Heavy Metals and Metalloids As a Cause for Protein Misfolding and Aggregation

Abstract
:1. Introduction
2. Impact of Heavy Metals on Native, Correctly Folded Proteins
3. Proteins Are Vulnerable during Folding, Be It In Vitro or In Vivo
4. Heavy Metals and Metalloids Interact with Functional Groups of Chemically Denatured Proteins and Inhibit Their In Vitro Refolding

| Kd’ at pH 7 * | Approximate pKa in proteins | |||
|---|---|---|---|---|
| Cd2+ | Hg2+ | Pb2+ | ||
| Thiol group | 2.5 µM | 0.063 nM | 13 µM | 9.4 |
| Imidazole group | 2.0 mM | 200 µM | 6.3 mM | 6.5 |
| Carboxyl group | 16 mM | 2.5 µM | 13 mM | 4.6 |
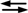 M + L, where ML is the 1:1 complex of the metal ion (M) and ligand (L) [45].
M + L, where ML is the 1:1 complex of the metal ion (M) and ligand (L) [45].| IC50 value (nM) | ||||
|---|---|---|---|---|
| Cd2+ | Hg2+ | Pb2+ | NaAsO2 | |
| Luciferase | ||||
| Spontaneous refolding | 66 ± 11 | 40 ± 3 | 63 ± 6 | 426 ± 4 |
| Chaperone-assisted refolding | 100 ± 5 | 53 ± 2 | 140 ± 11 | 297 ± 13 |
| Chaperone-mediated disaggregation | 280 ± 4 | 210 ± 16 | 325 ± 11 | 613 ± 31 |
| Lactate dehydrogenase | ||||
| Spontaneous refolding | 68 ± 2 | 58 ± 6 | 74 ± 9 | ND |
| Malate dehydrogenase | ||||
| Spontaneous refolding | 300 ± 45 | 290 ± 16 | 520 ± 44 | ND |
| Glucose-6-phosphate dehydrogenase | ||||
| Spontaneous refolding | 340 ± 15 | 230 ± 18 | >600 | ND |
5. Heavy Metals and Metalloids Interfere with Protein Folding and Induce Protein Aggregation in Living Cells
6. How Cells Deal with Heavy Metal- and Metalloid-Induced Protein Misfolding and Aggregation
7. Metal-Induced Protein Aggregation and Protein Misfolding Disorders
8. Perspectives
Acknowledgments
Conflicts of Interest
References
- Tamás, M.J.; Martinoia, E. Molecular Biology of Metal Homeostasis and Detoxification: From Microbes to Man; Springer Verlag: Heidelberg, Germany, 2005. [Google Scholar]
- Fraústo da Silva, J.J.R.; Williams, R.J.P. The Biological Chemistry of the Elements: The Inorganic Chemistry of Life; Clarendon Press: Oxford, UK, 1993. [Google Scholar]
- Lemire, J.A.; Harrison, J.J.; Turner, R.J. Antimicrobial activity of metals: Mechanisms, molecular targets and applications. Nat. Rev. Microbiol. 2013, 11, 371–384. [Google Scholar] [CrossRef]
- Beyersmann, D.; Hartwig, A. Carcinogenic metal compounds: Recent insight into molecular and cellular mechanisms. Arch. Toxicol. 2008, 82, 493–512. [Google Scholar] [CrossRef]
- Wysocki, R.; Tamás, M.J. How Saccharomyces cerevisiae copes with toxic metals and metalloids. FEMS Microbiol. Rev. 2010, 34, 925–951. [Google Scholar] [CrossRef]
- Sharma, S.K.; Goloubinoff, P.; Christen, P. Non-Native Proteins as Newly Identified Targets of Heavy Metals and Metalloids. In Cellular Effects of Heavy Metals; Bánfalvi, G., Ed.; Springer: Heidelberg, Germany, 2011; pp. 263–274. [Google Scholar]
- Sharma, S.K.; Goloubinoff, P.; Christen, P. Heavy metal ions are potent inhibitors of protein folding. Biochem. Biophys. Res. Commun. 2008, 372, 341–345. [Google Scholar] [CrossRef]
- Ramadan, D.; Rancy, P.C.; Nagarkar, R.P.; Schneider, J.P.; Thorpe, C. Arsenic(III) species inhibit oxidative protein folding in vitro. Biochemistry 2009, 48, 424–432. [Google Scholar] [CrossRef]
