co-cultured fibroblasts | [229] |
| Ganglioside GD2 elevated in small cell lung cancer | [230,231] | |
| Anti-GD2 antibody shown to suppress cell growth | ||
| and induced apoptosis in small cell lung cancer cells | ||
| Increased levels of ganglioside GD3 in small cell lung cancer | [232] | |
| Increased levels of Fucosyl-GM1 in small cell lung cancer | [232,233,234] | |
| Medulloblastoma | Ganglioside GD1a, GM2 and GM3 shed into the | [235] |
| microenvironment of Daoy medulloblastoma cell line | ||
| Melanoma | Ganglioside GM2 elevated compared to normal melanocytes | [236] |
| Increased levels of ganglioside GD2. Deposited in adhesion plaques, implicating GD2 as an adhesion mechanism in melanoma | [237, 238] | |
| Ganglioside GD3 is a predominant species found in melanoma, | [238,239] | |
| specifically deposited in adhesion plaques | ||
| Neuroblastoma | Abundant expression of ganglioside GD2 | [240,241] |
| Pancreatic Cancer | Elevated expression of ganglioside GB3 in pancreatic adenocarcinomas | [223, 224] |
| Renal Cell Carcinoma | Increased levels of lactosylceramide in granular cells | [242] |
| and decreased levels in clear cells | ||
| Ganglioside GM3 elevated Wilms tumour | [242, 243] | |
| and in granular cells of renal cell carcinoma | ||
| Expression of ganglioside GD3 mediates apoptosis of | [244] | |
| activated T-cells in renal cell carcinoma | ||
| Retinoblastoma | Increased levels of ganglioside GD2 | [245] |
5. Beyond the Single Enzyme: How is Sphingolipid Metabolism Reconfigured in Cancer?
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Canals, D.; Perry, D.M.; Jenkins, R.W.; Hannun, Y.A. Drug targeting of sphingolipid metabolism: Sphingomyelinases and ceramidases. Br. J. Pharmacol. 2011, 163, 694–712. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Hill, R.A.; Li, Y.T. Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv. Cancer Res. 2013, 117, 59–89. [Google Scholar] [CrossRef]
- Adan-Gokbulut, A.; Kartal-Yandim, M.; Iskender, G.; Baran, Y. Novel agents targeting bioactive sphingolipids for the treatment of cancer. Curr. Med. Chem. 2013, 20, 108–122. [Google Scholar]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef]
- Sonnino, S.; Prinetti, A.; Mauri, L.; Chigorno, V.; Tettamanti, G. Dynamic and structural properties of sphingolipids as driving forces for the formation of membrane domains. Chem. Rev. 2006, 106, 2111–2125. [Google Scholar] [CrossRef]
- Vadas, M.; Xia, P.; McCaughan, G.; Gamble, J. The role of sphingosine kinase 1 in cancer: Oncogene or non-oncogene addiction? Biochim. Biophys. Acta 2008, 1781, 442–447. [Google Scholar] [CrossRef]
- Henry, B.; Moller, C.; Dimanche-Boitrel, M.T.; Gulbins, E.; Becker, K.A. Targeting the ceramide system in cancer. Cancer Lett. 2013, 332, 286–294. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Many ceramides. J. Biol. Chem. 2011, 286, 27855–27862. [Google Scholar] [CrossRef]
- Rex, K.; Jeffries, S.; Brown, M.L.; Carlson, T.; Coxon, A.; Fajardo, F.; Frank, B.; Gustin, D.; Kamb, A.; Kassner, P.D.; et al. Sphingosine kinase activity is not required for tumor cell viability. PLoS One 2013, 8, e68328. [Google Scholar] [CrossRef]
- Kharel, Y.; Mathews, T.P.; Gellett, A.M.; Tomsig, J.L.; Kennedy, P.C.; Moyer, M.L.; Macdonald, T.L.; Lynch, K.R. Sphingosine kinase type 1 inhibition reveals rapid turnover of circulating sphingosine 1-phosphate. Biochem. J. 2011, 440, 345–353. [Google Scholar] [CrossRef]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef]
- Merrill, A.H., Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Lopez, P.H.; Schnaar, R.L. Gangliosides in cell recognition and membrane protein regulation. Curr. Opin. Struct. Biol. 2009, 19, 549–557. [Google Scholar] [CrossRef]
- Lahiri, S.; Lee, H.; Mesicek, J.; Fuks, Z.; Haimovitz-Friedman, A.; Kolesnick, R.N.; Futerman, A.H. Kinetic characterization of mammalian ceramide synthases: Determination of K(m) values towards sphinganine. FEBS Lett. 2007, 581, 5289–5294. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem. J. 2005, 390, 263–271. [Google Scholar] [CrossRef]
- Park, J.W.; Park, W.J.; Futerman, A.H. Ceramide synthases as potential targets for therapeutic intervention in human diseases. Biochim. Biophys. Acta 2013. [Google Scholar] [CrossRef]
- Hama, H. Fatty acid 2-Hydroxylation in mammalian sphingolipid biology. Biochim. Biophys. Acta 2010, 1801, 405–414. [Google Scholar]
- Xiong, Y.; Lee, H.J.; Mariko, B.; Lu, Y.C.; Dannenberg, A.J.; Haka, A.S.; Maxfield, F.R.; Camerer, E.; Proia, R.L.; Hla, T. Sphingosine kinases are not required for inflammatory responses in macrophages. J. Biol. Chem. 2013, 288, 32563–32573. [Google Scholar] [CrossRef]
- Lavieu, G.; Scarlatti, F.; Sala, G.; Carpentier, S.; Levade, T.; Ghidoni, R.; Botti, J.; Codogno, P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J. Biol. Chem. 2006, 281, 8518–8527. [Google Scholar]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef]
- Sims, K.; Haynes, C.A.; Kelly, S.; Allegood, J.C.; Wang, E.; Momin, A.; Leipelt, M.; Reichart, D.; Glass, C.K.; Sullards, M.C.; et al. Kdo2-lipid A, a TLR4-specific agonist, induces de novo sphingolipid biosynthesis in RAW264.7 macrophages, which is essential for induction of autophagy. J. Biol. Chem. 2010, 285, 38568–38579. [Google Scholar] [CrossRef]
- Kajimoto, T.; Okada, T.; Miya, S.; Zhang, L.; Nakamura, S. Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multivesicular endosomes. Nat. Commun. 2013. [Google Scholar] [CrossRef]
- Lacour, S.; Hammann, A.; Grazide, S.; Lagadic-Gossmann, D.; Athias, A.; Sergent, O.; Laurent, G.; Gambert, P.; Solary, E.; Dimanche-Boitrel, M.T. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res. 2004, 64, 3593–3598. [Google Scholar] [CrossRef]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003, 300, 1155–1159. [Google Scholar] [CrossRef]
- Schutze, S.; Potthoff, K.; Machleidt, T.; Berkovic, D.; Wiegmann, K.; Kronke, M. TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell 1992, 71, 765–776. [Google Scholar] [CrossRef]
- Grassme, H.; Jekle, A.; Riehle, A.; Schwarz, H.; Berger, J.; Sandhoff, K.; Kolesnick, R.; Gulbins, E. CD95 signaling via ceramide-rich membrane rafts. J. Biol. Chem. 2001, 276, 20589–20596. [Google Scholar] [CrossRef]
- Adam, D.; Wiegmann, K.; Adam-Klages, S.; Ruff, A.; Kronke, M. A novel cytoplasmic domain of the p55 tumor necrosis factor receptor initiates the neutral sphingomyelinase pathway. J. Biol. Chem. 1996, 271, 14617–14622. [Google Scholar] [CrossRef]
- Ji, C.; Yang, B.; Yang, Y.L.; He, S.H.; Miao, D.S.; He, L.; Bi, Z.G. Exogenous cell-permeable C6 ceramide sensitizes multiple cancer cell lines to doxorubicin-induced apoptosis by promoting AMPK activation and mTORC1 inhibition. Oncogene 2010, 29, 6557–6568. [Google Scholar] [CrossRef]
- Stancevic, B.; Kolesnick, R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010, 584, 1728–1740. [Google Scholar] [CrossRef]
- Gao, X.; Lowry, P.R.; Zhou, X.; Depry, C.; Wei, Z.; Wong, G.W.; Zhang, J. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc. Natl. Acad. Sci. USA 2011, 108, 14509–14514. [Google Scholar]
- Chalfant, C.E.; Szulc, Z.; Roddy, P.; Bielawska, A.; Hannun, Y.A. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J. Lipid Res. 2004, 45, 496–506. [Google Scholar]
- Wang, G.; Silva, J.; Krishnamurthy, K.; Tran, E.; Condie, B.G.; Bieberich, E. Direct binding to ceramide activates protein kinase Czeta before the formation of a pro-apoptotic complex with PAR-4 in differentiating stem cells. J. Biol. Chem. 2005, 280, 26415–26424. [Google Scholar]
- Zhang, Y.; Yao, B.; Delikat, S.; Bayoumy, S.; Lin, X.H.; Basu, S.; McGinley, M.; Chan-Hui, P.Y.; Lichenstein, H.; Kolesnick, R. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell 1997, 89, 63–72. [Google Scholar] [CrossRef]
- Colombini, M. Membrane channels formed by ceramide. Handb. Exp. Pharmacol. 2013, 215, 109–126. [Google Scholar] [CrossRef]
