Misfolding of Amyloidogenic Proteins and Their Interactions with Membranes

Abstract
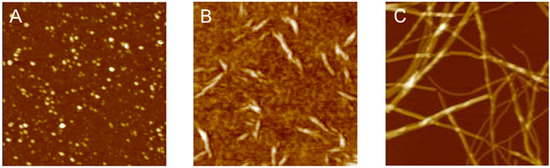
:
{kind=link}
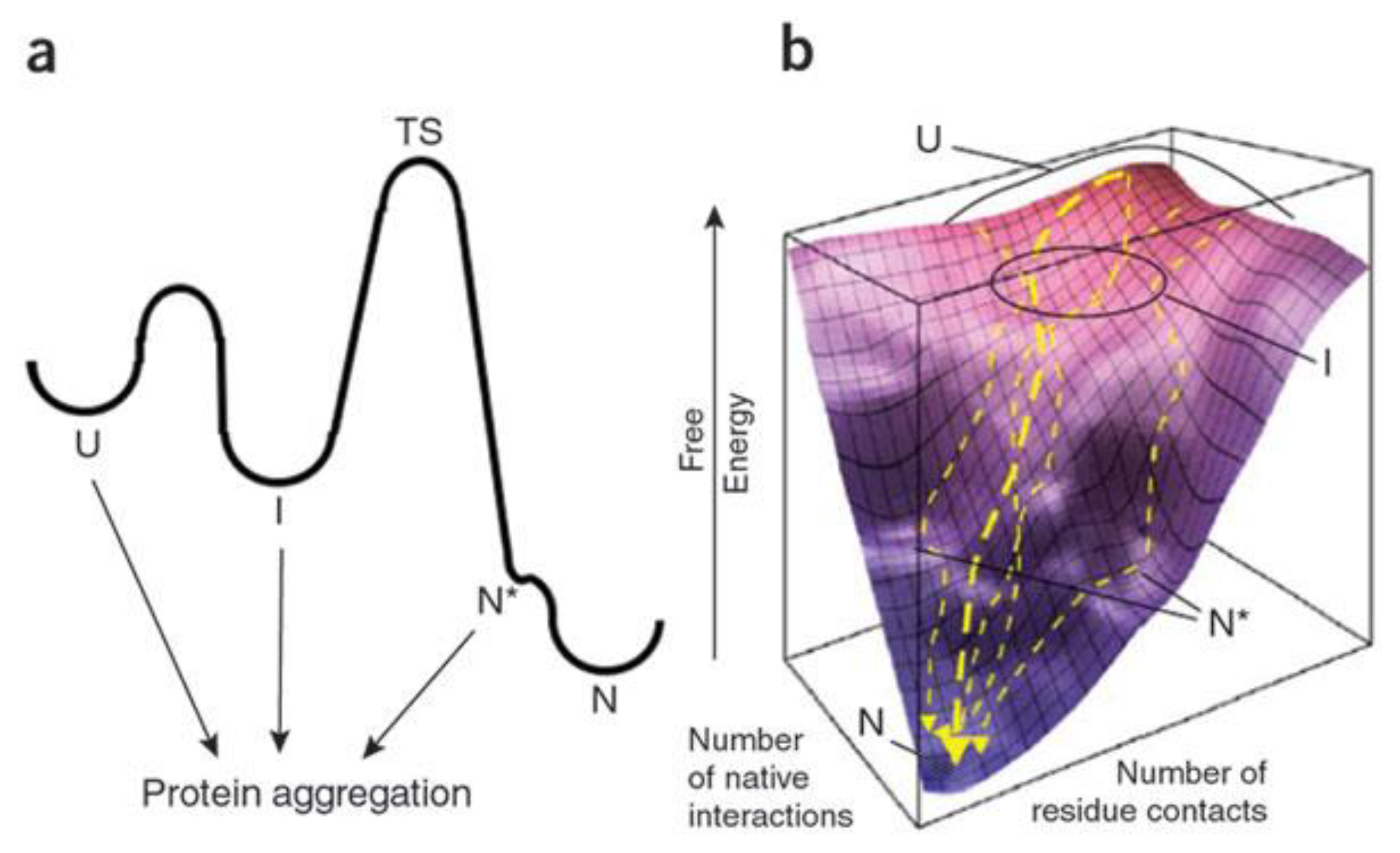{kind=link}
