Refolding Techniques for Recovering Biologically Active Recombinant Proteins from Inclusion Bodies

Abstract
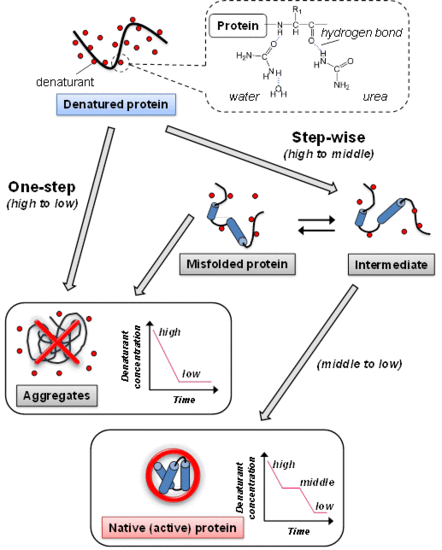
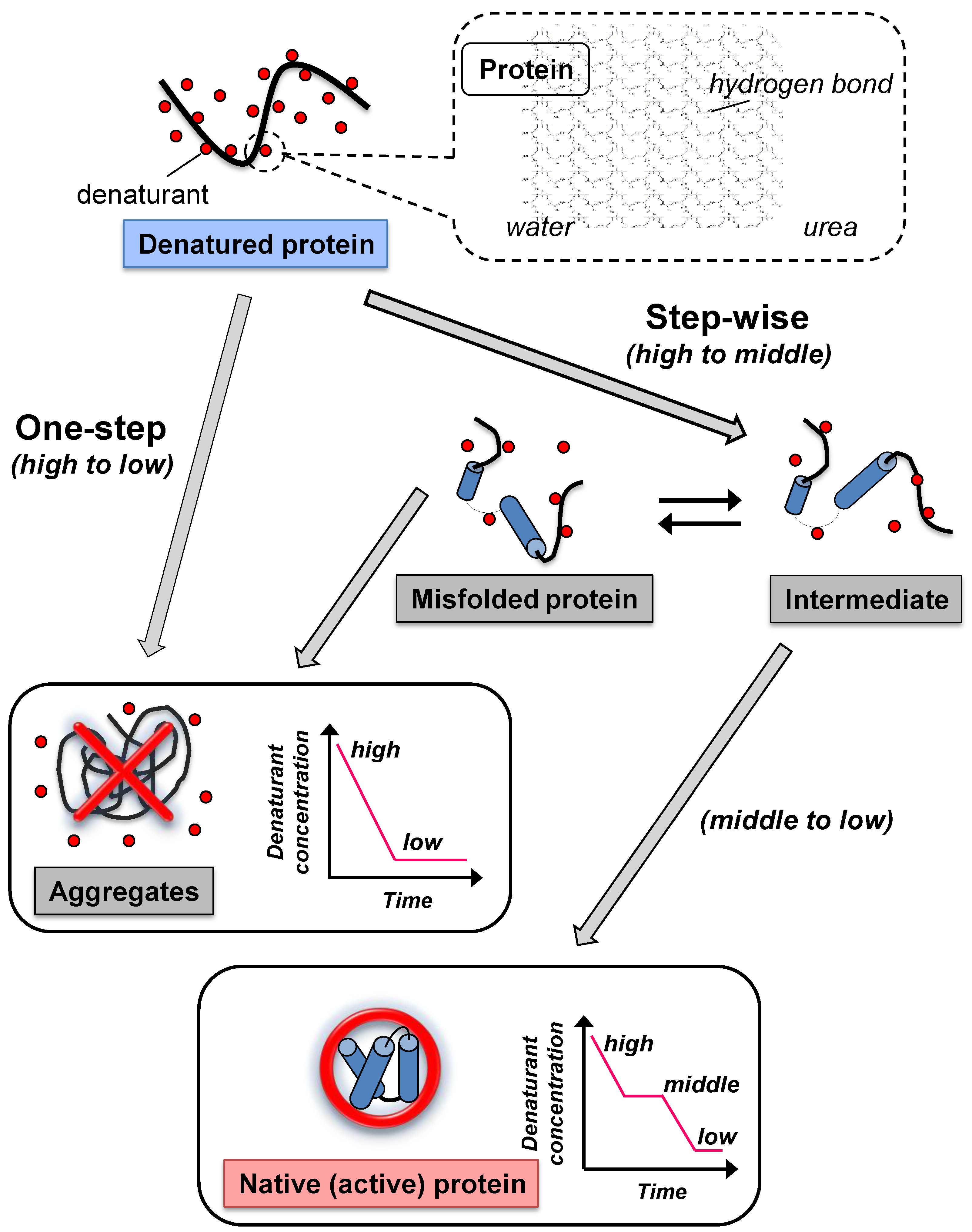
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Recovery yield | Concentration of denatured protein | Time | Temperature |
|---|---|---|---|---|
| Dialysis | ≤40% [13] | 1–10 mg/mL | 1–2 days | 4 °C |
| Dilution (chemical additive) | ≥80% [14] | 1–10 mg/mL | 1–2 days | 4 °C |
| Microfluidic chip | ≥70% [15] | ≥250 μg/mL | ≥10 min | ≥25 °C |
2. Conventional Refolding Methods to Remove Denaturants from Denatured Protein
2.1. Dialysis
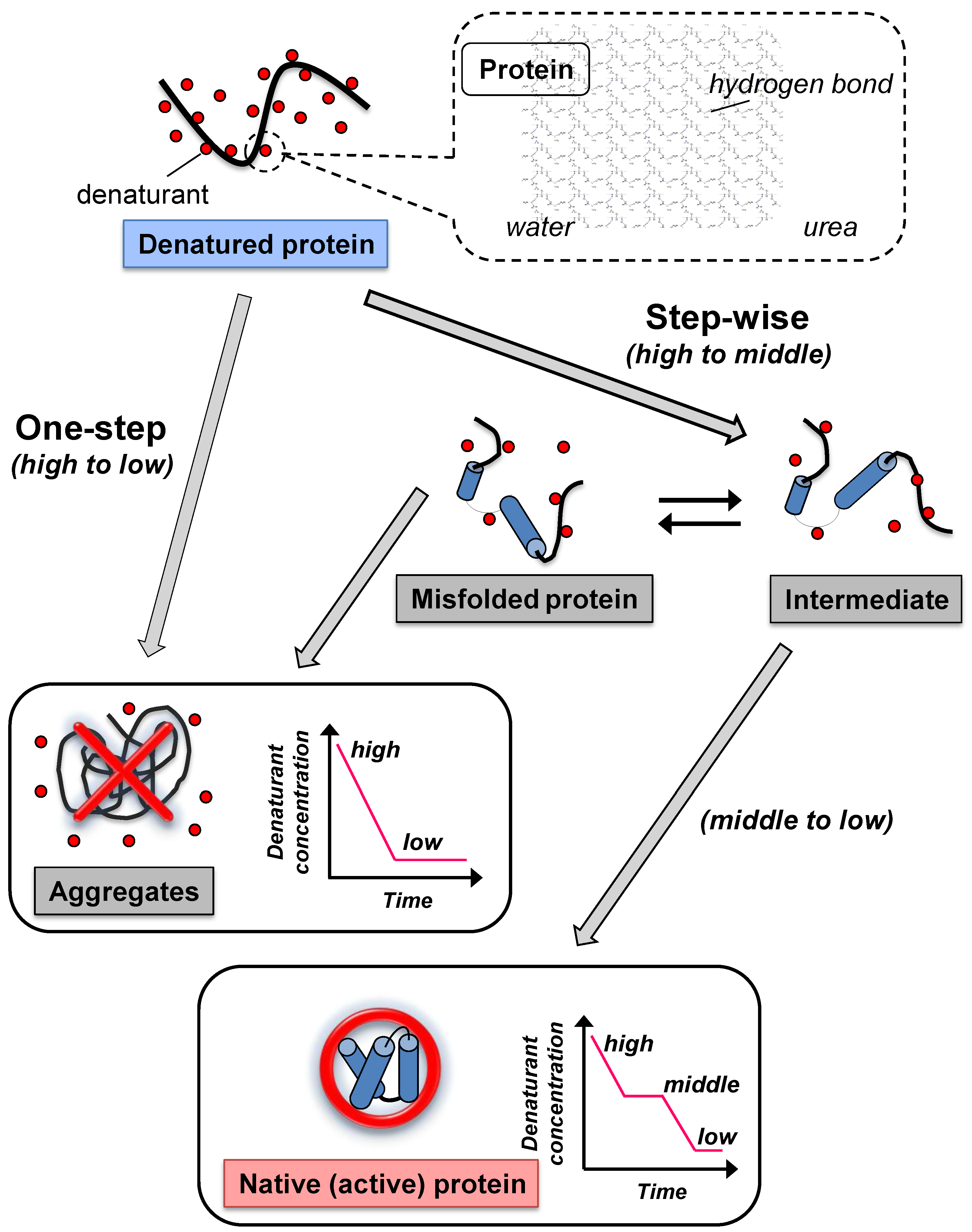
2.2. Dilution
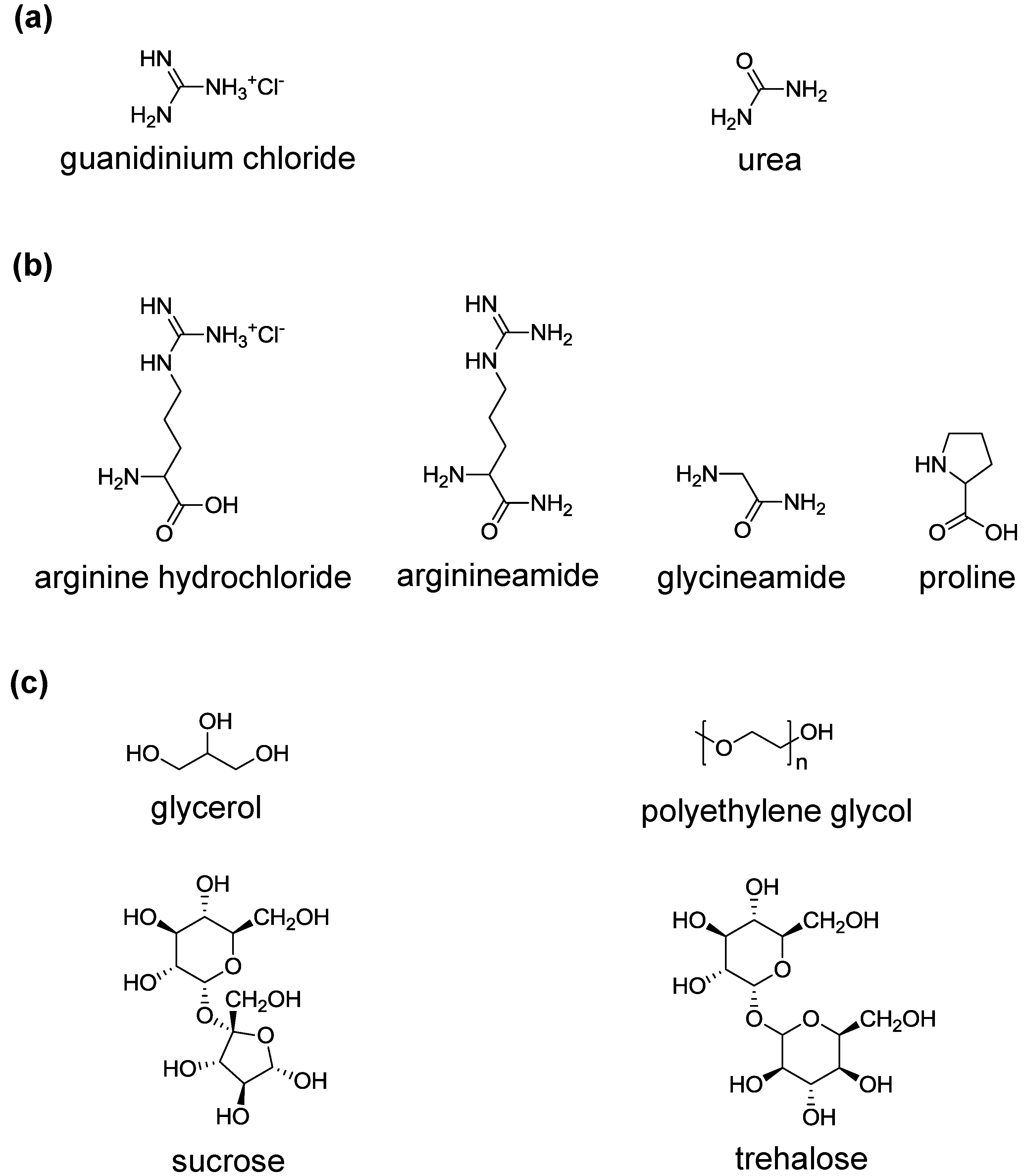
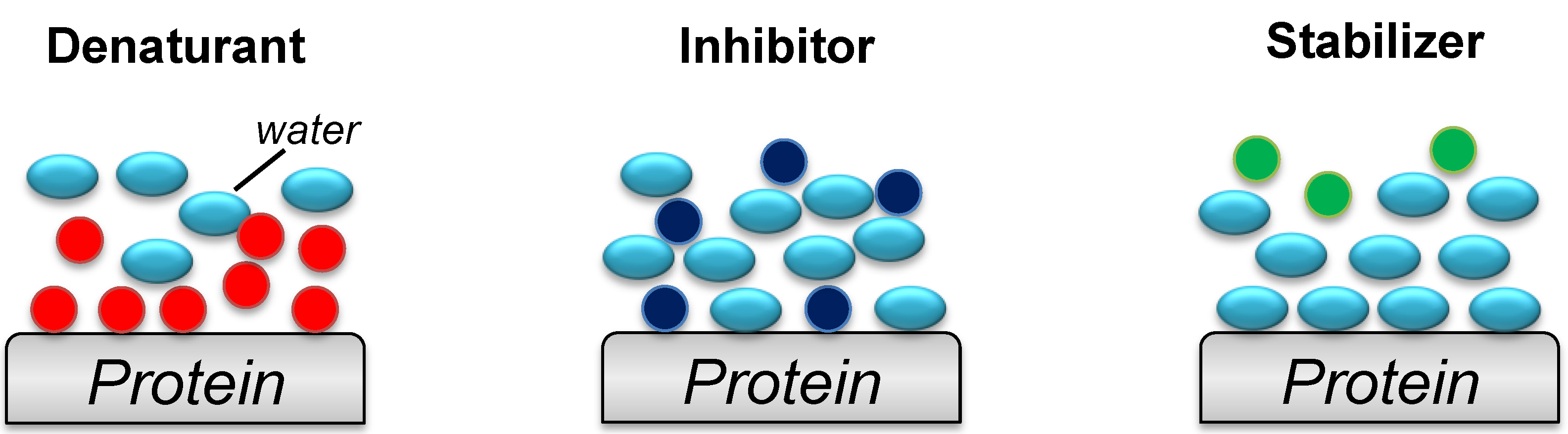
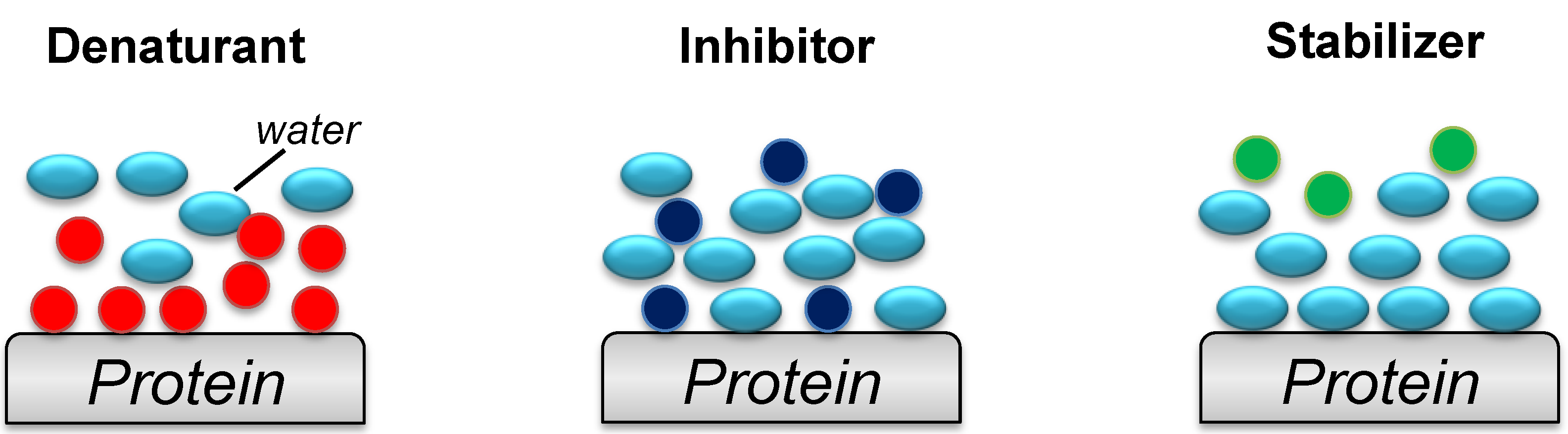
3. Protein Refolding Using Chemical Additives
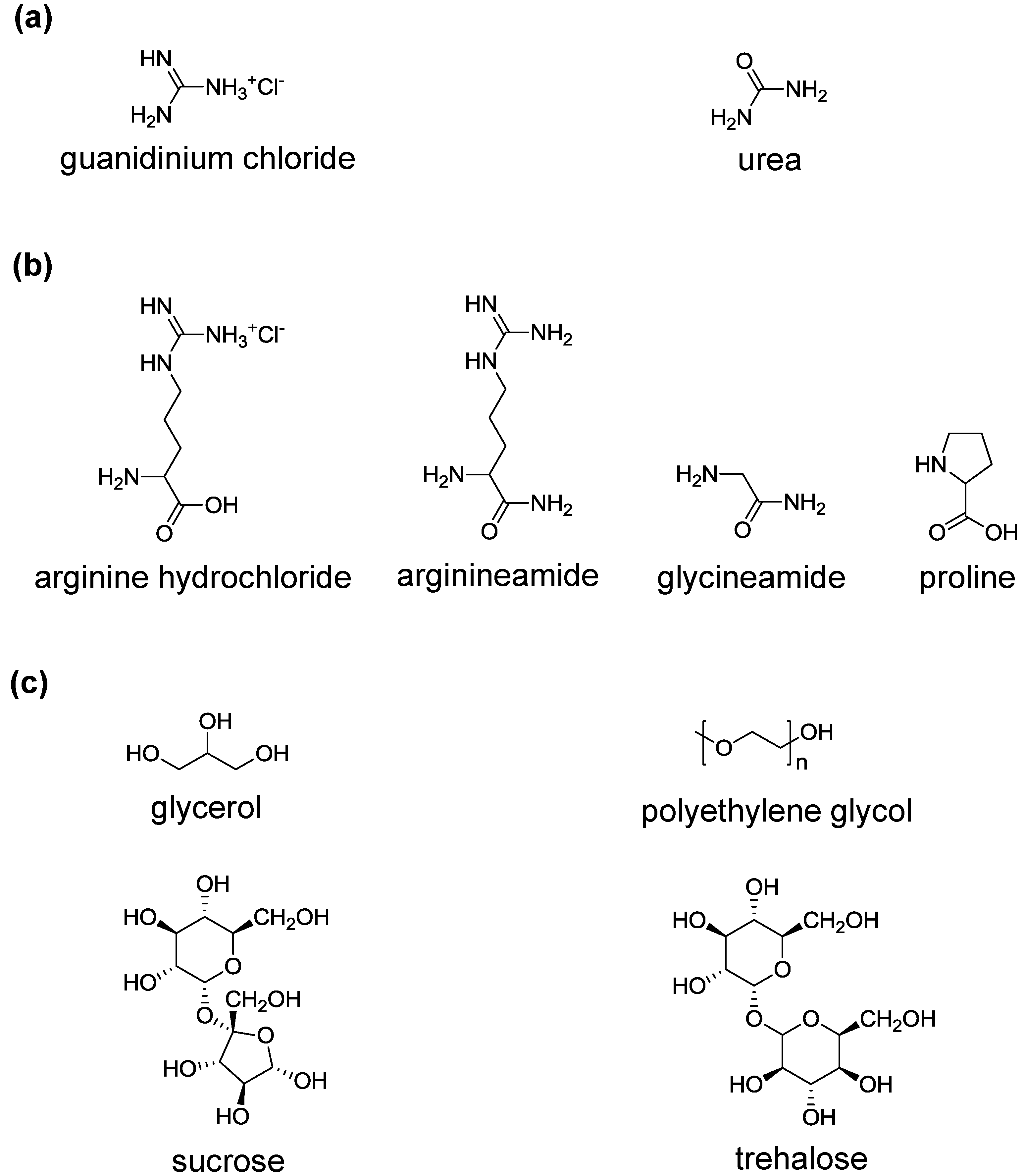
3.1. Amino Acids
3.2. Glycerol, Polyethylene Glycol and Sugars
3.3. Cyclodextrins
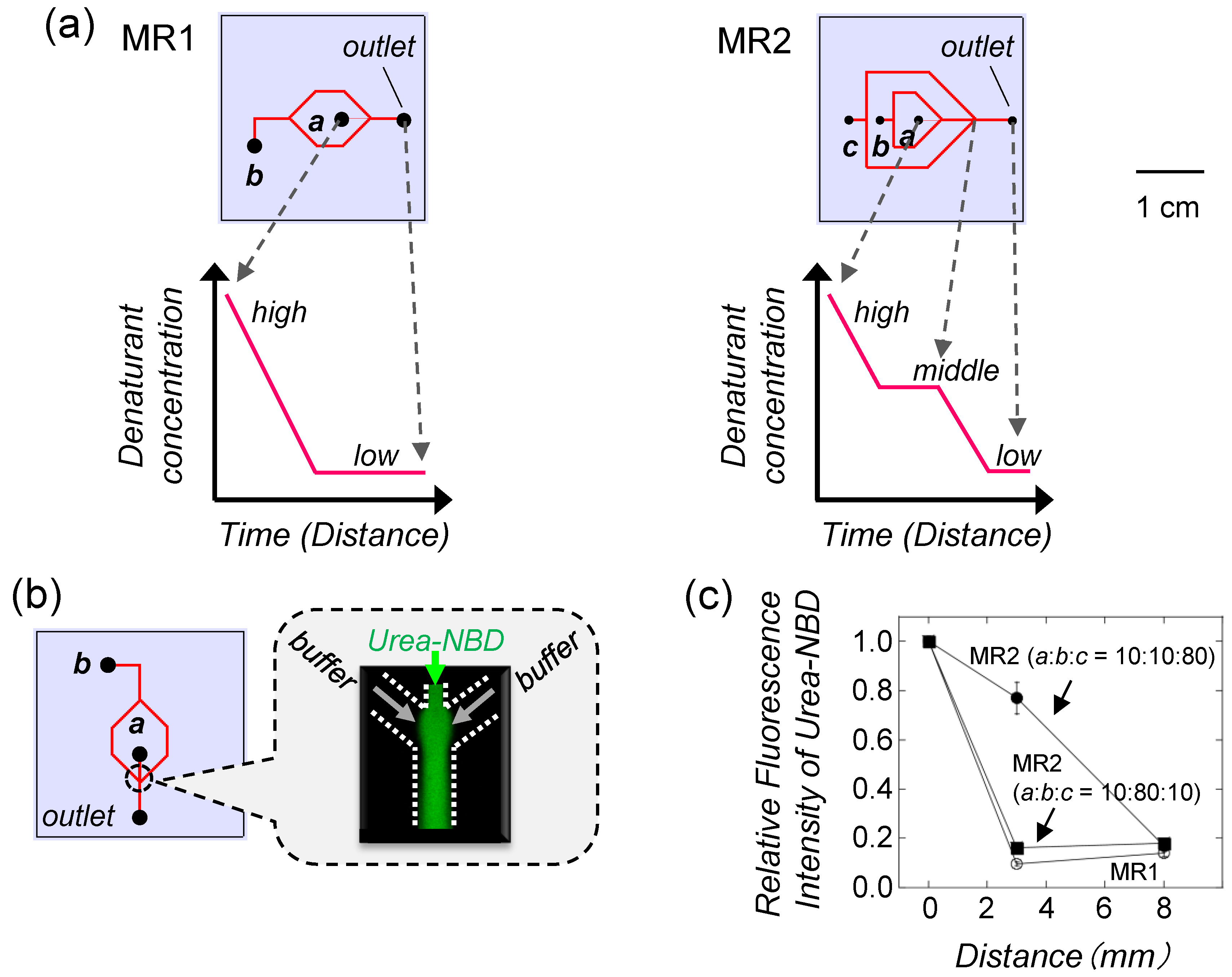
4. Decreasing Denaturant Concentration by Laminar Flow in Microfluidic Chips
4.1. Design of Microfluidic Chips
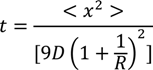
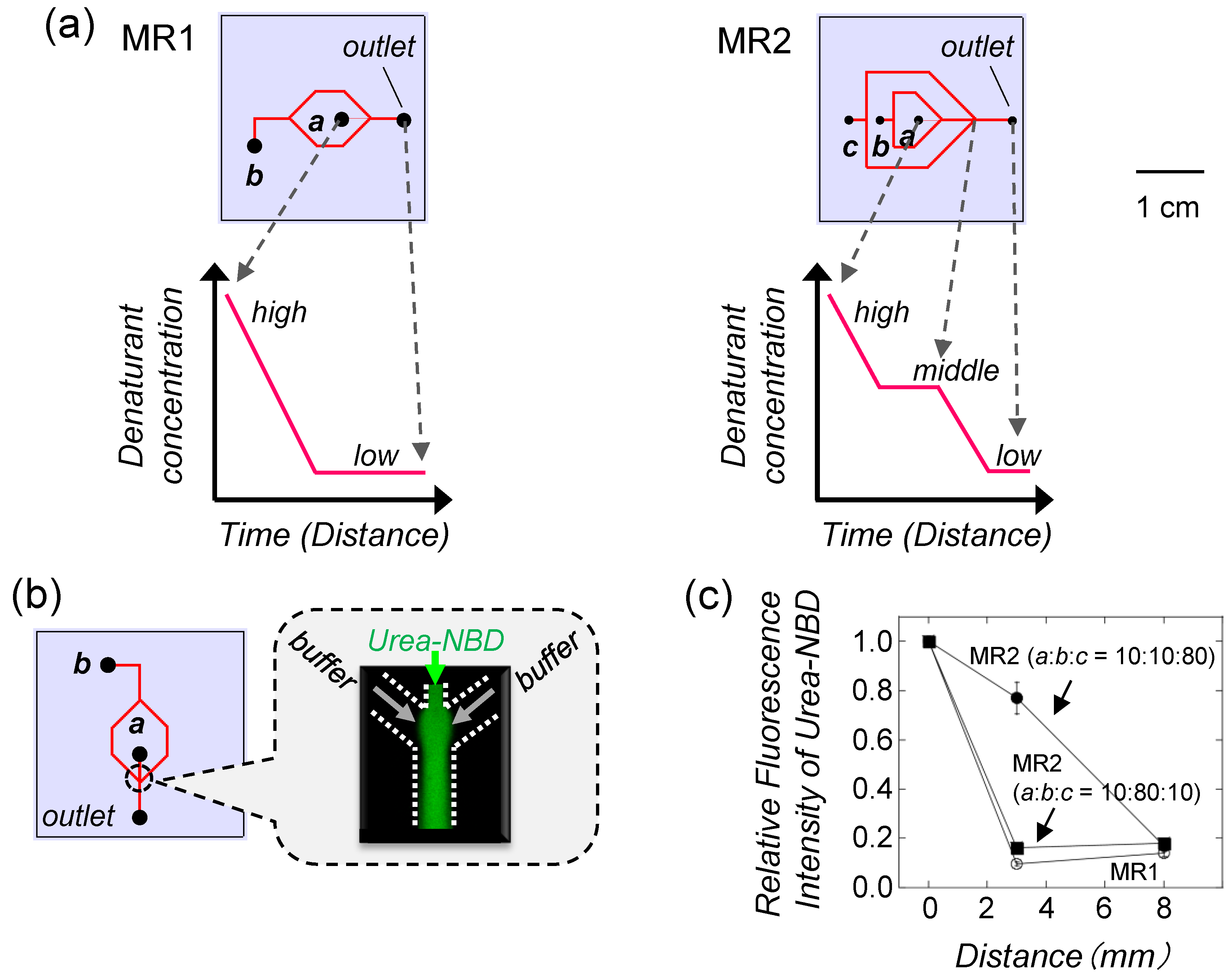
4.2. Protein Refolding Using Microfluidic Chips
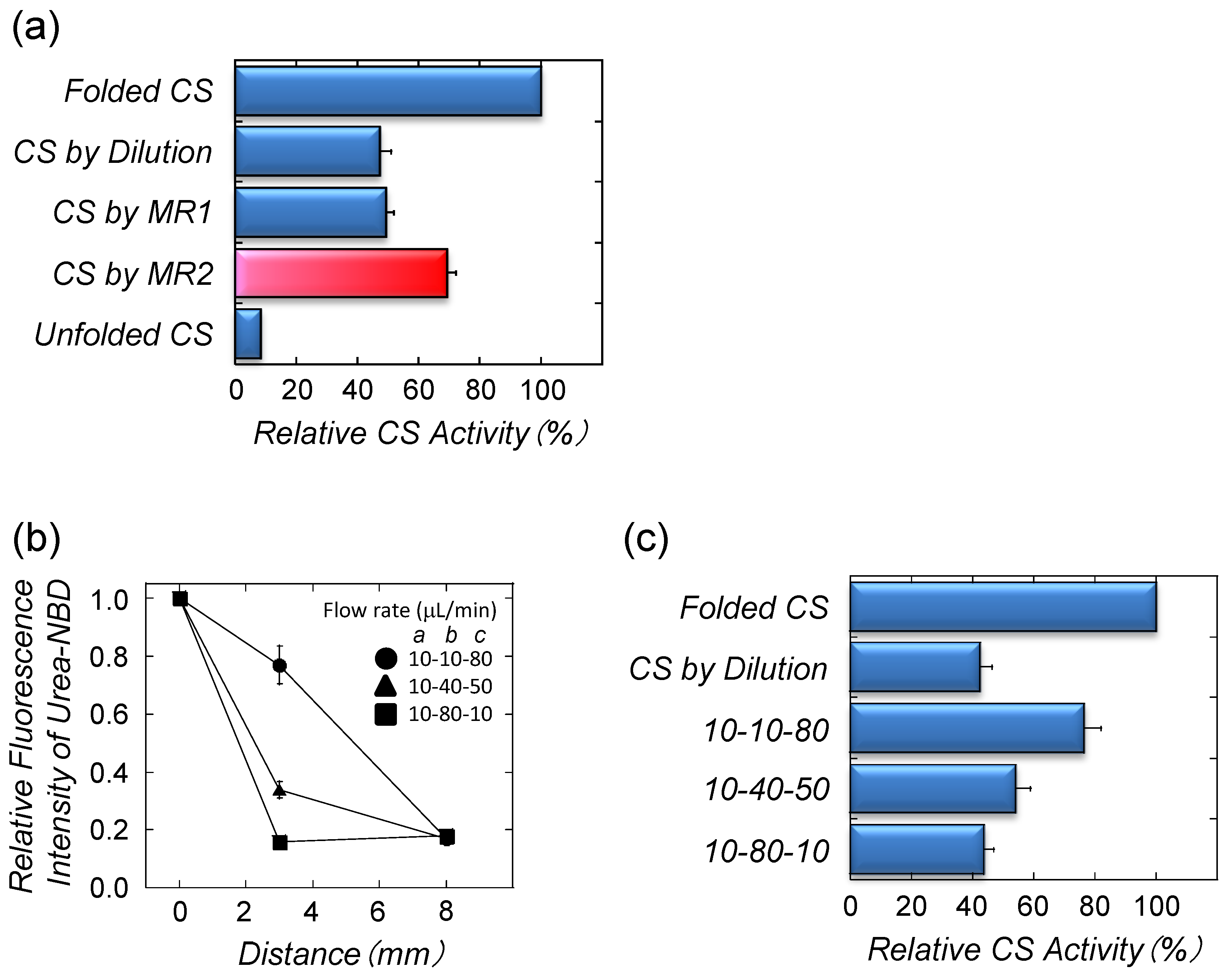
4.3. Refolding of Recombinant Protein from Inclusion Body by Microfluidic Chips
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Swartz, J.R. Advances in Escherichia coli production of therapeutic proteins. Curr. Opin. Biotechnol. 2001, 12, 195–201. [Google Scholar] [CrossRef]
- Clark, E.D.B. Protein refolding for industrial processes. Curr. Opin. Biotechnol. 2001, 12, 202–207. [Google Scholar] [CrossRef]
- Baneyx, F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 1999, 10, 411–421. [Google Scholar] [CrossRef]
- Baneyx, F.; Mujacic, M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 2004, 22, 1399–1408. [Google Scholar] [CrossRef]