- Yin, X.; Zafrullah, M.; Lee, H.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. A ceramide-binding C1 domain mediates kinase suppressor of ras membrane translocation. Cell. Physiol. Biochem. 2009, 24, 219–230. [Google Scholar] [CrossRef]
- Tagaram, H.R.; Divittore, N.A.; Barth, B.M.; Kaiser, J.M.; Avella, D.; Kimchi, E.T.; Jiang, Y.; Isom, H.C.; Kester, M.; Staveley-O’Carroll, K.F. Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut 2011, 60, 695–701. [Google Scholar] [CrossRef]
- Liu, X.; Ryland, L.; Yang, J.; Liao, A.; Aliaga, C.; Watts, R.; Tan, S.F.; Kaiser, J.; Shanmugavelandy, S.S.; Rogers, A.; et al. Targeting of survivin by nanoliposomal ceramide induces complete remission in a rat model of NK-LGL leukemia. Blood 2010, 116, 4192–4201. [Google Scholar] [CrossRef]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar] [CrossRef]
- Ginkel, C.; Hartmann, D.; vom Dorp, K.; Zlomuzica, A.; Farwanah, H.; Eckhardt, M.; Sandhoff, R.; Degen, J.; Rabionet, M.; Dere, E.; et al. Ablation of neuronal ceramide synthase 1 in mice decreases ganglioside levels and expression of myelin-associated glycoprotein in oligodendrocytes. J. Biol. Chem. 2012, 287, 41888–41902. [Google Scholar] [CrossRef]
- Imgrund, S.; Hartmann, D.; Farwanah, H.; Eckhardt, M.; Sandhoff, R.; Degen, J.; Gieselmann, V.; Sandhoff, K.; Willecke, K. Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J. Biol. Chem. 2009, 284, 33549–33560. [Google Scholar] [CrossRef]
- Sassa, T.; Ohno, Y.; Suzuki, S.; Nomura, T.; Nishioka, C.; Kashiwagi, T.; Hirayama, T.; Akiyama, M.; Taguchi, R.; Shimizu, H.; et al. Impaired epidermal permeability barrier in mice lacking elovl1, the gene responsible for very-long-chain fatty acid production. Mol. Cell. Biol. 2013, 33, 2787–2796. [Google Scholar] [CrossRef]
- Pewzner-Jung, Y.; Brenner, O.; Braun, S.; Laviad, E.L.; Ben-Dor, S.; Feldmesser, E.; Horn-Saban, S.; Amann-Zalcenstein, D.; Raanan, C.; Berkutzki, T.; et al. A critical role for ceramide synthase 2 in liver homeostasis: II. insights into molecular changes leading to hepatopathy. J. Biol. Chem. 2010, 285, 10911–10923. [Google Scholar] [CrossRef]
- Pewzner-Jung, Y.; Park, H.; Laviad, E.L.; Silva, L.C.; Lahiri, S.; Stiban, J.; Erez-Roman, R.; Brügger, B.; Sachsenheimer, T.; Wieland, F.; et al. A critical role for ceramide synthase 2 in liver homeostasis: I. alterations in lipid metabolic pathways. J. Biol. Chem. 2010, 285, 10902–10910. [Google Scholar] [CrossRef]
- Bieberich, E.; MacKinnon, S.; Silva, J.; Noggle, S.; Condie, B.G. Regulation of cell death in mitotic neural progenitor cells by asymmetric distribution of prostate apoptosis response 4 (PAR-4) and simultaneous elevation of endogenous ceramide. J. Cell Biol. 2003, 162, 469–479. [Google Scholar] [CrossRef]
- Krishnamurthy, K.; Wang, G.; Silva, J.; Condie, B.G.; Bieberich, E. Ceramide regulates atypical PKCzeta/lambda-mediated cell polarity in primitive ectoderm cells. A novel function of sphingolipids in morphogenesis. J. Biol. Chem. 2007, 282, 3379–3390. [Google Scholar] [CrossRef]
- Wang, G.; Krishnamurthy, K.; Chiang, Y.W.; Dasgupta, S.; Bieberich, E. Regulation of neural progenitor cell motility by ceramide and potential implications for mouse brain development. J. Neurochem. 2008, 106, 718–733. [Google Scholar] [CrossRef]
- Nakahara, K.; Ohkuni, A.; Kitamura, T.; Abe, K.; Naganuma, T.; Ohno, Y.; Zoeller, R.A.; Kihara, A. The Sjogren-Larsson syndrome gene encodes a hexadecenal dehydrogenase of the sphingosine 1-phosphate degradation pathway. Mol. Cell 2012, 46, 461–471. [Google Scholar] [CrossRef]
- Ohkuni, A.; Ohno, Y.; Kihara, A. Identification of acyl-CoA synthetases involved in the mammalian sphingosine 1-phosphate metabolic pathway. Biochem. Biophys. Res. Commun. 2013, 442, 195–201. [Google Scholar] [CrossRef]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef]
- Rosen, H.; Gonzalez-Cabrera, P.J.; Sanna, M.G.; Brown, S. Sphingosine 1-phosphate receptor signaling. Annu. Rev. Biochem. 2009, 78, 743–768. [Google Scholar] [CrossRef]
- Choi, J.W.; Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta 2013, 1831, 20–32. [Google Scholar]
- Woodcock, J.M.; Ma, Y.; Coolen, C.; Pham, D.; Jones, C.; Lopez, A.F.; Pitson, S.M. Sphingosine and FTY720 directly bind pro-survival 14-3-3 proteins to regulate their function. Cell. Signal. 2010, 22, 1291–1299. [Google Scholar] [CrossRef]
- Habrukowich, C.; Han, D.K.; Le, A.; Rezaul, K.; Pan, W.; Ghosh, M.; Li, Z.; Dodge-Kafka, K.; Jiang, X.; Bittman, R.; et al. Sphingosine interaction with acidic leucine-rich nuclear phosphoprotein-32A (ANP32A) regulates PP2A activity and cyclooxygenase (COX)-2 expression in human endothelial cells. J. Biol. Chem. 2010, 285, 26825–26831. [Google Scholar]
- Hannun, Y.A.; Bell, R.M. Lysosphingolipids inhibit protein kinase C: Implications for the sphingolipidoses. Science 1987, 235, 670–674. [Google Scholar]
- Hamaguchi, A.; Suzuki, E.; Murayama, K.; Fujimura, T.; Hikita, T.; Iwabuchi, K.; Handa, K.; Withers, D.A.; Masters, S.C.; Fu, H.; et al. Sphingosine-dependent protein kinase-1, directed to 14-3-3, is identified as the kinase domain of protein kinase C delta. J. Biol. Chem. 2003, 278, 41557–41565. [Google Scholar] [CrossRef]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 2005, 25, 11113–11121. [Google Scholar] [CrossRef]
- Bektas, M.; Allende, M.L.; Lee, B.G.; Chen, W.; Amar, M.J.; Remaley, A.T.; Saba, J.D.; Proia, R.L. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem. 2010, 285, 10880–10889. [Google Scholar]
- Allende, M.L.; Bektas, M.; Lee, B.G.; Bonifacino, E.; Kang, J.; Tuymetova, G.; Chen, W.; Saba, J.D.; Proia, R.L. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J. Biol. Chem. 2011, 286, 7348–7358. [Google Scholar] [CrossRef]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef]
- Chae, S.S.; Paik, J.H.; Furneaux, H.; Hla, T. Requirement for sphingosine 1-phosphate receptor-1 in tumor angiogenesis demonstrated by in vivo RNA interference. J. Clin. Invest. 2004, 114, 1082–1089. [Google Scholar] [CrossRef]
- Albinet, V.; Bats, M.L.; Huwiler, A.; Rochaix, P.; Chevreau, C.; Segui, B.; Levade, T.; Andrieu-Abadie, N. Dual role of sphingosine kinase-1 in promoting the differentiation of dermal fibroblasts and the dissemination of melanoma cells. Oncogene 2013. [Google Scholar] [CrossRef]
- Weigert, A.; Weis, N.; Brune, B. Regulation of macrophage function by sphingosine-1-phosphate. Immunobiology 2009, 214, 748–760. [Google Scholar] [CrossRef]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef]
- Sorensen, S.D.; Nicole, O.; Peavy, R.D.; Montoya, L.M.; Lee, C.J.; Murphy, T.J.; Traynelis, S.F.; Hepler, J.R. Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol. Pharmacol. 2003, 64, 1199–1209. [Google Scholar] [CrossRef]
- Kimura, T.; Watanabe, T.; Sato, K.; Kon, J.; Tomura, H.; Tamama, K.; Kuwabara, A.; Kanda, T.; Kobayashi, I.; Ohta, H.; et al. Sphingosine 1-phosphate stimulates proliferation and migration of human endothelial cells possibly through the lipid receptors, Edg-1 and Edg-3. Biochem. J. 2000, 348, 71–76. [Google Scholar] [CrossRef]
- Morita, Y.; Perez, G.I.; Paris, F.; Miranda, S.R.; Ehleiter, D.; Haimovitz-Friedman, A.; Fuks, Z.; Xie, Z.; Reed, J.C.; Schuchman, E.H.; et al. Oocyte apoptosis is suppressed by disruption of the acid sphingomyelinase gene or by sphingosine-1-phosphate therapy. Nat. Med. 2000, 6, 1109–1114. [Google Scholar] [CrossRef]
- Schnitzer, S.E.; Weigert, A.; Zhou, J.; Brune, B. Hypoxia enhances sphingosine kinase 2 activity and provokes sphingosine-1-phosphate-mediated chemoresistance in A549 lung cancer cells. Mol. Cancer Res. 2009, 7, 393–401. [Google Scholar] [CrossRef]
- Kimura, A.; Ohmori, T.; Kashiwakura, Y.; Ohkawa, R.; Madoiwa, S.; Mimuro, J.; Shimazaki, K.; Hoshino, Y.; Yatomi, Y.; Sakata, Y. Antagonism of sphingosine 1-phosphate receptor-2 enhances migration of neural progenitor cells toward an area of brain. Stroke 2008, 39, 3411–3417. [Google Scholar] [CrossRef]