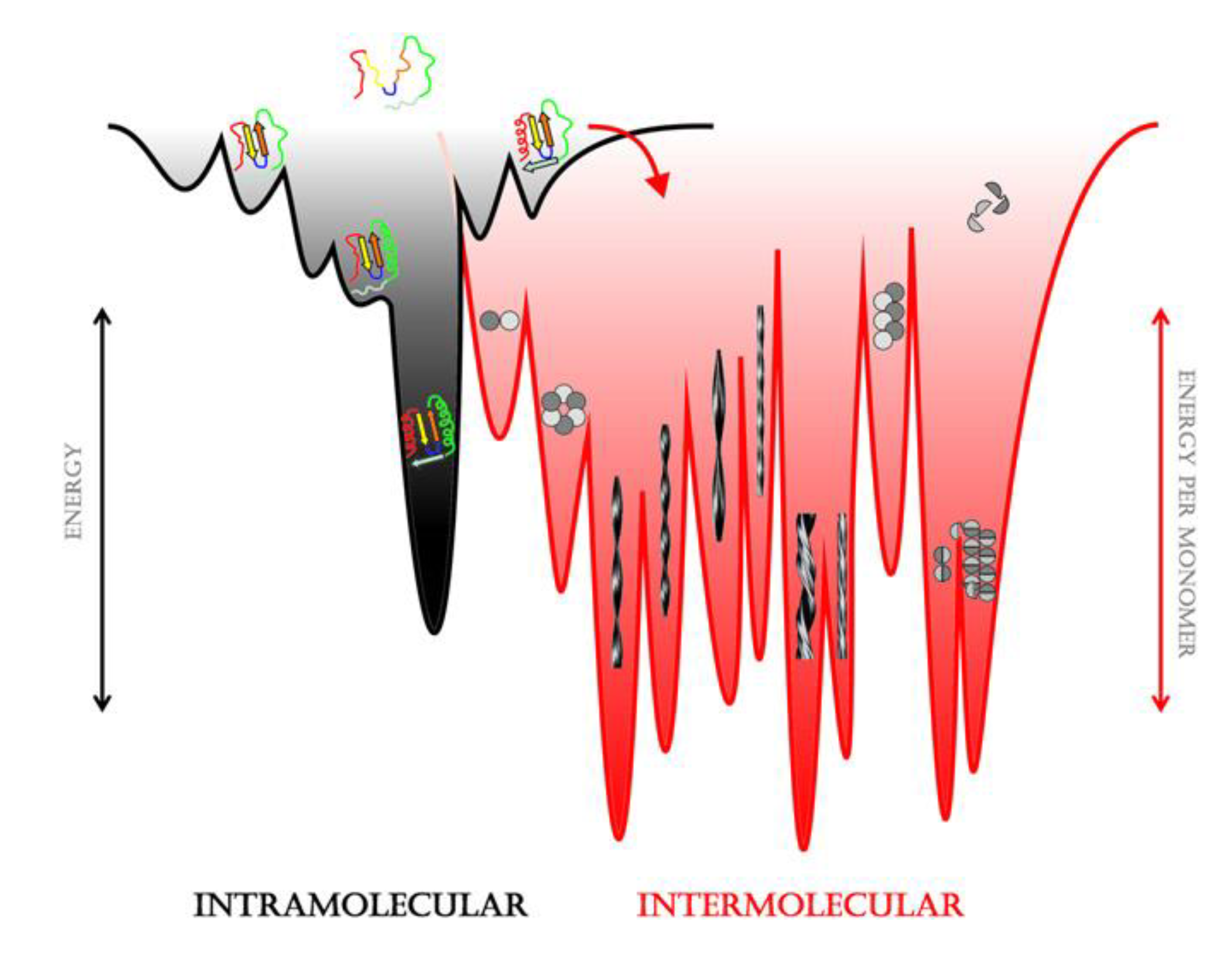{kind=link}
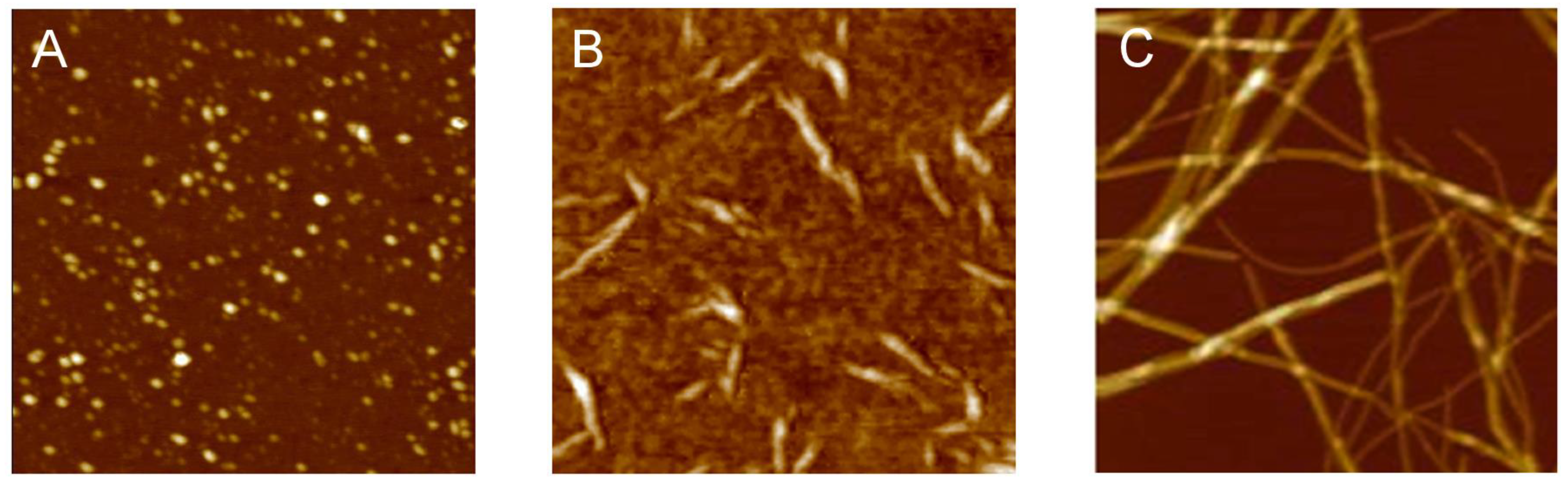{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