- Prasad, S.; Khadatare, P.B.; Roy, I. Effect of chemical chaperones in improving the solubility of recombinant proteins in Escherichia coli. Appl. Environ. Microbiol. 2011, 77, 4603–4609. [Google Scholar] [CrossRef]
- Li, M.; Su, Z.-G.; Janson, J.-C. In vitro protein refolding by chromatographic procedures. Protein Expr. Purif. 2004, 33, 1–10. [Google Scholar] [CrossRef]
- Sakono, M.; Kawashima, Y.; Ichinose, H.; Maruyama, T.; Kamiya, N.; Goto, M. Direct refolding of inclusion bodies using reversed micelles. Biotechnol. Prog. 2004, 20, 1783–1787. [Google Scholar]
- Nara, T.Y.; Togashi, H.; Sekikawa, C.; Kawakami, M.; Yaginuma, N.; Sakaguchi, K.; Mizukami, F.; Tsunoda, T. Use of zeolite to refold a disulfide-bonded protein. Colloids Surf. B Biointerfaces. 2009, 68, 68–73. [Google Scholar] [CrossRef]
- Zhi, W.; Landry, S.J.; Gierasch, L.M.; Srere, P.A. Renaturation of citrate synthase: Influence of denaturant and folding assistants. Protein Sci. 1992, 1, 522–529. [Google Scholar]
- Eiberle, M.K.; Jungbauer, A. Technical refolding of proteins: Do we have freedom to operate? Biotechnol. J. 2010, 5, 547–559. [Google Scholar] [CrossRef]
- Gautam, S.; Dubey, P.; Rather, G.M.; Gupta, M.N. Non-chromatographic strategies for protein refolding. Recent Pat. Biotechnol. 2012, 6, 57–68. [Google Scholar]
- Machold, C.; Schlegl, R.; Buchinger, W.; Jungbauer, A. Matrix assisted refolding of proteins by ion exchange chromatography. J. Biotechnol. 2005, 117, 83–97. [Google Scholar] [CrossRef]
- Zhi, W.; Landry, S.J.; Gierasch, L.M.; Srere, P.A. Renaturation of citrate synthase: Influence of denaturant and folding assistants. Protein Sci. 1992, 1, 522–529. [Google Scholar]
- Reddy, K.R.C.; Lilie, H.; Rudolph, R.; Lange, C. l-Arginine increases the solubility of unfolded species of hen egg white lysozyme. Protein Sci. 2005, 14, 929–935. [Google Scholar]
- Yamaguchi, H.; Miyazaki, M.; Briones-Nagata, M.P.; Maeda, H. Refolding of difficult-to-fold proteins by a gradual decrease of denaturant using microfluidic chips. J. Biochem. 2010, 147, 895–903. [Google Scholar]