- Sato, K.; Malchinkhuu, E.; Horiuchi, Y.; Mogi, C.; Tomura, H.; Tosaka, M.; Yoshimoto, Y.; Kuwabara, A.; Okajima, F. HDL-like lipoproteins in cerebrospinal fluid affect neural cell activity through lipoprotein-associated sphingosine 1-phosphate. Biochem. Biophys. Res. Commun. 2007, 359, 649–654. [Google Scholar] [CrossRef]
- Malchinkhuu, E.; Sato, K.; Horiuchi, Y.; Mogi, C.; Ohwada, S.; Ishiuchi, S.; Saito, N.; Kurose, H.; Tomura, H.; Okajima, F. Role of p38 mitogen-activated kinase and c-Jun terminal kinase in migration response to lysophosphatidic acid and sphingosine-1-phosphate in glioma cells. Oncogene 2005, 24, 6676–6688. [Google Scholar] [CrossRef]
- Sanchez, T.; Thangada, S.; Wu, M.T.; Kontos, C.D.; Wu, D.; Wu, H.; Hla, T. PTEN as an effector in the signaling of antimigratory G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 4312–4317. [Google Scholar]
- Miller, A.V.; Alvarez, S.E.; Spiegel, S.; Lebman, D.A. Sphingosine kinases and sphingosine-1-phosphate are critical for transforming growth factor beta-induced extracellular signal-regulated kinase 1 and 2 activation and promotion of migration and invasion of esophageal cancer cells. Mol. Cell. Biol. 2008, 28, 4142–4151. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Allende, M.L.; Dreier, J.L.; Mandala, S.; Proia, R.L. Expression of the sphingosine 1-phosphate receptor, S1P1, on T-cells controls thymic emigration. J. Biol. Chem. 2004, 279, 15396–15401. [Google Scholar] [CrossRef]
- Sanna, M.G.; Wang, S.K.; Gonzalez-Cabrera, P.J.; Don, A.; Marsolais, D.; Matheu, M.P.; Wei, S.H.; Parker, I.; Jo, E.; Cheng, W.C.; et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat. Chem. Biol. 2006, 2, 434–441. [Google Scholar] [CrossRef]
- Song, L.; Xiong, H.; Li, J.; Liao, W.; Wang, L.; Wu, J.; Li, M. Sphingosine kinase-1 enhances resistance to apoptosis through activation of PI3K/Akt/NF-kappaB pathway in human non-small cell lung cancer. Clin. Cancer Res. 2011, 17, 1839–1849. [Google Scholar] [CrossRef]
- Ruckhaberle, E.; Rody, A.; Engels, K.; Gaetje, R.; von Minckwitz, G.; Schiffmann, S.; Grösch, S.; Geisslinger, G.; Holtrich, U.; Karn, T.; et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res. Treat. 2008, 112, 41–52. [Google Scholar] [CrossRef]
- Watson, C.; Long, J.S.; Orange, C.; Tannahill, C.L.; Mallon, E.; McGlynn, L.M.; Pyne, S.; Pyne, N.J.; Edwards, J. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am. J. Pathol. 2010, 177, 2205–2215. [Google Scholar] [CrossRef]
- Li, W.; Yu, C.P.; Xia, J.T.; Zhang, L.; Weng, G.X.; Zheng, H.Q.; Kong, Q.L.; Hu, L.J.; Zeng, M.S.; Zeng, Y.X.; et al. Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin. Cancer Res. 2009, 15, 1393–1399. [Google Scholar] [CrossRef]
- Guan, H.; Liu, L.; Cai, J.; Liu, J.; Ye, C.; Li, M.; Li, Y. Sphingosine kinase 1 is overexpressed and promotes proliferation in human thyroid cancer. Mol. Endocrinol. 2011, 25, 1858–1866. [Google Scholar] [CrossRef]
- Malavaud, B.; Pchejetski, D.; Mazerolles, C.; de Paiva, G.R.; Calvet, C.; Doumerc, N.; Pitson, S.; Rischmann, P.; Cuvillier, O. Sphingosine kinase-1 activity and expression in human prostate cancer resection specimens. Eur. J. Cancer 2010, 46, 3417–3424. [Google Scholar] [CrossRef]
- Sinha, U.K.; Schorn, V.J.; Hochstim, C.; Chinn, S.B.; Zhu, S.; Masood, R. Increased radiation sensitivity of head and neck squamous cell carcinoma with sphingosine kinase 1 inhibition. Head Neck 2011, 33, 178–188. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: Roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 2005, 64, 695–705. [Google Scholar] [CrossRef]
- Li, J.; Guan, H.Y.; Gong, L.Y.; Song, L.B.; Zhang, N.; Wu, J.; Yuan, J.; Zheng, Y.J.; Huang, Z.S.; Li, M. Clinical significance of sphingosine kinase-1 expression in human astrocytomas progression and overall patient survival. Clin. Cancer Res. 2008, 14, 6996–7003. [Google Scholar]
- Xia, P.; Gamble, J.R.; Wang, L.; Pitson, S.M.; Moretti, P.A.; Wattenberg, B.W.; D’Andrea, R.J.; Vadas, M.A. An oncogenic role of sphingosine kinase. Curr. Biol. 2000, 10, 1527–1530. [Google Scholar] [CrossRef]
- Pitson, S.M.; Xia, P.; Leclercq, T.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Wattenberg, B.W.; Vadas, M.A. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J. Exp. Med. 2005, 201, 49–54. [Google Scholar] [CrossRef]
- Kawamori, T.; Kaneshiro, T.; Okumura, M.; Maalouf, S.; Uflacker, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009, 23, 405–414. [Google Scholar]
- Kohno, M.; Momoi, M.; Oo, M.L.; Paik, J.H.; Lee, Y.M.; Venkataraman, K.; Ai, Y.; Ristimaki, A.P.; Fyrst, H.; Sano, H.; et al. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol. Cell. Biol. 2006, 26, 7211–7223. [Google Scholar] [CrossRef]
- Shirai, K.; Kaneshiro, T.; Wada, M.; Furuya, H.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M.; Ogretmen, B.; Kawamori, T. A role of sphingosine kinase 1 in head and neck carcinogenesis. Cancer Prev. Res. 2011, 4, 454–462. [Google Scholar] [CrossRef]
- Heffernan-Stroud, L.A.; Helke, K.L.; Jenkins, R.W.; de Costa, A.M.; Hannun, Y.A.; Obeid, L.M. Defining a role for sphingosine kinase 1 in p53-dependent tumors. Oncogene 2012, 31, 1166–1175. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Selvam, S.P.; Mehrotra, S.; Kawamori, T.; Snider, A.J.; Obeid, L.M.; Shao, Y.; Sabbadini, R.; Ogretmen, B. Communication between host organism and cancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. EMBO Mol. Med. 2012, 4, 761–775. [Google Scholar] [CrossRef]
- Pchejetski, D.; Golzio, M.; Bonhoure, E.; Calvet, C.; Doumerc, N.; Garcia, V.; Mazerolles, C.; Rischmann, P.; Teissié, J.; Malavaud, B.; et al. Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 2005, 65, 11667–11675. [Google Scholar] [CrossRef]
- Salas, A.; Ponnusamy, S.; Senkal, C.E.; Meyers-Needham, M.; Selvam, S.P.; Saddoughi, S.A.; Apohan, E.; Sentelle, R.D.; Smith, C.; Gault, C.R.; et al. Sphingosine kinase-1 and sphingosine 1-phosphate receptor 2 mediate Bcr-Abl1 stability and drug resistance by modulation of protein phosphatase 2A. Blood 2011, 117, 5941–5952. [Google Scholar] [CrossRef]
- Kapitonov, D.; Allegood, J.C.; Mitchell, C.; Hait, N.C.; Almenara, J.A.; Adams, J.K.; Zipkin, R.E.; Dent, P.; Kordula, T.; Milstien, S.; et al. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009, 69, 6915–6923. [Google Scholar] [CrossRef]
- Visentin, B.; Vekich, J.A.; Sibbald, B.J.; Cavalli, A.L.; Moreno, K.M.; Matteo, R.G.; Garland, W.A.; Lu, Y.; Yu, S.; Hall, H.S.; et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 2006, 9, 225–238. [Google Scholar] [CrossRef]
- French, K.J.; Upson, J.J.; Keller, S.N.; Zhuang, Y.; Yun, J.K.; Smith, C.D. Antitumor activity of sphingosine kinase inhibitors. J. Pharmacol. Exp. Ther. 2006, 318, 596–603. [Google Scholar] [CrossRef]
- French, K.J.; Schrecengost, R.S.; Lee, B.D.; Zhuang, Y.; Smith, S.N.; Eberly, J.L.; Yun, J.K.; Smith, C.D. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003, 63, 5962–5969. [Google Scholar]
- Paugh, S.W.; Paugh, B.S.; Rahmani, M.; Kapitonov, D.; Almenara, J.A.; Kordula, T.; Milstien, S.; Adams, J.K.; Zipkin, R.E.; Grant, S.; et al. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood 2008, 112, 1382–1391. [Google Scholar] [CrossRef]
- Doll, F.; Pfeilschifter, J.; Huwiler, A. The epidermal growth factor stimulates sphingosine kinase-1 expression and activity in the human mammary carcinoma cell line MCF7. Biochim. Biophys. Acta 2005, 1738, 72–81. [Google Scholar]
- Estrada-Bernal, A.; Lawler, S.E.; Nowicki, M.O.; Ray Chaudhury, A.; van Brocklyn, J.R. The role of sphingosine kinase-1 in EGFRvIII-regulated growth and survival of glioblastoma cells. J. Neurooncol. 2010, 102, 353–366. [Google Scholar]
- Li, Q.F.; Huang, W.R.; Duan, H.F.; Wang, H.; Wu, C.T.; Wang, L.S. Sphingosine kinase-1 mediates BCR/ABL-induced upregulation of Mcl-1 in chronic myeloid leukemia cells. Oncogene 2007, 26, 7904–7908. [Google Scholar]