Protein Unfolding and Misfolding
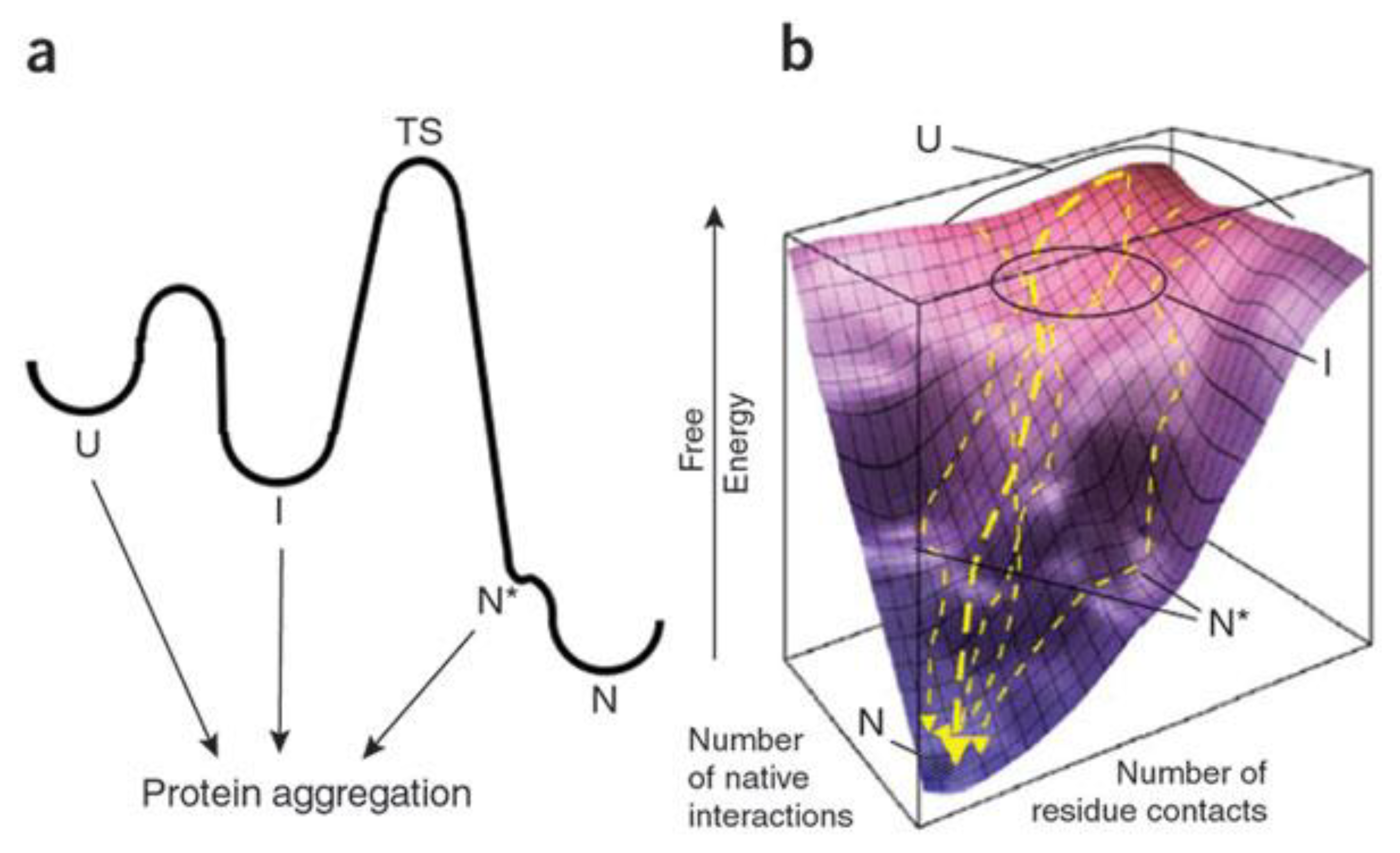