- Jacobson, T.; Navarrete, C.; Sharma, S.K.; Sideri, T.C.; Ibstedt, S.; Priya, S.; Grant, C.M.; Christen, P.; Goloubinoff, P.; Tamás, M.J. Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J. Cell Sci. 2012, 125, 5073–5083. [Google Scholar] [CrossRef] [Green Version]
- Holland, S.; Lodwig, E.; Sideri, T.; Reader, T.; Clarke, I.; Gkargkas, K.; Hoyle, D.C.; Delneri, D.; Oliver, S.G.; Avery, S.V. Application of the comprehensive set of heterozygous yeast deletion mutants to elucidate the molecular basis of cellular chromium toxicity. Genome Biol. 2007, 8, R268. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef]
- Breydo, L.; Uversky, V.N. Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics 2011, 3, 1163–1180. [Google Scholar] [CrossRef]
- Alies, B.; Hureau, C.; Faller, P. The role of metal ions in amyloid formation: General principles from model peptides. Metallomics 2013, 5, 183–192. [Google Scholar] [CrossRef]
- Caudle, W.M.; Guillot, T.S.; Lazo, C.R.; Miller, G.W. Industrial toxicants and Parkinson’s disease. Neurotoxicology 2012, 33, 178–188. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Lee, S.; Liu, Y.; Lim, M.H. Untangling amyloid-beta, tau, and metals in Alzheimer’s disease. ACS Chem. Biol. 2013, 8, 856–865. [Google Scholar] [CrossRef]
- Bourassa, M.W.; Miller, L.M. Metal imaging in neurodegenerative diseases. Metallomics 2012, 4, 721–738. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788. [Google Scholar] [CrossRef]
- Buchberger, A.; Bukau, B.; Sommer, T. Protein quality control in the cytosol and the endoplasmic reticulum: Brothers in arms. Mol. Cell 2010, 40, 238–252. [Google Scholar] [CrossRef]
- Waldron, K.J.; Rutherford, J.C.; Ford, D.; Robinson, N.J. Metalloproteins and metal sensing. Nature 2009, 460, 823–830. [Google Scholar] [CrossRef]
- Bleackley, M.R.; Macgillivray, R.T. Transition metal homeostasis: From yeast to human disease. Biometals 2011, 24, 785–809. [Google Scholar] [CrossRef]
- Finney, L.A.; O’Halloran, T.V. Transition metal speciation in the cell: Insights from the chemistry of metal ion receptors. Science 2003, 300, 931–936. [Google Scholar] [CrossRef]
- Reyes-Caballero, H.; Campanello, G.C.; Giedroc, D.P. Metalloregulatory proteins: Metal selectivity and allosteric switching. Biophys. Chem. 2011, 156, 103–114. [Google Scholar] [CrossRef]
- Naganuma, A.; Miura, N.; Kaneko, S.; Mishina, T.; Hosoya, S.; Miyairi, S.; Furuchi, T.; Kuge, S. GFAT as a target molecule of methylmercury toxicity in Saccharomyces cerevisiae. FASEB J. 2000, 14, 968–972. [Google Scholar]
- Chrestensen, C.A.; Starke, D.W.; Mieyal, J.J. Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J. Biol. Chem. 2000, 275, 26556–26565. [Google Scholar] [CrossRef]
- Hartwig, A. Zinc finger proteins as potential targets for toxic metal ions: Differential effects on structure and function. Antioxid. Redox Signal. 2001, 3, 625–634. [Google Scholar] [CrossRef]
- Faller, P.; Kienzler, K.; Krieger-Liszkay, A. Mechanism of Cd2+ toxicity: Cd2+ inhibits photoactivation of Photosystem II by competitive binding to the essential Ca2+ site. Biochim. Biophys. Acta 2005, 1706, 158–164. [Google Scholar] [CrossRef]