- Gault, C.R.; Eblen, S.T.; Neumann, C.A.; Hannun, Y.A.; Obeid, L.M. Oncogenic K-Ras regulates bioactive sphingolipids in a sphingosine kinase 1-dependent manner. J. Biol. Chem. 2012, 287, 31794–31803. [Google Scholar]
- Billich, A.; Bornancin, F.; Mechtcheriakova, D.; Natt, F.; Huesken, D.; Baumruker, T. Basal and induced sphingosine kinase 1 activity in A549 carcinoma cells: Function in cell survival and IL-1beta and TNF-alpha induced production of inflammatory mediators. Cell. Signal. 2005, 17, 1203–1217. [Google Scholar] [CrossRef]
- Paugh, B.S.; Bryan, L.; Paugh, S.W.; Wilczynska, K.M.; Alvarez, S.M.; Singh, S.K.; Kapitonov, D.; Rokita, H.; Wright, S.; Griswold-Prenner, I.; et al. Interleukin-1 regulates the expression of sphingosine kinase 1 in glioblastoma cells. J. Biol. Chem. 2009, 284, 3408–3417. [Google Scholar] [CrossRef]
- Xia, P.; Wang, L.; Moretti, P.A.; Albanese, N.; Chai, F.; Pitson, S.M.; D’Andrea, R.J.; Gamble, J.R.; Vadas, M.A. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J. Biol. Chem. 2002, 277, 7996–8003. [Google Scholar] [CrossRef]
- Anelli, V.; Gault, C.R.; Cheng, A.B.; Obeid, L.M. Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. Role of hypoxia-inducible factors 1 and 2. J. Biol. Chem. 2008, 283, 3365–3375. [Google Scholar]
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642. [Google Scholar] [CrossRef]
- Sukocheva, O.; Wang, L.; Verrier, E.; Vadas, M.A.; Xia, P. Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology 2009, 150, 4484–4492. [Google Scholar] [CrossRef]
- Baran, Y.; Salas, A.; Senkal, C.E.; Gunduz, U.; Bielawski, J.; Obeid, L.M.; Ogretmen, B. Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in K562 human chronic myeloid leukemia cells. J. Biol. Chem. 2007, 282, 10922–10934. [Google Scholar] [CrossRef]
- Antoon, J.W.; White, M.D.; Burow, M.E.; Beckman, B.S. Dual inhibition of sphingosine kinase isoforms ablates TNF-induced drug resistance. Oncol. Rep. 2012, 27, 1779–1786. [Google Scholar]
- Sukocheva, O.; Wadham, C.; Holmes, A.; Albanese, N.; Verrier, E.; Feng, F.; Bernal, A.; Derian, C.K.; Ullrich, A.; Vadas, M.A.; et al. Estrogen transactivates EGFR via the sphingosine 1-phosphate receptor Edg-3: The role of sphingosine kinase-1. J. Cell Biol. 2006, 173, 301–310. [Google Scholar] [CrossRef]
- Long, J.S.; Fujiwara, Y.; Edwards, J.; Tannahill, C.L.; Tigyi, G.; Pyne, S.; Pyne, N.J. Sphingosine 1-phosphate receptor 4 uses HER2 (ERBB2) to regulate extracellular signal regulated kinase-1/2 in MDA-MB-453 breast cancer cells. J. Biol. Chem. 2010, 285, 35957–35966. [Google Scholar]
- Guan, H.; Song, L.; Cai, J.; Huang, Y.; Wu, J.; Yuan, J.; Li, J.; Li, M. Sphingosine kinase 1 regulates the Akt/FOXO3a/Bim pathway and contributes to apoptosis resistance in glioma cells. PLoS One 2011, 6, e19946. [Google Scholar]
- Takuwa, Y.; Okamoto, Y.; Yoshioka, K.; Takuwa, N. Sphingosine-1-phosphate signaling in physiology and diseases. Biofactors 2012, 38, 329–337. [Google Scholar] [CrossRef]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef]
- Yoshida, Y.; Nakada, M.; Harada, T.; Tanaka, S.; Furuta, T.; Hayashi, Y.; Kita, D.; Uchiyama, N.; Hayashi, Y.; Hamada, J. The expression level of sphingosine-1-phosphate receptor type 1 is related to MIB-1 labeling index and predicts survival of glioblastoma patients. J. Neurooncol. 2010, 98, 41–47. [Google Scholar] [CrossRef]
- Yoshida, Y.; Nakada, M.; Sugimoto, N.; Harada, T.; Hayashi, Y.; Kita, D.; Uchiyama, N.; Hayashi, Y.; Yachie, A.; Takuwa, Y.; et al. Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int. J. Cancer 2010, 126, 2341–2352. [Google Scholar]
- Nagahashi, M.; Ramachandran, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, R.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012, 72, 726–735. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Stapleton, S.C.; Yang, M.T.; Cha, S.S.; Choi, C.K.; Galie, P.A.; Chen, C.S. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 6712–6717. [Google Scholar]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A metabolic shift favouring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef]
- Anelli, V.; Gault, C.R.; Snider, A.J.; Obeid, L.M. Role of sphingosine kinase-1 in paracrine/transcellular angiogenesis and lymphangiogenesis in vitro. FASEB J. 2010, 24, 2727–2738. [Google Scholar] [CrossRef]
- Yoon, C.M.; Hong, B.S.; Moon, H.G.; Lim, S.; Suh, P.G.; Kim, Y.K.; Chae, C.B.; Gho, Y.S. Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/Gi/PLC/Ca2+ signaling pathways. Blood 2008, 112, 1129–1138. [Google Scholar] [CrossRef]
- Jang, C.; Koh, Y.J.; Lim, N.K.; Kang, H.J.; Kim, D.H.; Park, S.K.; Lee, G.M.; Jeon, C.J.; Koh, G.Y. Angiopoietin-2 exocytosis is stimulated by sphingosine-1-phosphate in human blood and lymphatic endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 401–407. [Google Scholar] [CrossRef]
- Tornquist, K. Sphingosine 1-phosphate and cancer: Lessons from thyroid cancer cells. Biomolecules 2013, 3, 303–315. [Google Scholar] [CrossRef]
- Du, W.; Takuwa, N.; Yoshioka, K.; Okamoto, Y.; Gonda, K.; Sugihara, K.; Fukamizu, A.; Asano, M.; Takuwa, Y. S1P(2), the G protein-coupled receptor for sphingosine-1-phosphate, negatively regulates tumor angiogenesis and tumor growth in vivo in mice. Cancer Res. 2010, 70, 772–781. [Google Scholar] [CrossRef]
- Edsall, L.C.; van Brocklyn, J.R.; Cuvillier, O.; Kleuser, B.; Spiegel, S. N,N-Dimethylsphingosine is a potent competitive inhibitor of sphingosine kinase but not of protein kinase C: Modulation of cellular levels of sphingosine 1-phosphate and ceramide. Biochemistry 1998, 37, 12892–12898. [Google Scholar] [CrossRef]
- Endo, K.; Igarashi, Y.; Nisar, M.; Zhou, Q.H.; Hakomori, S. Cell membrane signaling as target in cancer therapy: Inhibitory effect of N,N-dimethyl and N,N,N-trimethyl sphingosine derivatives on in vitro and in vivo growth of human tumor cells in nude mice. Cancer Res. 1991, 51, 1613–1618. [Google Scholar]
- Jiang, Y.; Divittore, N.A.; Young, M.M.; Jia, Z.; Xie, K.; Ritty, T.M.; Kester, M.; Fox, T.E. Altered sphingolipid metabolism in patients with metastatic pancreatic Cancer. Biomolecules 2013, 3, 435–448. [Google Scholar] [CrossRef]
- Sutphen, R.; Xu, Y.; Wilbanks, G.D.; Fiorica, J.; Grendys, E.C., Jr.; LaPolla, J.P.; Arango, H.; Hoffman, M.S.; Martino, M.; Wakeley, K.; et al. Lysophospholipids are potential biomarkers of ovarian cancer. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 1185–1191. [Google Scholar]
- Schwab, S.R.; Pereira, J.P.; Matloubian, M.; Xu, Y.; Huang, Y.; Cyster, J.G. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005, 309, 1735–1739. [Google Scholar] [CrossRef]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef]
- Nunes, J.; Naymark, M.; Sauer, L.; Muhammad, A.; Keun, H.; Sturge, J.; Stebbing, J.; Waxman, J.; Pchejetski, D. Circulating sphingosine-1-phosphate and erythrocyte sphingosine kinase-1 activity as novel biomarkers for early prostate cancer detection. Br. J. Cancer 2012, 106, 909–915. [Google Scholar] [CrossRef]
- Igarashi, N.; Okada, T.; Hayashi, S.; Fujita, T.; Jahangeer, S.; Nakamura, S. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J. Biol. Chem. 2003, 278, 46832–46839. [Google Scholar]
- Liu, H.; Toman, R.E.; Goparaju, S.K.; Maceyka, M.; Nava, V.E.; Sankala, H.; Payne, S.G.; Bektas, M.; Ishii, I.; Chun, J.; et al. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J. Biol. Chem. 2003, 278, 40330–40336. [Google Scholar] [CrossRef]
- Gao, P.; Smith, C.D. Ablation of sphingosine kinase-2 inhibits tumor cell proliferation and migration. Mol. Cancer Res. 2011, 9, 1509–1519. [Google Scholar] [CrossRef]
- Antoon, J.W.; White, M.D.; Meacham, W.D.; Slaughter, E.M.; Muir, S.E.; Elliott, S.; Rhodes, L.V.; Ashe, H.B.; Wiese, T.E.; Smith, C.D.; et al. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology 2010, 151, 5124–5135. [Google Scholar] [CrossRef]
- White, M.D.; Chan, L.; Antoon, J.W.; Beckman, B.S. Targeting ovarian cancer and chemoresistance through selective inhibition of sphingosine kinase-2 with ABC294640. Anticancer Res. 2013, 33, 3573–3579. [Google Scholar]