- Marston, F.A.O. The purification of eukaryotic polypeptides synthesized in Escherichia coli. Biochem. J. 1986, 240, 1–12. [Google Scholar]
- Tsumoto, K.; Ejima, D.; Kumagai, I.; Arakawa, T. Practical considerations in refolding proteins from inclusion bodies. Protein Expr. Purif. 2003, 28, 1–8. [Google Scholar] [CrossRef]
- Umetsu, M.; Tsumoto, K.; Hara, M.; Ashish, K.; Goda, S.; Adschiri, T.; Kumagai, I. How additives influence the refolding of immunoglobulin-folded proteins in a stepwise dialysis system. Spectroscopic evidence for highly efficient refolding of a single-chain Fv fragment. J. Biol. Chem. 2003, 278, 8979–8987. [Google Scholar]
- Ho, J.G.S.; Middelberg, A.P.J.; Ramage, P.; Kocher, H.P. The likelihood of aggregation during protein renaturation can be assessed using the second virial coefficient. Protein Sci. 2003, 12, 708–716. [Google Scholar]
- Liu, W.; Cellmer, T.; Keerl, D.; Prausnitz, J.M.; Blanch, H.W. Interactions of lysozyme in guanidinium chloride solutions from static and dynamic light-scattering measurements. Biotchnol. Bioeng. 2005, 90, 482–490. [Google Scholar]
- Hevehan, D.L.; Clark, E.D.B. Oxidative renaturation of lysozyme at high concentrations. Biotechnol. Bioeng. 1997, 54, 221–230. [Google Scholar] [CrossRef]
- Orsini, G.; Goldberg, M.E. The renaturation of reduced chymotrypsinogen A in guanidine HCl. Refolding versus aggregation. J. Biol. Chem. 1978, 253, 3453–3458. [Google Scholar]
- Yamaguchi, S.; Yamamoto, E.; Mannen, T.; Nagamune, T. Protein refolding using chemical refolding additives. Biotechnol. J. 2013, 8, 17–31. [Google Scholar]
- Kudou, M.; Yumioka, R.; Ejima, D.; Arakawa, T.; Tsumoto, K. A novel protein refolding system using lauroyl-l-glutamate as a solubilizing detergent and arginine as a folding assisting agent. Protein Expr. Purif. 2011, 75, 46–54. [Google Scholar] [CrossRef]
- Ohtake, S.; Kita, Y.; Arakawa, T. Interactions of formulation excipients with proteins in solution and in the dried state. Adv. Drug Deliv. Rev. 2011, 63, 1053–1073. [Google Scholar]