- Jin, Y.H.; Clark, A.B.; Slebos, R.J.; Al-Refai, H.; Taylor, J.A.; Kunkel, T.A.; Resnick, M.A.; Gordenin, D.A. Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat. Genet. 2003, 34, 326–329. [Google Scholar] [CrossRef]
- Banerjee, S.; Flores-Rozas, H. Cadmium inhibits mismatch repair by blocking the ATPase activity of the MSH2-MSH6 complex. Nucleic Acids Res. 2005, 33, 1410–1419. [Google Scholar] [CrossRef]
- Shen, S.; Li, X.F.; Cullen, W.R.; Weinfeld, M.; Le, X.C. Arsenic binding to proteins. Chem. Rev. 2013, 113, 7769–7792. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, F.; Shim, J.Y.; Kirk, K.L.; Anderson, D.E.; Chen, X. Identification of arsenic-binding proteins in human breast cancer cells. Cancer Lett. 2007, 255, 95–106. [Google Scholar] [CrossRef]
- Lu, J.; Chew, E.H.; Holmgren, A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. USA 2007, 104, 12288–12293. [Google Scholar] [CrossRef]
- Zhang, X.W.; Yan, X.J.; Zhou, Z.R.; Yang, F.F.; Wu, Z.Y.; Sun, H.B.; Liang, W.X.; Song, A.X.; Lallemand-Breitenbach, V.; Jeanne, M.; et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science 2010, 328, 240–243. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de Thé, H. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Samikkannu, T.; Chen, C.H.; Yih, L.H.; Wang, A.S.; Lin, S.Y.; Chen, T.C.; Jan, K.Y. Reactive oxygen species are involved in arsenic trioxide inhibition of pyruvate dehydrogenase activity. Chem. Res. Toxicol. 2003, 16, 409–414. [Google Scholar] [CrossRef]
- Brandt, F.; Etchells, S.A.; Ortiz, J.O.; Elcock, A.H.; Hartl, F.U.; Baumeister, W. The native 3D organization of bacterial polysomes. Cell 2009, 136, 261–271. [Google Scholar] [CrossRef]
- Sharma, S.K.; de los Rios, P.; Christen, P.; Lustig, A.; Goloubinoff, P. The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat. Chem. Biol. 2010, 6, 914–920. [Google Scholar]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar]
- Robertson, A.D.; Murphy, K.P. Protein structure and the energetics of protein stability. Chem. Rev. 1997, 97, 1251–1268. [Google Scholar] [CrossRef]
- Wedemeyer, W.J.; Welker, E.; Scheraga, H.A. Proline cis-trans isomerization and protein folding. Biochemistry 2002, 41, 14637–14644. [Google Scholar] [CrossRef]
- Gurd, F.R.; Wilcox, P.E. Complex formation between metallic cations and proteins, peptides and amino acids. Adv. Protein Chem. 1956, 11, 311–427. [Google Scholar] [CrossRef]
- Vallee, B.L.; Ulmer, D.D. Biochemical effects of mercury, cadmium, and lead. Annu. Rev. Biochem. 1972, 41, 91–128. [Google Scholar] [CrossRef]
- Kägi, J.H.R.; Hapke, H.-J. Biochemical Interactions of Mercury, Cadmium and Lead. In Changing Metal Cycles and Human Health; Nriagu, J.O., Ed.; Springer Berlin: Dahlem Konferenzen, Germany, 1984; pp. 237–250. [Google Scholar]
- Dennis, P.P.; Bremer, H. Differential rate of ribosomal protein synthesis in Escherichia coli B/r. J. Mol. Biol. 1974, 84, 407–422. [Google Scholar] [CrossRef]
- Piques, M.; Schulze, W.X.; Hohne, M.; Usadel, B.; Gibon, Y.; Rohwer, J.; Stitt, M. Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol. Syst. Biol. 2009, 5, 314. [Google Scholar]
- Jacobson, T.; Ashouri, A.; Tanghe, R.; Sharma, S.K.; Priya, S.; Goloubinoff, P.; Christen, P.; Tamás, M.J. Cadmium interferes with protein folding and increases chaperone load in yeast. 2014; unpublished work, manuscript in preparation. [Google Scholar]