- Beljanski, V.; Knaak, C.; Zhuang, Y.; Smith, C.D. Combined anticancer effects of sphingosine kinase inhibitors and sorafenib. Invest. New Drugs 2010, 29, 11323–1142. [Google Scholar]
- Beljanski, V.; Knaak, C.; Smith, C.D. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J. Pharmacol. Exp. Ther. 2010, 333, 454–464. [Google Scholar]
- French, K.J.; Zhuang, Y.; Maines, L.W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J.J.; Green, C.L.; Keller, S.N.; Smith, C.D. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [Google Scholar] [CrossRef]
- Chumanevich, A.A.; Poudyal, D.; Cui, X.; Davis, T.; Wood, P.A.; Smith, C.D.; Hofseth, L.J. Suppression of colitis-driven colon cancer in mice by a novel small molecule inhibitor of sphingosine kinase. Carcinogenesis 2010, 31, 1787–1793. [Google Scholar] [CrossRef]
- Liu, K.; Guo, T.L.; Hait, N.C.; Allegood, J.; Parikh, H.I.; Xu, W.; Kellogg, G.E.; Grant, S.; Spiegel, S.; Zhang, S. Biological characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a selective sphingosine kinase-2 inhibitor and anticancer agent. PLoS One 2013, 8, e56471. [Google Scholar] [CrossRef]
- Kharel, Y.; Raje, M.; Gao, M.; Gellett, A.M.; Tomsig, J.L.; Lynch, K.R.; Santos, W.L. Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1-phosphate. Biochem. J. 2012, 447, 149–157. [Google Scholar] [CrossRef]
- Sankala, H.M.; Hait, N.C.; Paugh, S.W.; Shida, D.; Lepine, S.; Elmore, L.W.; Dent, P.; Milstien, S.; Spiegel, S. Involvement of sphingosine kinase 2 in p53-independent induction of p21 by the chemotherapeutic drug doxorubicin. Cancer Res. 2007, 67, 10466–10474. [Google Scholar] [CrossRef]
- Hofmann, L.P.; Ren, S.; Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine kinase 1 and 2 regulate the capacity of mesangial cells to resist apoptotic stimuli in an opposing manner. Biol. Chem. 2008, 389, 1399–1407. [Google Scholar]
- Oskouian, B.; Sooriyakumaran, P.; Borowsky, A.D.; Crans, A.; Dillard-Telm, L.; Tam, Y.Y.; Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 17384–17389. [Google Scholar] [CrossRef]
- Brizuela, L.; Ader, I.; Mazerolles, C.; Bocquet, M.; Malavaud, B.; Cuvillier, O. First evidence of sphingosine 1-phosphate lyase protein expression and activity downregulation in human neoplasm: Implication for resistance to therapeutics in prostate cancer. Mol. Cancer Ther. 2012, 11, 1841–1851. [Google Scholar] [CrossRef]
- Colie, S.; van Veldhoven, P.P.; Kedjouar, B.; Bedia, C.; Albinet, V.; Sorli, S.C.; Garcia, V.; Djavaheri-Mergny, M.; Bauvy, C.; Codogno, P.; et al. Disruption of sphingosine 1-phosphate lyase confers resistance to chemotherapy and promotes oncogenesis through Bcl-2/Bcl-xL upregulation. Cancer Res. 2009, 69, 9346–9353. [Google Scholar] [CrossRef]
- Petersen, N.H.; Olsen, O.D.; Groth-Pedersen, L.; Ellegaard, A.M.; Bilgin, M.; Redmer, S.; Ostenfeld, M.S.; Ulanet, D.; Dovmark, T.H.; Lønborg, A.; et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 2013, 24, 379–393. [Google Scholar] [CrossRef]
- Savic, R.; He, X.; Fiel, I.; Schuchman, E.H. Recombinant human acid sphingomyelinase as an adjuvant to sorafenib treatment of experimental liver cancer. PLoS One 2013, 8, e65620. [Google Scholar]
- Riboni, L.; Campanella, R.; Bassi, R.; Villani, R.; Gaini, S.M.; Martinelli-Boneschi, F.; Viani, P.; Tettamanti, G. Ceramide levels are inversely associated with malignant progression of human glial tumors. Glia 2002, 39, 105–113. [Google Scholar] [CrossRef]
- Karahatay, S.; Thomas, K.; Koybasi, S.; Senkal, C.E.; Elojeimy, S.; Liu, X.; Bielawski, J.; Day, T.A.; Gillespie, M.B.; Sinha, D.; et al. Clinical relevance of ceramide metabolism in the pathogenesis of human head and neck squamous cell carcinoma (HNSCC): Attenuation of C(18)-ceramide in HNSCC tumors correlates with lymphovascular invasion and nodal metastasis. Cancer Lett. 2007, 256, 101–111. [Google Scholar] [CrossRef]
- Koybasi, S.; Senkal, C.E.; Sundararaj, K.; Spassieva, S.; Bielawski, J.; Osta, W.; Day, T.A.; Jiang, J.C.; Jazwinski, S.M.; Hannun, Y.A.; et al. Defects in cell growth regulation by C18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J. Biol. Chem. 2004, 279, 44311–44319. [Google Scholar] [CrossRef]
- Senkal, C.E.; Ponnusamy, S.; Bielawski, J.; Hannun, Y.A.; Ogretmen, B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. 2010, 24, 296–308. [Google Scholar] [CrossRef]
- Senkal, C.E.; Ponnusamy, S.; Manevich, Y.; Meyers-Needham, M.; Saddoughi, S.A.; Mukhopadyay, A.; Dent, P.; Bielawski, J.; Ogretmen, B. Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J. Biol. Chem. 2011, 286, 42446–42458. [Google Scholar]
- Schiffmann, S.; Sandner, J.; Birod, K.; Wobst, I.; Angioni, C.; Ruckhäberle, E.; Kaufmann, M.; Ackermann, H.; Lötsch, J.; Schmidt, H.; et al. Ceramide synthases and ceramide levels are increased in breast cancer tissue. Carcinogenesis 2009, 30, 745–752. [Google Scholar] [CrossRef]
- Erez-Roman, R.; Pienik, R.; Futerman, A.H. Increased ceramide synthase 2 and 6 mRNA levels in breast cancer tissues and correlation with sphingosine kinase expression. Biochem. Biophys. Res. Commun. 2010, 391, 219–223. [Google Scholar] [CrossRef]
- Hartmann, D.; Lucks, J.; Fuchs, S.; Schiffmann, S.; Schreiber, Y.; Ferreiros, N.; Merkens, J.; Marschalek, R.; Geisslinger, G.; Grosch, S. Long chain ceramides and very long chain ceramides have opposite effects on human breast and colon cancer cell growth. Int. J. Biochem. Cell Biol. 2012, 44, 620–628. [Google Scholar] [CrossRef]
- Mesicek, J.; Lee, H.; Feldman, T.; Jiang, X.; Skobeleva, A.; Berdyshev, E.V.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cell. Signal. 2010, 22, 1300–1307. [Google Scholar] [CrossRef]
- White-Gilbertson, S.; Mullen, T.; Senkal, C.; Lu, P.; Ogretmen, B.; Obeid, L.; Voelkel-Johnson, C. Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene 2009, 28, 1132–1141. [Google Scholar] [CrossRef]
- Seelan, R.S.; Qian, C.; Yokomizo, A.; Bostwick, D.G.; Smith, D.I.; Liu, W. Human acid ceramidase is overexpressed but not mutated in prostate cancer. Genes Chromosomes Cancer 2000, 29, 137–146. [Google Scholar]
- Norris, J.S.; Bielawska, A.; Day, T.; El-Zawahri, A.; ElOjeimy, S.; Hannun, Y.; Holman, D.; Hyer, M.; Landon, C.; Lowe, S.; et al. Combined therapeutic use of AdGFPFasL and small molecule inhibitors of ceramide metabolism in prostate and head and neck cancers: A status report. Cancer Gene Ther. 2006, 13, 1045–1051. [Google Scholar] [CrossRef]
- Camacho, L.; Meca-Cortés, O.; Abad, J.L.; García, S.; Rubio, N.; Díaz, A.; Celià-Terrassa, T.; Cingolani, F.; Bermudo, R.; Fernández, P.L.; et al. Acid ceramidase as a therapeutic target in metastatic prostate cancer. J. Lipid Res. 2013, 54, 1207–1220. [Google Scholar] [CrossRef]
- Elojeimy, S.; Liu, X.; McKillop, J.C.; El-Zawahry, A.M.; Holman, D.H.; Cheng, J.Y.; Meacham, W.D.; Mahdy, A.E.; Saad, A.F.; Turner, L.S.; et al. Role of acid ceramidase in resistance to FasL: Therapeutic approaches based on acid ceramidase inhibitors and FasL gene therapy. Mol. Ther. 2007, 15, 1259–1263. [Google Scholar]
- Shah, M.V.; Zhang, R.; Irby, R.; Kothapalli, R.; Liu, X.; Arrington, T.; Frank, B.; Lee, N.H.; Loughran, T.P., Jr. Molecular profiling of LGL leukemia reveals role of sphingolipid signaling in survival of cytotoxic lymphocytes. Blood 2008, 112, 770–781. [Google Scholar] [CrossRef]
- Cheng, J.C.; Bai, A.; Beckham, T.H.; Marrison, S.T.; Yount, C.L.; Young, K.; Lu, P.; Bartlett, A.M.; Wu, B.X.; Keane, B.J.; et al. Radiation-induced acid ceramidase confers prostate cancer resistance and tumor relapse. J. Clin. Invest. 2013, 123, 4344–4358. [Google Scholar]
- Beckham, T.H.; Cheng, J.C.; Lu, P.; Marrison, S.T.; Norris, J.S.; Liu, X. Acid ceramidase promotes nuclear export of PTEN through sphingosine 1-phosphate mediated Akt signaling. PLoS One 2013, 8, e76593. [Google Scholar]