- Lin, W.J.; Traugh, J.A. Renaturation of casein kinase II from recombinant subunits produced in Escherichia coli: Purification and characterization of the reconstituted holoenzyme. Protein Expr. Purif. 1993, 3, 256–264. [Google Scholar]
- Arora, D.; Khanna, N. Method for increasing the yield of properly folded recombinant human gamma interferon from inclusion bodies. J. Biotechnol. 1996, 52, 127–133. [Google Scholar] [CrossRef]
- Bell, S.; Hansen, S.; Buchner, J. Refolding and structural characterization of the human p53 tumor suppressor protein. Biophys. Chem. 2002, 96, 243–257. [Google Scholar] [CrossRef]
- Asano, R.; Kudo, T.; Makabe, K.; Tsumoto, K.; Kumagai, I. Antitumor activity of interleukin-21 prepared by novel refolding procedure from inclusion bodies expressed in Escherichia coli. FEBS Lett. 2002, 528, 70–76. [Google Scholar] [CrossRef]
- Mande, S.C.; Sobhia, M.E. Structural characterization of protein-denaturant interactions: Crystal structures of hen egg-white lysozyme in complex with DMSO and guanidinium chloride. Protein Eng. 2000, 13, 133–141. [Google Scholar] [CrossRef]
- Ito, L.; Shiraki, K.; Matsuura, T.; Okumura, M.; Hasegawa, K.; Baba, S.; Yamaguchi, H.; Kumasaka, T. High-resolution X-ray analysis reveals binding of arginine to aromatic residues of lysozyme surface: Implication of suppression of protein aggregation by arginine. Protein. Eng. Des. Sel. 2011, 24, 269–274. [Google Scholar] [CrossRef]
- Matsuoka, T.; Hamada, H.; Matsumoto, K.; Shiraki, K. Indispensable structure of solution additives to prevent inactivation of lysozyme for heating and refolding. Biotechnol. Prog. 2009, 25, 1515–1524. [Google Scholar]
- Hamada, H.; Shiraki, K. l-Argininamide improves the refolding more effectively than l-arginine. J. Biotechnol. 2007, 130, 153–160. [Google Scholar]
- Ito, L.; Shiraki, K.; Makino, M.; Hasegawa, K.; Kumasaka, T. Glycine amide shielding on the aromatic surfaces of lysozyme: Implication for suppression of protein aggregation. FEBS Lett. 2011, 585, 555–560. [Google Scholar] [CrossRef] [Green Version]