- Rudolph, R.; Gerschitz, J.; Jaenicke, R. Effect of zinc(II) on the refolding and reactivation of liver alcohol dehydrogenase. Eur. J. Biochem. 1978, 87, 601–606. [Google Scholar] [CrossRef]
- Ibstedt, S.; Sideri, T.C.; Grant, C.M.; Tamás, M.J. Global analysis of protein aggregation in yeast during physiological conditions and arsenite stress. 2014; unpublished work, manuscript in preparation. [Google Scholar]
- Pan, X.; Reissman, S.; Douglas, N.R.; Huang, Z.; Yuan, D.S.; Wang, X.; McCaffery, J.M.; Frydman, J.; Boeke, J.D. Trivalent arsenic inhibits the functions of chaperonin complex. Genetics 2010, 186, 725–734. [Google Scholar] [CrossRef]
- Marino, S.M.; Li, Y.; Fomenko, D.E.; Agisheva, N.; Cerny, R.L.; Gladyshev, V.N. Characterization of surface-exposed reactive cysteine residues in Saccharomyces cerevisiae. Biochemistry 2010, 49, 7709–7721. [Google Scholar] [CrossRef]
- Medicherla, B.; Goldberg, A.L. Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J. Cell Biol. 2008, 182, 663–673. [Google Scholar] [CrossRef]
- Gardarin, A.; Chedin, S.; Lagniel, G.; Aude, J.C.; Godat, E.; Catty, P.; Labarre, J. Endoplasmic reticulum is a major target of cadmium toxicity in yeast. Mol. Microbiol. 2010, 76, 1034–1048. [Google Scholar] [CrossRef]
- Yokouchi, M.; Hiramatsu, N.; Hayakawa, K.; Kasai, A.; Takano, Y.; Yao, J.; Kitamura, M. Atypical, bidirectional regulation of cadmium-induced apoptosis via distinct signaling of unfolded protein response. Cell Death Differ. 2007, 14, 1467–1474. [Google Scholar] [CrossRef]
- Liu, F.; Inageda, K.; Nishitai, G.; Matsuoka, M. Cadmium induces the expression of Grp78, an endoplasmic reticulum molecular chaperone, in LLC-PK1 renal epithelial cells. Environ. Health Perspect. 2006, 114, 859–864. [Google Scholar] [CrossRef]
- Hiramatsu, N.; Kasai, A.; Du, S.; Takeda, M.; Hayakawa, K.; Okamura, M.; Yao, J.; Kitamura, M. Rapid, transient induction of ER stress in the liver and kidney after acute exposure to heavy metal: Evidence from transgenic sensor mice. FEBS Lett. 2007, 581, 2055–2059. [Google Scholar] [CrossRef]
- Biagioli, M.; Pifferi, S.; Ragghianti, M.; Bucci, S.; Rizzuto, R.; Pinton, P. Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium 2008, 43, 184–195. [Google Scholar]
- Bernales, S.; Papa, F.R.; Walter, P. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2006, 22, 487–508. [Google Scholar] [CrossRef]
- Bonilla, M.; Nastase, K.K.; Cunningham, K.W. Essential role of calcineurin in response to endoplasmic reticulum stress. EMBO J. 2002, 21, 2343–2353. [Google Scholar] [CrossRef]
- Sumner, E.R.; Shanmuganathan, A.; Sideri, T.C.; Willetts, S.A.; Houghton, J.E.; Avery, S.V. Oxidative protein damage causes chromium toxicity in yeast. Microbiology 2005, 151, 1939–1948. [Google Scholar] [CrossRef]
- Johnston, D.; Oppermann, H.; Jackson, J.; Levinson, W. Induction of four proteins in chick embryo cells by sodium arsenite. J. Biol. Chem. 1980, 255, 6975–6980. [Google Scholar]
- Levinson, W.; Oppermann, H.; Jackson, J. Transition series metals and sulfhydryl reagents induce the synthesis of four proteins in eukaryotic cells. Biochim. Biophys. Acta 1980, 606, 170–180. [Google Scholar] [CrossRef]