2.1. Amyloid Misfolding, Aggregation and Protein Conformation
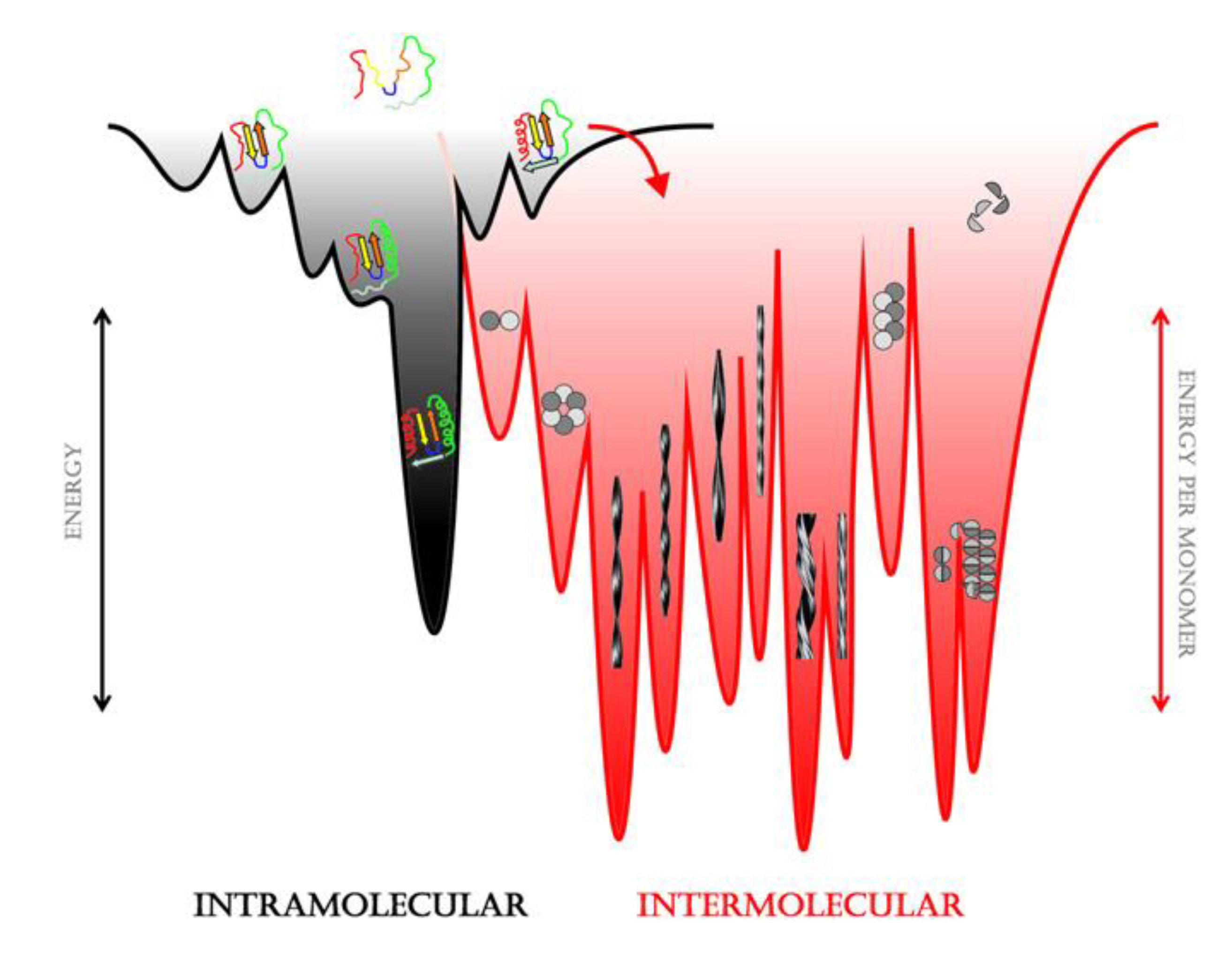