- Beckham, T.H.; Cheng, J.C.; Lu, P.; Shao, Y.; Troyer, D.; Lance, R.; Marrison, S.T.; Norris, J.S.; Liu, X. Acid ceramidase induces sphingosine kinase 1/S1P receptor 2-mediated activation of oncogenic Akt signaling. Oncogenesis 2013, 2, e49. [Google Scholar] [CrossRef]
- Beckham, T.H.; Lu, P.; Cheng, J.C.; Zhao, D.; Turner, L.S.; Zhang, X.; Hoffman, S.; Armeson, K.E.; Liu, A.; Marrison, T.; et al. Acid ceramidase-mediated production of sphingosine 1-phosphate promotes prostate cancer invasion through upregulation of cathepsin B. Int. J. Cancer 2012, 131, 2034–2043. [Google Scholar] [CrossRef]
- Hanker, L.C.; Karn, T.; Holtrich, U.; Gatje, R.; Rody, A.; Heinrich, T.; Ruckhaberle, E.; Engels, K. Acid ceramidase (AC)—A key enzyme of sphingolipid metabolism—Correlates with better prognosis in epithelial ovarian cancer. Int. J. Gynecol. Pathol. 2013, 32, 249–257. [Google Scholar] [CrossRef]
- Ruckhaberle, E.; Holtrich, U.; Engels, K.; Hanker, L.; Gatje, R.; Metzler, D.; Karn, T.; Kaufmann, M.; Rody, A. Acid ceramidase 1 expression correlates with a better prognosis in ER-positive breast cancer. Climacteric 2009, 12, 502–513. [Google Scholar]
- Gangoiti, P.; Arana, L.; Ouro, A.; Granado, M.H.; Trueba, M.; Gomez-Munoz, A. Activation of mTOR and RhoA is a major mechanism by which Ceramide 1-phosphate stimulates macrophage proliferation. Cell. Signal. 2011, 23, 27–34. [Google Scholar] [CrossRef]
- Granado, M.H.; Gangoiti, P.; Ouro, A.; Arana, L.; Gonzalez, M.; Trueba, M.; Gomez-Munoz, A. Ceramide 1-phosphate (C1P) promotes cell migration Involvement of a specific C1P receptor. Cell. Signal. 2009, 21, 405–412. [Google Scholar] [CrossRef]
- Pettus, B.J.; Bielawska, A.; Spiegel, S.; Roddy, P.; Hannun, Y.A.; Chalfant, C.E. Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J. Biol. Chem. 2003, 278, 38206–38213. [Google Scholar]
- Pettus, B.J.; Bielawska, A.; Subramanian, P.; Wijesinghe, D.S.; Maceyka, M.; Leslie, C.C.; Evans, J.H.; Freiberg, J.; Roddy, P.; Hannun, Y.A.; et al. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J. Biol. Chem. 2004, 279, 11320–11326. [Google Scholar] [CrossRef]
- Scott, K.F.; Sajinovic, M.; Hein, J.; Nixdorf, S.; Galettis, P.; Liauw, W.; de Souza, P.; Dong, Q.; Graham, G.G.; Russell, P.J. Emerging roles for phospholipase A2 enzymes in cancer. Biochimie 2010, 92, 601–610. [Google Scholar] [CrossRef]
- Mitra, P.; Maceyka, M.; Payne, S.G.; Lamour, N.; Milstien, S.; Chalfant, C.E.; Spiegel, S. Ceramide kinase regulates growth and survival of A549 human lung adenocarcinoma cells. FEBS Lett. 2007, 581, 735–740. [Google Scholar] [CrossRef]
- Bini, F.; Frati, A.; Garcia-Gil, M.; Battistini, C.; Granado, M.; Martinesi, M.; Mainardi, M.; Vannini, E.; Luzzati, F.; Caleo, M.; et al. New signalling pathway involved in the anti-proliferative action of vitamin D(3) and its analogues in human neuroblastoma cells. A role for ceramide kinase. Neuropharmacology 2012, 63, 524–537. [Google Scholar] [CrossRef]
- Barth, B.M.; Gustafson, S.J.; Hankins, J.L.; Kaiser, J.M.; Haakenson, J.K.; Kester, M.; Kuhn, T.B. Ceramide kinase regulates TNFalpha-stimulated NADPH oxidase activity and eicosanoid biosynthesis in neuroblastoma cells. Cell. Signal. 2012, 24, 1126–1133. [Google Scholar] [CrossRef]
- Ruckhaberle, E.; Karn, T.; Rody, A.; Hanker, L.; Gatje, R.; Metzler, D.; Holtrich, U.; Kaufmann, M. Gene expression of ceramide kinase, galactosyl ceramide synthase and ganglioside GD3 synthase is associated with prognosis in breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 1005–1013. [Google Scholar] [CrossRef]
- Duan, R.D. Alkaline sphingomyelinase: An old enzyme with novel implications. Biochim. Biophys. Acta 2006, 1761, 281–291. [Google Scholar]
- De Maria, R.; Rippo, M.R.; Schuchman, E.H.; Testi, R. Acidic sphingomyelinase (ASM) is necessary for fas-induced GD3 ganglioside accumulation and efficient apoptosis of lymphoid cells. J. Exp. Med. 1998, 187, 897–902. [Google Scholar] [CrossRef]
- Kim, W.J.; Okimoto, R.A.; Purton, L.E.; Goodwin, M.; Haserlat, S.M.; Dayyani, F.; Sweetser, D.A.; McClatchey, A.I.; Bernard, O.A.; Look, A.T.; et al. Mutations in the neutral sphingomyelinase gene SMPD3 implicate the ceramide pathway in human leukemias. Blood 2008, 111, 4716–4722. [Google Scholar]
- Hertervig, E.; Nilsson, A.; Nyberg, L.; Duan, R.D. Alkaline sphingomyelinase activity is decreased in human colorectal carcinoma. Cancer 1997, 79, 448–453. [Google Scholar] [CrossRef]
- Osawa, Y.; Suetsugu, A.; Matsushima-Nishiwaki, R.; Yasuda, I.; Saibara, T.; Moriwaki, H.; Seishima, M.; Kozawa, O. Liver acid sphingomyelinase inhibits growth of metastatic colon cancer. J. Clin. Invest. 2013, 123, 834–843. [Google Scholar]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef]
- Utermohlen, O.; Herz, J.; Schramm, M.; Kronke, M. Fusogenicity of membranes: The impact of acid sphingomyelinase on innate immune responses. Immunobiology 2008, 213, 307–314. [Google Scholar] [CrossRef]
- Magenau, A.; Benzing, C.; Proschogo, N.; Don, A.S.; Hejazi, L.; Karunakaran, D.; Jessup, W.; Gaus, K. Phagocytosis of IgG-coated polystyrene beads by macrophages induces and requires high membrane order. Traffic 2011, 12, 1730–1743. [Google Scholar] [CrossRef]
- Hendrich, A.B.; Michalak, K. Lipids as a target for drugs modulating multidrug resistance of cancer cells. Curr. Drug Targets 2003, 4, 23–30. [Google Scholar]
- Barcelo-Coblijn, G.; Martin, M.L.; de Almeida, R.F.; Noguera-Salva, M.A.; Marcilla-Etxenike, A.; Guardiola-Serrano, F.; Luth, A.; Kleuser, B.; Halver, J.E.; Escriba, P.V. Sphingomyelin and sphingomyelin synthase (SMS) in the malignant transformation of glioma cells and in 2-hydroxyoleic acid therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19569–19574. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Expression of glucosylceramide synthase, converting ceramide to glucosylceramide, confers adriamycin resistance in human breast cancer cells. J. Biol. Chem. 1999, 274, 1140–1146. [Google Scholar]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Hansen, N.; Cabot, M.C. Uncoupling ceramide glycosylation by transfection of glucosylceramide synthase antisense reverses adriamycin resistance. J. Biol. Chem. 2000, 275, 7138–7143. [Google Scholar] [CrossRef]
- Giussani, P.; Bassi, R.; Anelli, V.; Brioschi, L.; de Zen, F.; Riccitelli, E.; Caroli, M.; Campanella, R.; Gaini, S.M.; Viani, P.; et al. Glucosylceramide synthase protects glioblastoma cells against autophagic and apoptotic death induced by temozolomide and Paclitaxel. Cancer Invest. 2012, 30, 27–37. [Google Scholar] [CrossRef]
- Nicholson, K.M.; Quinn, D.M.; Kellett, G.L.; Warr, J.R. Preferential killing of multidrug-resistant KB cells by inhibitors of glucosylceramide synthase. Br. J. Cancer 1999, 81, 423–430. [Google Scholar] [CrossRef]
- Lucci, A.; Cho, W.I.; Han, T.Y.; Giuliano, A.E.; Morton, D.L.; Cabot, M.C. Glucosylceramide: A marker for multiple-drug resistant cancers. Anticancer Res. 1998, 18, 475–480. [Google Scholar]
- Lavie, Y.; Cao, H.; Bursten, S.L.; Giuliano, A.E.; Cabot, M.C. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J. Biol. Chem. 1996, 271, 19530–19536. [Google Scholar]
- Patwardhan, G.A.; Zhang, Q.J.; Yin, D.; Gupta, V.; Bao, J.; Senkal, C.E.; Ogretmen, B.; Cabot, M.C.; Shah, G.V.; Sylvester, P.W.; et al. A new mixed-backbone oligonucleotide against glucosylceramide synthase sensitizes multidrug-resistant tumors to apoptosis. PLoS One 2009, 4, e6938. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Gupta, V.; Patwardhan, G.A.; Bhinge, K.; Zhao, Y.; Bao, J.; Mehendale, H.; Cabot, M.C.; Li, Y.T.; Jazwinski, S.M. Glucosylceramide synthase upregulates MDR1 expression in the regulation of cancer drug resistance through cSrc and beta-catenin signaling. Mol. Cancer 2010. [Google Scholar] [CrossRef]
- Itoh, M.; Kitano, T.; Watanabe, M.; Kondo, T.; Yabu, T.; Taguchi, Y.; Iwai, K.; Tashima, M.; Uchiyama, T.; Okazaki, T. Possible role of ceramide as an indicator of chemoresistance: Decrease of the ceramide content via activation of glucosylceramide synthase and sphingomyelin synthase in chemoresistant leukemia. Clin. Cancer Res. 2003, 9, 415–423. [Google Scholar]