- Samuel, D.; Kumar, T.K.; Ganesh, G.; Jayaraman, G.; Yang, P.W.; Chang, M.M.; Trivedi, V.D.; Wang, S.L.; Hwang, K.C.; Chang, D.K.; Yu, C. Proline inhibits aggregation during protein refolding. Protein Sci. 2000, 9, 344–352. [Google Scholar]
- Timasheff, S.N. Protein hydration, thermodynamic binding, and preferential hydration. Biochemistry 2002, 41, 13473–13482. [Google Scholar] [CrossRef]
- Kohyama, K.; Matsumoto, T.; Imoto, T. Refolding of an unstable lysozyme by gradient removal of a solubilizer and gradient addition of a stabilizer. J. Biochem. 2010, 147, 427–431. [Google Scholar]
- Nian, R.; Kim, D.S.; Tan, L.; Kim, C.W.; Choe, W.S. Synergistic coordination of polyethylene glycol with ClpB/DnaKJE bichaperone for refolding of heat-denatured malate dehydrogenase. Biotechnol. Prog. 2009, 25, 1078–1085. [Google Scholar]
- Webb, S.D.; Cleland, J.L.; Carpenter, J.F.; Randolph, T.W. A new mechanism for decreasing aggregation of recombinant human interferon-gamma by a surfactant: Slowed dissolution of lyophilized formulations in a solution containing 0.03% polysorbate 20. J. Pharm. Sci. 2002, 91, 543–558. [Google Scholar]
- Akbari, N.; Khajeh, K.; Ghaemi, N.; Salemi, Z. Efficient refolding of recombinant lipase from Escherichia coli inclusion bodies by response surface methodology. Protein Expr. Purif. 2010, 70, 254–259. [Google Scholar] [CrossRef]
- Paul, S.; Punam, S.; Chaudhuri, T.K. Chaperone-assisted refolding of Escherichia coli maltodextrin glucosidase. FEBS J. 2007, 274, 6000–6010. [Google Scholar]
- Wang, F.; Liu, Y.; Li, J.; Ma, G.; Su, Z. On-column refolding of consensus interferon at high concentration with guanidine-hydrochloride and polyethylene glycol gradients. J. Chromatogr. A. 2006, 1115, 72–80. [Google Scholar] [CrossRef]
- Hart, R.A.; Lester, P.M.; Reifsnyder, D.H.; Ogez, J.R.; Builder, S.E. Large scale, in situ isolation of periplasmic IGF-I from E. coli. Bio/Technol. 1994, 12, 1113–1117. [Google Scholar] [CrossRef]