- Zheng, P.Z.; Wang, K.K.; Zhang, Q.Y.; Huang, Q.H.; Du, Y.Z.; Zhang, Q.H.; Xiao, D.K.; Shen, S.H.; Imbeaud, S.; Eveno, E.; et al. Systems analysis of transcriptome and proteome in retinoic acid/arsenic trioxide-induced cell differentiation/apoptosis of promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2005, 102, 7653–7658. [Google Scholar] [CrossRef]
- Thorsen, M.; Lagniel, G.; Kristiansson, E.; Junot, C.; Nerman, O.; Labarre, J.; Tamás, M.J. Quantitative transcriptome, proteome, and sulfur metabolite profiling of the Saccharomyces cerevisiae response to arsenite. Physiol. Genomics 2007, 30, 35–43. [Google Scholar] [CrossRef]
- Jin, Y.H.; Dunlap, P.E.; McBride, S.J.; Al-Refai, H.; Bushel, P.R.; Freedman, J.H. Global transcriptome and deletome profiles of yeast exposed to transition metals. PLoS Genet. 2008, 4, e1000053. [Google Scholar] [CrossRef]
- Haugen, A.C.; Kelley, R.; Collins, J.B.; Tucker, C.J.; Deng, C.; Afshari, C.A.; Brown, J.M.; Ideker, T.; van Houten, B. Integrating phenotypic and expression profiles to map arsenic-response networks. Genome Biol. 2004, 5, R95. [Google Scholar] [CrossRef] [Green Version]
- Kusakabe, T.; Nakajima, K.; Nakazato, K.; Suzuki, K.; Takada, H.; Satoh, T.; Oikawa, M.; Arakawa, K.; Nagamine, T. Changes of heavy metal, metallothionein and heat shock proteins in Sertoli cells induced by cadmium exposure. Toxicol. in Vitro: Int. J. Publ. Assoc. BIBRA 2008, 22, 1469–1475. [Google Scholar]
- Wagner, M.; Hermanns, I.; Bittinger, F.; Kirkpatrick, C.J. Induction of stress proteins in human endothelial cells by heavy metal ions and heat shock. Am. J. Physiol. 1999, 277, L1026–L1033. [Google Scholar]
- Sanchez, Y.; Taulien, J.; Borkovich, K.A.; Lindquist, S. Hsp104 is required for tolerance to many forms of stress. EMBO J. 1992, 11, 2357–2364. [Google Scholar]
- Stanhill, A.; Haynes, C.M.; Zhang, Y.; Min, G.; Steele, M.C.; Kalinina, J.; Martinez, E.; Pickart, C.M.; Kong, X.P.; Ron, D. An arsenite-inducible 19S regulatory particle-associated protein adapts proteasomes to proteotoxicity. Mol. Cell 2006, 23, 875–885. [Google Scholar] [CrossRef]
- Kirkpatrick, D.S.; Dale, K.V.; Catania, J.M.; Gandolfi, A.J. Low-level arsenite causes accumulation of ubiquitinated proteins in rabbit renal cortical slices and HEK293 cells. Toxicol. Appl. Pharmacol. 2003, 186, 101–109. [Google Scholar] [CrossRef]
- Yun, C.; Stanhill, A.; Yang, Y.; Zhang, Y.; Haynes, C.M.; Xu, C.F.; Neubert, T.A.; Mor, A.; Philips, M.R.; Ron, D. Proteasomal adaptation to environmental stress links resistance to proteotoxicity with longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2008, 105, 7094–7099. [Google Scholar] [CrossRef]
- Gsponer, J.; Babu, M.M. Cellular strategies for regulating functional and nonfunctional protein aggregation. Cell Rep. 2012, 2, 1425–1437. [Google Scholar] [CrossRef]
- Thorsen, M.; Perrone, G.G.; Kristiansson, E.; Traini, M.; Ye, T.; Dawes, I.W.; Nerman, O.; Tamás, M.J. Genetic basis of arsenite and cadmium tolerance in Saccharomyces cerevisiae. BMC Genomics 2009, 10, 105. [Google Scholar] [CrossRef]
- Sanchez, Y.; Lindquist, S.L. HSP104 required for induced thermotolerance. Science 1990, 248, 1112–1115. [Google Scholar]