- Huang, W.C.; Tsai, C.C.; Chen, C.L.; Chen, T.Y.; Chen, Y.P.; Lin, Y.S.; Lu, P.J.; Lin, C.M.; Wang, S.H.; Tsao, C.W.; et al. Glucosylceramide synthase inhibitor PDMP sensitizes chronic myeloid leukemia T315I mutant to Bcr-Abl inhibitor and cooperatively induces glycogen synthase kinase-3-regulated apoptosis. FASEB J. 2011, 25, 3661–3673. [Google Scholar] [CrossRef]
- Chai, L.; McLaren, R.P.; Byrne, A.; Chuang, W.L.; Huang, Y.; Dufault, M.R.; Pacheco, J.; Madhiwalla, S.; Zhang, X.; Zhang, M.; et al. The chemosensitizing activity of inhibitors of glucosylceramide synthase is mediated primarily through modulation of P-gp function. Int. J. Oncol. 2011, 38, 701–711. [Google Scholar]
- Gouaze, V.; Liu, Y.Y.; Prickett, C.S.; Yu, J.Y.; Giuliano, A.E.; Cabot, M.C. Glucosylceramide synthase blockade down-regulates P-glycoprotein and resensitizes multidrug-resistant breast cancer cells to anticancer drugs. Cancer Res. 2005, 65, 3861–3867. [Google Scholar]
- Xie, P.; Shen, Y.F.; Shi, Y.P.; Ge, S.M.; Gu, Z.H.; Wang, J.; Mu, H.J.; Zhang, B.; Qiao, W.Z.; Xie, K.M. Overexpression of glucosylceramide synthase in associated with multidrug resistance of leukemia cells. Leuk Res. 2008, 32, 475–480. [Google Scholar] [CrossRef]
- Gupta, V.; Bhinge, K.N.; Hosain, S.B.; Xiong, K.; Gu, X.; Shi, R.; Ho, M.Y.; Khoo, K.H.; Li, S.C.; Li, Y.T.; et al. Ceramide glycosylation by glucosylceramide synthase selectively maintains the properties of breast cancer stem cells. J. Biol. Chem. 2012, 287, 37195–37205. [Google Scholar]
- Sun, C.C.; Zhang, Z.; Zhang, S.Y.; Li, J.; Li, Z.L.; Kong, C.Z. Up-regulation of glucosylceramide synthase in urinary bladder neoplasms. Urol. Oncol. 2012, 30, 444–449. [Google Scholar]
- Ruckhaberle, E.; Karn, T.; Hanker, L.; Gatje, R.; Metzler, D.; Holtrich, U.; Kaufmann, M.; Rody, A. Prognostic relevance of glucosylceramide synthase (GCS) expression in breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 81–90. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Patwardhan, G.A.; Xie, P.; Gu, X.; Giuliano, A.E.; Cabot, M.C. Glucosylceramide synthase, a factor in modulating drug resistance, is overexpressed in metastatic breast carcinoma. Int. J. Oncol. 2011, 39, 425–431. [Google Scholar]
- Birkle, S.; Zeng, G.; Gao, L.; Yu, R.K.; Aubry, J. Role of tumor-associated gangliosides in cancer progression. Biochimie 2003, 85, 455–463. [Google Scholar] [CrossRef]
- Furukawa, K.; Hamamura, K.; Ohkawa, Y.; Ohmi, Y.; Furukawa, K. Disialyl gangliosides enhance tumor phenotypes with differential modalities. Glycoconj. J. 2012, 29, 579–584. [Google Scholar] [CrossRef]
- Durrant, L.G.; Noble, P.; Spendlove, I. Immunology in the clinic review series; focus on cancer: Glycolipids as targets for tumour immunotherapy. Clin. Exp. Immunol. 2012, 167, 206–215. [Google Scholar] [CrossRef]
- Chang, W.W.; Lee, C.H.; Lee, P.; Lin, J.; Hsu, C.W.; Hung, J.T.; Lin, J.J.; Yu, J.C.; Shao, L.E.; Yu, J.; et al. Expression of Globo H and SSEA3 in breast cancer stem cells and the involvement of fucosyl transferases 1 and 2 in Globo H synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 11667–11672. [Google Scholar] [CrossRef]
- Siddiqui, B.; Whitehead, J.S.; Kim, Y.S. Glycosphingolipids in human colonic adenocarcinoma. J. Biol. Chem. 1978, 253, 2168–2175. [Google Scholar]
- Kakugawa, Y.; Wada, T.; Yamaguchi, K.; Yamanami, H.; Ouchi, K.; Sato, I.; Miyagi, T. Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc. Natl. Acad. Sci. USA 2002, 99, 10718–10723. [Google Scholar] [CrossRef]
- Kovbasnjuk, O.; Mourtazina, R.; Baibakov, B.; Wang, T.; Elowsky, C.; Choti, M.A.; Kane, A.; Donowitz, M. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 19087–19092. [Google Scholar] [CrossRef]
- Distler, U.; Souady, J.; Hülsewig, M.; Drmić-Hofman, I.; Haier, J.; Friedrich, A.W.; Karch, H.; Senninger, N.; Dreisewerd, K.; Berkenkamp, S.; et al. Shiga toxin receptor Gb3Cer/CD77: Tumor-association and promising therapeutic target in pancreas and colon cancer. PLoS One 2009, 4, e6813. [Google Scholar] [CrossRef]
- Sung, C.C.; Pearl, D.K.; Coons, S.W.; Scheithauer, B.W.; Johnson, P.C.; Yates, A.J. Gangliosides as diagnostic markers of human astrocytomas and primitive neuroectodermal tumors. Cancer 1994, 74, 3010–3022. [Google Scholar] [CrossRef]
- Comas, T.C.; Tai, T.; Kimmel, D.; Scheithauer, B.W.; Burger, P.C.; Pearl, D.K.; Jewell, S.D.; Yates, A.J. Immunohistochemical staining for ganglioside GD1b as a diagnostic and prognostic marker for primary human brain tumors. Neuro Oncol. 1999, 1, 261–267. [Google Scholar]
- Berra, B.; Gaini, S.M.; Riboni, L. Correlation between ganglioside distribution and histological grading of human astrocytomas. Int. J. Cancer 1985, 36, 363–366. [Google Scholar]
- Fredman, P.; von Holst, H.; Collins, V.P.; Dellheden, B.; Svennerholm, L. Expression of gangliosides GD3 and 3'-isoLM1 in autopsy brains from patients with malignant tumors. J. Neurochem. 1993, 60, 99–105. [Google Scholar] [CrossRef]
- Nakamura, K.; Hanibuchi, M.; Yano, S.; Tanaka, Y.; Fujino, I.; Inoue, M.; Takezawa, T.; Shitara, K.; Sone, S.; Hanai, N. Apoptosis induction of human lung cancer cell line in multicellular heterospheroids with humanized antiganglioside GM2 monoclonal antibody. Cancer Res. 1999, 59, 5323–5330. [Google Scholar]
- Cheresh, D.A.; Rosenberg, J.; Mujoo, K.; Hirschowitz, L.; Reisfeld, R.A. Biosynthesis and expression of the disialoganglioside GD2, a relevant target antigen on small cell lung carcinoma for monoclonal antibody-mediated cytolysis. Cancer Res. 1986, 46, 5112–5118. [Google Scholar]
- Yoshida, S.; Fukumoto, S.; Kawaguchi, H.; Sato, S.; Ueda, R.; Furukawa, K. Ganglioside G(D2) in small cell lung cancer cell lines: Enhancement of cell proliferation and mediation of apoptosis. Cancer Res. 2001, 61, 4244–4252. [Google Scholar]
- Brezicka, T.; Bergman, B.; Olling, S.; Fredman, P. Reactivity of monoclonal antibodies with ganglioside antigens in human small cell lung cancer tissues. Lung Cancer 2000, 28, 29–36. [Google Scholar] [CrossRef]
- Brezicka, F.T.; Olling, S.; Bergman, B.; Berggren, H.; Engstrom, C.P.; Hammarstrom, S.; Holmgren, J.; Larsson, S.; Lindholm, L. Coexpression of ganglioside antigen Fuc-GM1, neural-cell adhesion molecule, carcinoembryonic antigen, and carbohydrate tumor-associated antigen CA 50 in lung cancer. Tumour Biol. 1992, 13, 308–315. [Google Scholar] [CrossRef]
- Brezicka, F.T.; Olling, S.; Nilsson, O.; Bergh, J.; Holmgren, J.; Sorenson, S.; Yngvason, F.; Lindholm, L. Immunohistological detection of fucosyl-GM1 ganglioside in human lung cancer and normal tissues with monoclonal antibodies. Cancer Res. 1989, 49, 1300–1305. [Google Scholar]
- Chang, F.; Li, R.; Ladisch, S. Shedding of gangliosides by human medulloblastoma cells. Exp. Cell Res. 1997, 234, 341–346. [Google Scholar] [CrossRef]
- Tsuchida, T.; Saxton, R.E.; Morton, D.L.; Irie, R.F. Gangliosides of human melanoma. J. Natl. Cancer Inst. 1987, 78, 45–54. [Google Scholar]
- Thurin, J.; Thurin, M.; Herlyn, M.; Elder, D.E.; Steplewski, Z.; Clark, W.H., Jr.; Koprowski, H. GD2 ganglioside biosynthesis is a distinct biochemical event in human melanoma tumor progression. FEBS Lett. 1986, 208, 17–22. [Google Scholar]
- Cheresh, D.A.; Harper, J.R.; Schulz, G.; Reisfeld, R.A. Localization of the gangliosides GD2 and GD3 in adhesion plaques and on the surface of human melanoma cells. Proc. Natl. Acad. Sci. USA 1984, 81, 5767–5771. [Google Scholar] [CrossRef]
- Tsuchida, T.; Saxton, R.E.; Morton, D.L.; Irie, R.F. Gangliosides of human melanoma. Cancer 1989, 63, 1166–1174. [Google Scholar] [CrossRef]
- Mujoo, K.; Cheresh, D.A.; Yang, H.M.; Reisfeld, R.A. Disialoganglioside GD2 on human neuroblastoma cells: Target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor growth. Cancer Res. 1987, 47, 1098–1104. [Google Scholar]