- Bajorunaite, E.; Cirkovas, A.; Radzevicius, K.; Larsen, K.L.; Sereikaite, J.; Bumelis, V.A. Anti-aggregatory effect of cyclodextrins in the refolding process of recombinant growth hormones from Escherichia coli inclusion bodies. Int. J. Biol. Macromol. 2009, 44, 428–434. [Google Scholar] [CrossRef]
- Sharma, L.; Sharma, A. Influence of cyclodextrin ring substituents on folding-related aggregation of bovine carbonic anhydrase. Eur. J. Biochem. 2001, 268, 2456–2463. [Google Scholar] [CrossRef]
- Vandevenne, M.; Gaspard, G.; Belgsir, E.M.; Ramnath, M.; Cenatiempo, Y.; Marechal, D.; Dumoulin, M.; Frere, J.M.; Matagne, A.; Galleni, M.; Filee, P. Effects of monopropanediamino-β-cyclodextrin on the denaturation process of the hybrid protein BlaPChBD. Biochim. Biophys. Acta. 2011, 1814, 1146–1153. [Google Scholar] [CrossRef]
- Daugherty, D.L.; Rozema, D.; Hanson, P.E.; Gellman, S.H. Artificial chaperone-assisted refolding of citrate synthase. J. Biol. Chem. 1998, 273, 33961–33971. [Google Scholar]
- Rozema, D.; Gellman, S.H. Artificial chaperone-assisted refolding of carbonic anhydrase B. J. Biol. Chem. 1996, 271, 3478–3487. [Google Scholar]
- Machida, S.; Ogawa, S.; Xiaohua, S.; Takaha, T.; Fujii, K.; Hayashi, K. Cycloamylose as an efficient artificial chaperone for protein refolding. FEBS Lett. 2000, 486, 131–135. [Google Scholar] [CrossRef]
- Haeberle, S.; Zengerle, R.K. Microfluidic platforms for lab-on-a-chip applications. Lab Chip 2007, 7, 1094–1110. [Google Scholar]
- Ohno, K.; Tachikawa, K.; Manz, A. Microfluidics: Applications for analytical purposes in chemistry and biochemistry. Electrophoresis 2008, 29, 4443–4453. [Google Scholar] [CrossRef]
- Asanomi, Y.; Yamaguchi, H.; Miyazaki, M.; Maeda, H. Enzyme-immobilized microfluidic process reactors. Molecules 2011, 16, 6041–6059. [Google Scholar]
- Yamaguchi, H.; Maeki, M.; Yamashita, K.; Nakamura, H.; Miyazaki, M.; Maeda, H. Controlling one protein crystal growth by droplet-based microfluidic system. J. Biochem. 2013, 153, 339–346. [Google Scholar] [CrossRef]