- Weibezahn, J.; Tessarz, P.; Schlieker, C.; Zahn, R.; Maglica, Z.; Lee, S.; Zentgraf, H.; Weber-Ban, E.U.; Dougan, D.A.; Tsai, F.T.; et al. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 2004, 119, 653–665. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Navarrete, C.; Tamás, M.J. Arsenic-induced protein aggregates are primarily cleared via proteasomal degradation. 2014; unpublished work. [Google Scholar]
- Di, Y.; Tamás, M.J. Regulation of the arsenic-responsive transcription factor Yap8p involves the ubiquitin-proteasome pathway. J. Cell Sci. 2007, 120, 256–264. [Google Scholar] [CrossRef]
- Momose, Y.; Iwahashi, H. Bioassay of cadmium using a DNA microarray: Genome-wide expression patterns of Saccharomyces cerevisiae response to cadmium. Environ. Toxicol. Chem. 2001, 20, 2353–2360. [Google Scholar] [CrossRef]
- Vido, K.; Spector, D.; Lagniel, G.; Lopez, S.; Toledano, M.B.; Labarre, J. A proteome analysis of the cadmium response in Saccharomyces cerevisiae. J. Biol. Chem. 2001, 276, 8469–8474. [Google Scholar] [CrossRef]
- Fauchon, M.; Lagniel, G.; Aude, J.C.; Lombardia, L.; Soularue, P.; Petat, C.; Marguerie, G.; Sentenac, A.; Werner, M.; Labarre, J. Sulfur sparing in the yeast proteome in response to sulfur demand. Mol. Cell 2002, 9, 713–723. [Google Scholar] [CrossRef]
- Thorsen, M.; Jacobson, T.; Vooijs, R.; Navarrete, C.; Bliek, T.; Schat, H.; Tamás, M.J. Glutathione serves an extracellular defence function to decrease arsenite accumulation and toxicity in yeast. Mol. Microbiol. 2012, 84, 1177–1188. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Rastgou Talemi, S.; Jacobson, T.; Garla, V.; Navarrete, C.; Wagner, A.; Tamás, M.J.; Schaber, J. Mathematical modeling of arsenic transport, intracellular distribution and detoxification processes in yeast. 2014, unpublished work, submitted. [Google Scholar]
- Tartaglia, G.G.; Pechmann, S.; Dobson, C.M.; Vendruscolo, M. Life on the edge: A link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci. 2007, 32, 204–206. [Google Scholar] [CrossRef]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef]
- Gong, G.; O’Bryant, S.E. The arsenic exposure hypothesis for Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 311–316. [Google Scholar] [CrossRef]
- Singh, A.P.; Goel, R.K.; Kaur, T. Mechanisms pertaining to arsenic toxicity. Toxicol. Int. 2011, 18, 87–93. [Google Scholar] [CrossRef]
- Hinault, M.P.; Farina-Henriquez-Cuendet, A.; Goloubinoff, P. Molecular chaperones and associated cellular clearance mechanisms against toxic protein conformers in Parkinson’s disease. Neurodegener. Dis. 2011, 8, 397–412. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tamás, M.J.; Sharma, S.K.; Ibstedt, S.; Jacobson, T.; Christen, P. Heavy Metals and Metalloids As a Cause for Protein Misfolding and Aggregation. Biomolecules 2014, 4, 252-267. https://doi.org/10.3390/biom4010252
Tamás MJ, Sharma SK, Ibstedt S, Jacobson T, Christen P. Heavy Metals and Metalloids As a Cause for Protein Misfolding and Aggregation. Biomolecules. 2014; 4(1):252-267. https://doi.org/10.3390/biom4010252
Chicago/Turabian StyleTamás, Markus J., Sandeep K. Sharma, Sebastian Ibstedt, Therese Jacobson, and Philipp Christen. 2014. "Heavy Metals and Metalloids As a Cause for Protein Misfolding and Aggregation" Biomolecules 4, no. 1: 252-267. https://doi.org/10.3390/biom4010252