- Wu, Z.L.; Schwartz, E.; Seeger, R.; Ladisch, S. Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res. 1986, 46, 440–443. [Google Scholar]
- Saito, S.; Orikasa, S.; Ohyama, C.; Satoh, M.; Fukushi, Y. Changes in glycolipids in human renal-cell carcinoma and their clinical significance. Int. J. Cancer 1991, 49, 329–334. [Google Scholar] [CrossRef]
- Sakakibara, N.; Gasa, S.; Kamio, K.; Makita, A.; Nonomura, K.; Togashi, M.; Koyanagi, T.; Hatae, Y.; Takeda, K. Distinctive glycolipid patterns in Wilms’ tumor and renal cell carcinoma. Cancer Lett. 1991, 57, 187–192. [Google Scholar] [CrossRef]
- Sa, G.; Das, T.; Moon, C.; Hilston, C.M.; Rayman, P.A.; Rini, B.I.; Tannenbaum, C.S.; Finke, J.H. GD3, an overexpressed tumor-derived ganglioside, mediates the apoptosis of activated but not resting T cells. Cancer Res. 2009, 69, 3095–3104. [Google Scholar] [CrossRef]
- Portoukalian, J.; David, M.J.; Gain, P.; Richard, M. Shedding of GD2 ganglioside in patients with retinoblastoma. Int. J. Cancer 1993, 53, 948–951. [Google Scholar] [CrossRef]
- Lo, A.S.; Ma, Q.; Liu, D.L.; Junghans, R.P. Anti-GD3 chimeric sFv-CD28/T-cell receptor zeta designer T cells for treatment of metastatic melanoma and other neuroectodermal tumors. Clin. Cancer Res. 2010, 16, 2769–2780. [Google Scholar] [CrossRef]
- Chapman, P.B.; Wu, D.; Ragupathi, G.; Lu, S.; Williams, L.; Hwu, W.J.; Johnson, D.; Livingston, P.O. Sequential immunization of melanoma patients with GD3 ganglioside vaccine and anti-idiotypic monoclonal antibody that mimics GD3 ganglioside. Clin. Cancer Res. 2004, 10, 4717–4723. [Google Scholar] [CrossRef]
- Webb, T.J.; Li, X.; Giuntoli, R.L., 2nd; Lopez, P.H.; Heuser, C.; Schnaar, R.L.; Tsuji, M.; Kurts, C.; Oelke, M.; Schneck, J.P. Molecular identification of GD3 as a suppressor of the innate immune response in ovarian cancer. Cancer Res. 2012, 72, 3744–3752. [Google Scholar] [CrossRef]
- De Maria, R.; Lenti, L.; Malisan, F.; d’Agostino, F.; Tomassini, B.; Zeuner, A.; Rippo, M.R.; Testi, R. Requirement for GD3 ganglioside in CD95- and ceramide-induced apoptosis. Science 1997, 277, 1652–1655. [Google Scholar] [CrossRef]
- Omran, O.M.; Saqr, H.E.; Yates, A.J. Molecular mechanisms of GD3-induced apoptosis in U-1242 MG glioma cells. Neurochem. Res. 2006, 31, 1171–1180. [Google Scholar] [CrossRef]
- Oblinger, J.L.; Pearl, D.K.; Boardman, C.L.; Saqr, H.; Prior, T.W.; Scheithauer, B.W.; Jenkins, R.B.; Burger, P.C.; Yates, A.J. Diagnostic and prognostic value of glycosyltransferase mRNA in glioblastoma multiforme patients. Neuropathol. Appl. Neurobiol. 2006, 32, 410–418. [Google Scholar] [CrossRef]
- Saqr, H.E.; Omran, O.; Dasgupta, S.; Yu, R.K.; Oblinger, J.L.; Yates, A.J. Endogenous GD3 ganglioside induces apoptosis in U-1242 MG glioma cells. J. Neurochem. 2006, 96, 1301–1314. [Google Scholar] [CrossRef]
- Coetzee, T.; Fujita, N.; Dupree, J.; Shi, R.; Blight, A.; Suzuki, K.; Suzuki, K.; Popko, B. Myelination in the absence of galactocerebroside and sulfatide: Normal structure with abnormal function and regional instability. Cell 1996, 86, 209–219. [Google Scholar] [CrossRef]
- Dupree, J.L.; Coetzee, T.; Blight, A.; Suzuki, K.; Popko, B. Myelin galactolipids are essential for proper node of Ranvier formation in the CNS. J. Neurosci. 1998, 18, 1642–1649. [Google Scholar]
- Liu, Y.; Chen, Y.; Momin, A.; Shaner, R.; Wang, E.; Bowen, N.J.; Matyunina, L.V.; Walker, L.D.; McDonald, J.F.; Sullards, M.C.; et al. Elevation of sulfatides in ovarian cancer: An integrated transcriptomic and lipidomic analysis including tissue-imaging mass spectrometry. Mol. Cancer 2010, 9, 186. [Google Scholar] [CrossRef]
- Kiguchi, K.; Takamatsu, K.; Tanaka, J.; Nozawa, S.; Iwamori, M.; Nagai, Y. Glycosphingolipids of various human ovarian tumors: A significantly high expression of I3SO3GalCer and Lewis antigen in mucinous cystadenocarcinoma. Cancer Res. 1992, 52, 416–421. [Google Scholar]
- Makhlouf, A.M.; Fathalla, M.M.; Zakhary, M.A.; Makarem, M.H. Sulfatides in ovarian tumors: Clinicopathological correlates. Int. J. Gynecol. Cancer 2004, 14, 89–93. [Google Scholar]
- Morichika, H.; Hamanaka, Y.; Tai, T.; Ishizuka, I. Sulfatides as a predictive factor of lymph node metastasis in patients with colorectal adenocarcinoma. Cancer 1996, 78, 43–47. [Google Scholar] [CrossRef]
- Aruffo, A.; Kolanus, W.; Walz, G.; Fredman, P.; Seed, B. CD62/P-selectin recognition of myeloid and tumor cell sulfatides. Cell 1991, 67, 35–44. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Patwardhan, G.A.; Bhinge, K.; Gupta, V.; Gu, X.; Jazwinski, S.M. Suppression of glucosylceramide synthase restores p53-dependent apoptosis in mutant p53 cancer cells. Cancer Res. 2011, 71, 2276–2285. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Heemers, H.; Deboel, L.; Foufelle, F.; Heyns, W.; Verhoeven, G. Stimulation of tumor-associated fatty acid synthase expression by growth factor activation of the sterol regulatory element-binding protein pathway. Oncogene 2000, 19, 5173–5181. [Google Scholar] [CrossRef]
- Louie, S.M.; Roberts, L.S.; Mulvihill, M.M.; Luo, K.; Nomura, D.K. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim. Biophys. Acta 2013, 1831, 1566–1572. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Yu, J.Y.; Yin, D.; Patwardhan, G.A.; Gupta, V.; Hirabayashi, Y.; Holleran, W.M.; Giuliano, A.E.; Jazwinski, S.M.; Gouaze-Andersson, V.; et al. A role for ceramide in driving cancer cell resistance to doxorubicin. FASEB J. 2008, 22, 2541–2551. [Google Scholar] [CrossRef]
- Alvarez-Vasquez, F.; Sims, K.J.; Cowart, L.A.; Okamoto, Y.; Voit, E.O.; Hannun, Y.A. Simulation and validation of modelled sphingolipid metabolism in Saccharomyces cerevisiae. Nature 2005, 433, 425–430. [Google Scholar] [CrossRef]
- Alvarez-Vasquez, F.; Sims, K.J.; Voit, E.O.; Hannun, Y.A. Coordination of the dynamics of yeast sphingolipid metabolism during the diauxic shift. Theor. Biol. Med. Model. 2007, 4, 42. [Google Scholar] [CrossRef]
- Chen, P.W.; Fonseca, L.L.; Hannun, Y.A.; Voit, E.O. Coordination of rapid sphingolipid responses to heat stress in yeast. PLoS Comput. Biol. 2013, 9, e1003078. [Google Scholar] [CrossRef]
- Gupta, S.; Maurya, M.R.; Merrill, A.H., Jr.; Glass, C.K.; Subramaniam, S. Integration of lipidomics and transcriptomics data towards a systems biology model of sphingolipid metabolism. BMC Syst. Biol. 2011. [Google Scholar] [CrossRef]
- Mora, R.; Dokic, I.; Kees, T.; Huber, C.M.; Keitel, D.; Geibig, R.; Brugge, B.; Zentgraf, H.; Brady, N.R.; Regnier-Vigouroux, A. Sphingolipid rheostat alterations related to transformation can be exploited for specific induction of lysosomal cell death in murine and human glioma. Glia 2010, 58, 1364–1383. [Google Scholar]
- Momin, A.A.; Park, H.; Portz, B.J.; Haynes, C.A.; Shaner, R.L.; Kelly, S.L.; Jordan, I.K.; Merrill, A.H., Jr. A method for visualization of “omic” datasets for sphingolipid metabolism to predict potentially interesting differences. J. Lipid Res. 2011, 52, 1073–1083. [Google Scholar] [CrossRef]
- Vital-Lopez, F.G.; Wallqvist, A.; Reifman, J. Bridging the gap between gene expression and metabolic phenotype via kinetic models. BMC Syst. Biol. 2013. [Google Scholar] [CrossRef]
- Dickson, M.A.; Carvajal, R.D.; Merrill, A.H., Jr.; Gonen, M.; Cane, L.M.; Schwartz, G.K. A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clin. Cancer Res. 2011, 17, 2484–2492. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Don, A.S.; Lim, X.Y.; Couttas, T.A. Re-Configuration of Sphingolipid Metabolism by Oncogenic Transformation. Biomolecules 2014, 4, 315-353. https://doi.org/10.3390/biom4010315
Don AS, Lim XY, Couttas TA. Re-Configuration of Sphingolipid Metabolism by Oncogenic Transformation. Biomolecules. 2014; 4(1):315-353. https://doi.org/10.3390/biom4010315
Chicago/Turabian StyleDon, Anthony S., Xin Y. Lim, and Timothy A. Couttas. 2014. "Re-Configuration of Sphingolipid Metabolism by Oncogenic Transformation" Biomolecules 4, no. 1: 315-353. https://doi.org/10.3390/biom4010315