2.2. Factors Contributing to Unfolding/Misfolding
3. Amyloid Aggregation
3.1. The Different Steps of the Aggregation Process
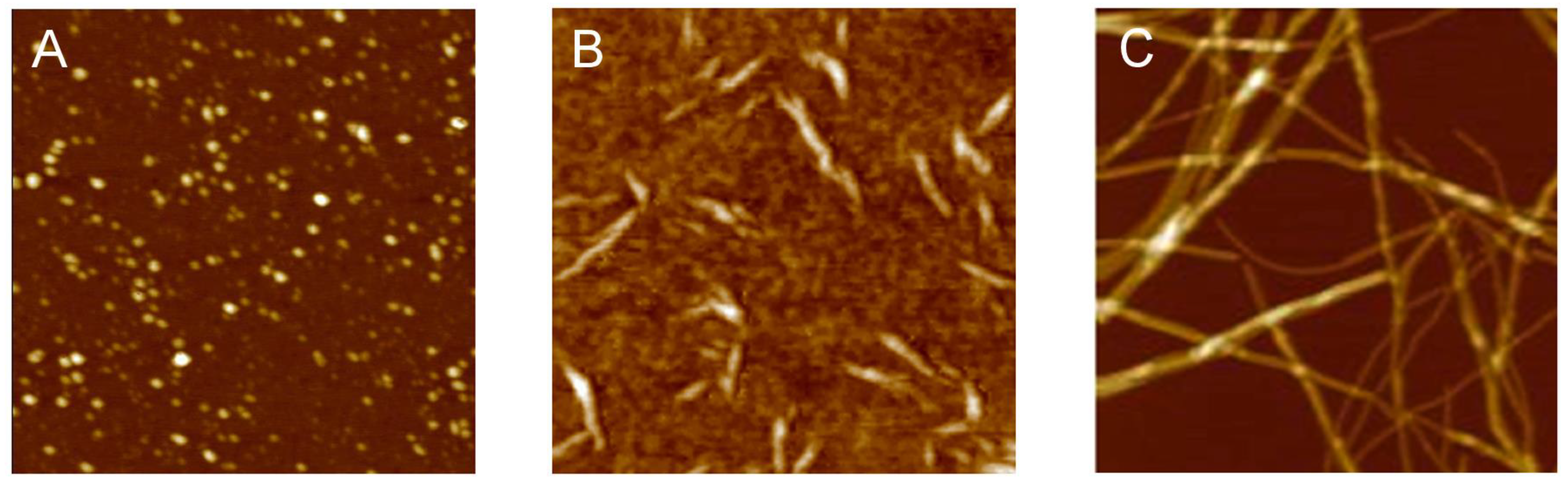
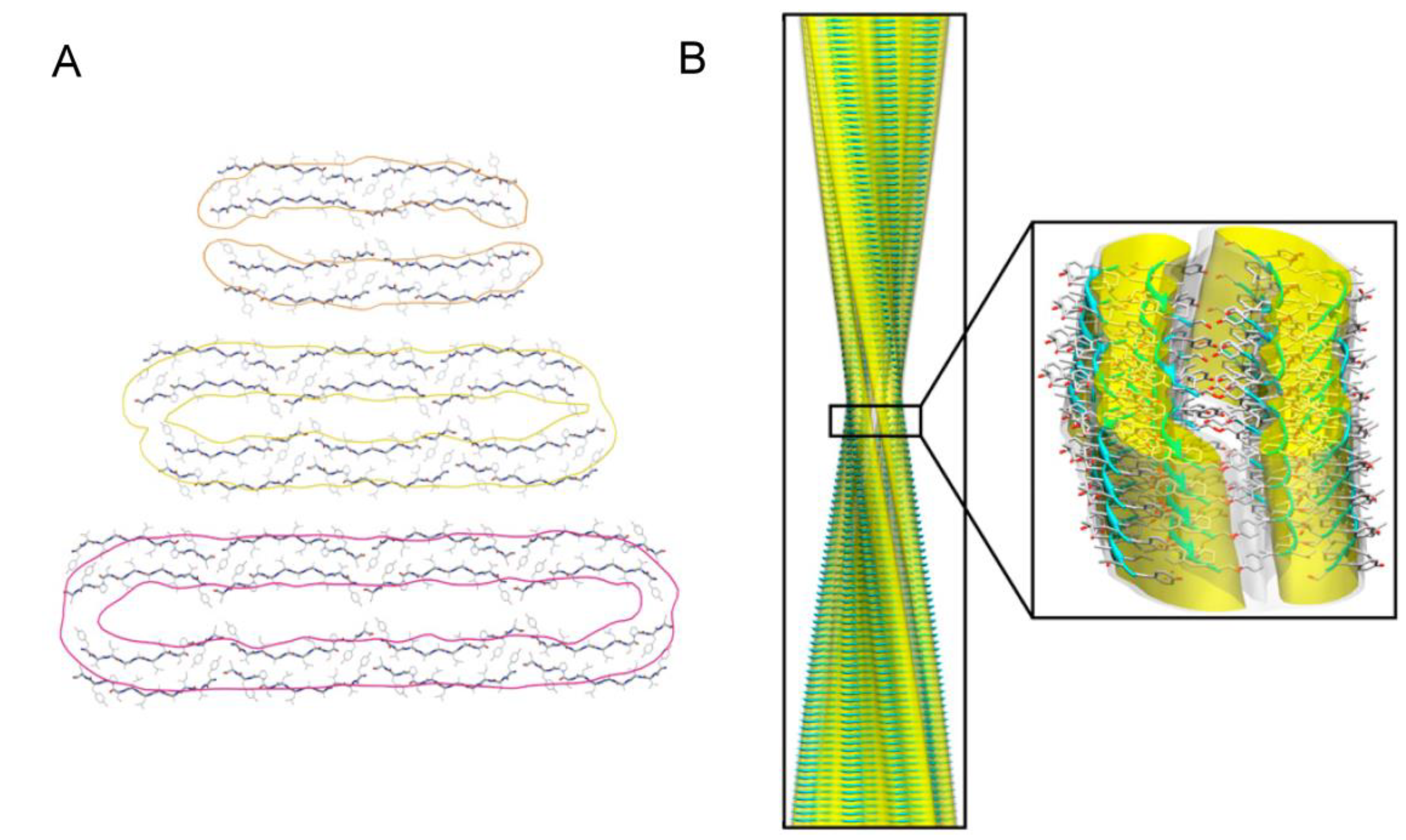