- Lapidus, L.J.; Yao, S.; Mcgarrity, K.S.; Hertzog, D.E.; Tubman, E.; Bakajin, O. Protein hydrophobic collapse and early folding steps observed in a microfluidic mixer. Biophys. J. 2007, 93, 218–224. [Google Scholar]
- Kane, A.S.; Hoffmann, A.; Baumgärtel, P.; Seckler, R.; Reichardt, G.; Horsley, D.A.; Schuler, B.; Bakajin, O. Microfluidic mixers for the investigation of rapid protein folding kinetics using synchrotron radiation circular dichroism spectroscopy. Anal. Chem. 2008, 80, 9534–9541. [Google Scholar]
- Hertzog, D.E.; Michalet, X.; Jäger, M.; Kong, X.; Santiago, J.G.; Weiss, S.; Bakajin, O. Femtomole mixer for microsecond kinetic studies of protein folding. Anal. Chem. 2004, 76, 7169–7178. [Google Scholar]
- Kerby, M.B.; Lee, J.; Ziperstein, J.; Tripathi, A. Kinetic measurements of protein conformation in a microchip. Biotechnol. Prog. 2006, 22, 1416–1425. [Google Scholar]
- Yamamoto, E.; Yamaguchi, S.; Sasaki, N.; Kim, H.-B.; Kitamori, T.; Nagamune, T. Artificial chaperone-assisted refolding in a microchannel. Bioprocess Biosyst. Eng. 2010, 33, 171–177. [Google Scholar]
- Karnik, R.; Gu, F.; Basto, P.; Cannizzaro, C.; Dean, L.; Kyei-Manu, W.; Langer, R.; Farokhzad, O.C. Microfluidic platform for controlled synthesis of polymeric nanoparticles. Nano Lett. 2008, 8, 2906–2912. [Google Scholar]
- Hong, S.; Tsou, P.H.; Chou, C.K.; Yamaguchi, H.; Su, C.B.; Hung, M.C.; Kameoka, J. Microfluidic three-dimensional hydrodynamic flow focusing for the rapid protein concentration analysis. Biomicrofluidics 2012, 6, 24132. [Google Scholar] [CrossRef]
- Dayel, M.J.; Hom, E.F.; Verkman, A.S. Diffusion of green fluorescent protein in the aqueous-phase lumen of endoplasmic reticulum. Biophys. J. 1999, 76, 2843–2851. [Google Scholar]
- Gosting, L.J.; Akeley, D.F. A study of the diffusion of urea in water at 25° with the Gouy interference method. J. Am. Chem. Soc. 1952, 74, 2058–2060. [Google Scholar]
- Jahn, A.; Vreeland, W.N.; deVoe, D.L.; Locascio, L.E.; Gaitan, M. Microfluidic directed formation of liposomes of controlled size. Langmuir 2007, 23, 6289–6293. [Google Scholar]
- Jin, L.; Pluskey, S.; Petrella, E.C.; Cantin, S.M.; Gorga, J.C.; Rynkiewicz, M.J.; Pandey, P.; Strickler, J.E.; Babine, R.E.; Weaver, D.T.; Seidl, K.J. The three-dimensional structure of the ZAP-70 kinase domain in complex with staurosporine. J. Biol. Chem. 2004, 279, 42818–42825. [Google Scholar]
- Visco, C.; Magistrelli, G.; Bosotti, R.; Perego, R.; Rusconi, L.; Toma, S.; Zamai, M.; Acuto, O.; Isacchi, A. Activation of Zap-70 tyrosine kinase due to a structural rearrangement induced by tyrosine phosphorylation and/or ITAM binding. Biochemistry 2000, 39, 2784–2791. [Google Scholar]
- Swietnicki, W. Folding aggregated proteins into functionally active forms. Curr. Opin. Biotechnol. 2006, 17, 367–372. [Google Scholar] [CrossRef]
- Ordidge, G.C.; Mannall, G.; Liddell, J.; Dalby, P.A.; Micheletti, M. A generic hierarchical screening method for the analysis of microscale refolds using an automated robotic platform. Biotechnol. Prog. 2012, 28, 435–444. [Google Scholar]
- Nian, R.; Kim, D.S.; Tan, L.; Kim, C.W.; Choe, W.S. Synergistic coordination of polyethylene glycol with ClpB/DnaKJE bichaperone for refolding of heat-denatured malate dehydrogenase. Biotechnol. Prog. 2009, 25, 1078–1085. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yamaguchi, H.; Miyazaki, M. Refolding Techniques for Recovering Biologically Active Recombinant Proteins from Inclusion Bodies. Biomolecules 2014, 4, 235-251. https://doi.org/10.3390/biom4010235
Yamaguchi H, Miyazaki M. Refolding Techniques for Recovering Biologically Active Recombinant Proteins from Inclusion Bodies. Biomolecules. 2014; 4(1):235-251. https://doi.org/10.3390/biom4010235
Chicago/Turabian StyleYamaguchi, Hiroshi, and Masaya Miyazaki. 2014. "Refolding Techniques for Recovering Biologically Active Recombinant Proteins from Inclusion Bodies" Biomolecules 4, no. 1: 235-251. https://doi.org/10.3390/biom4010235