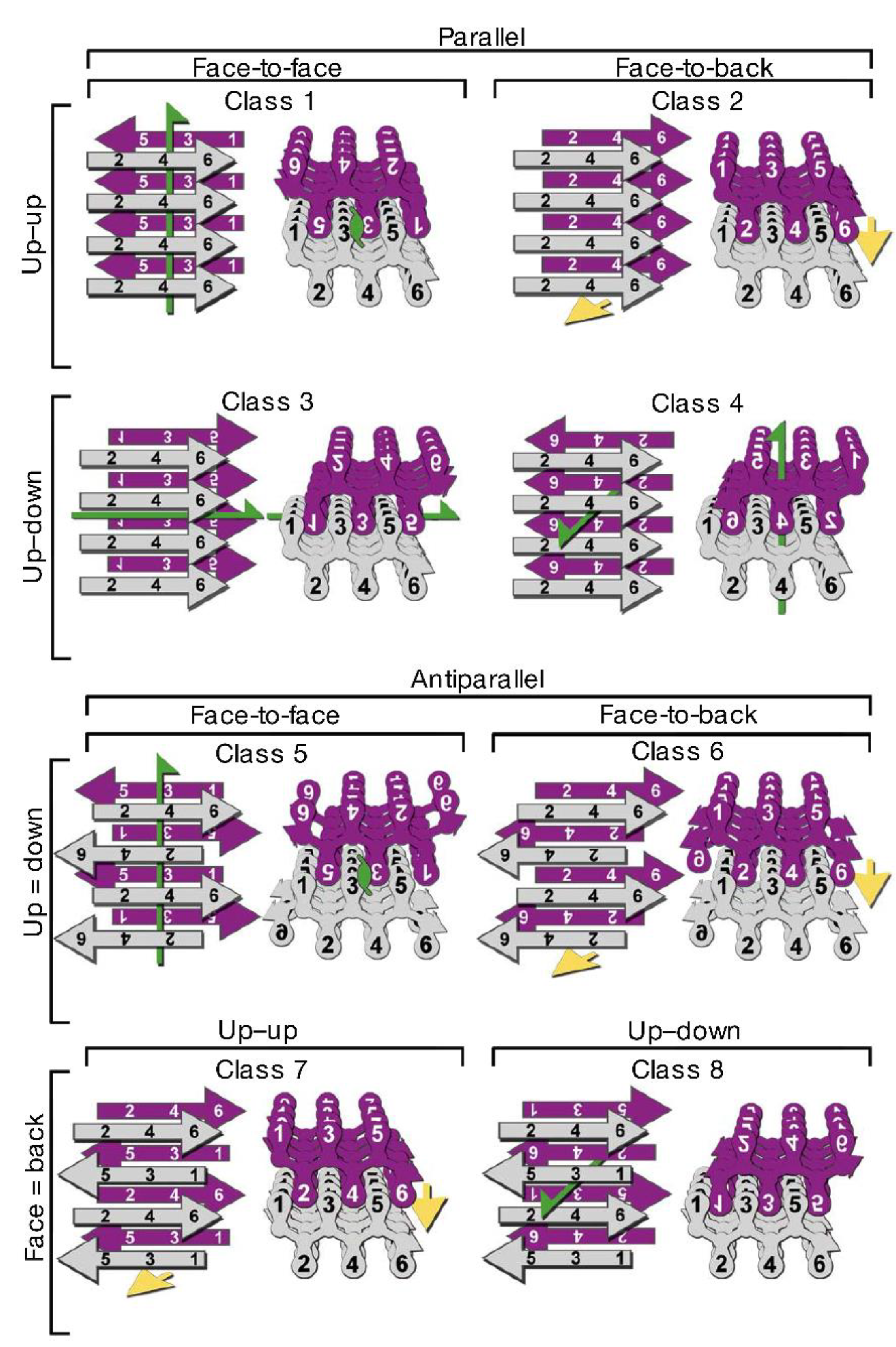
3.2. Polymorphism of Amyloid Aggregates
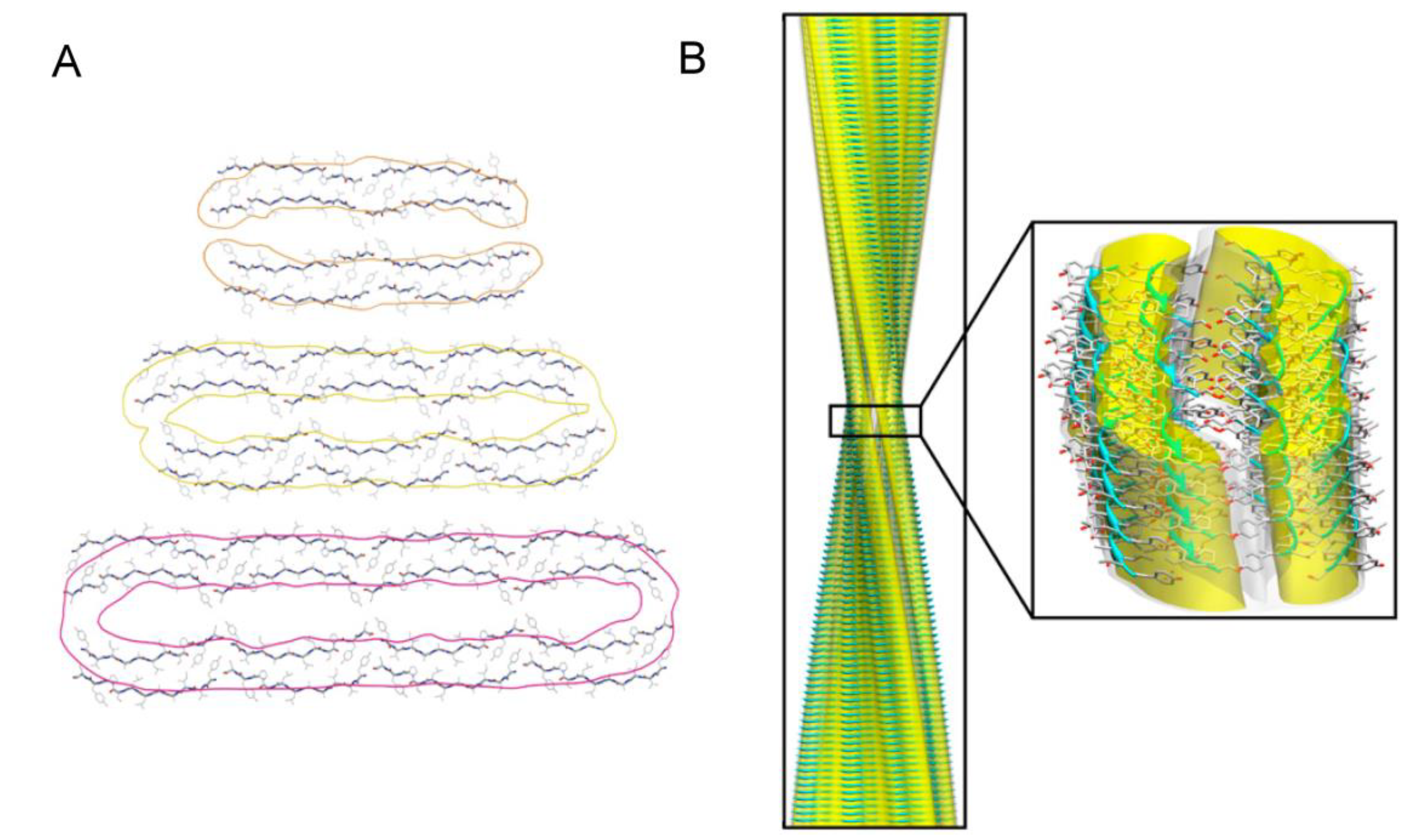
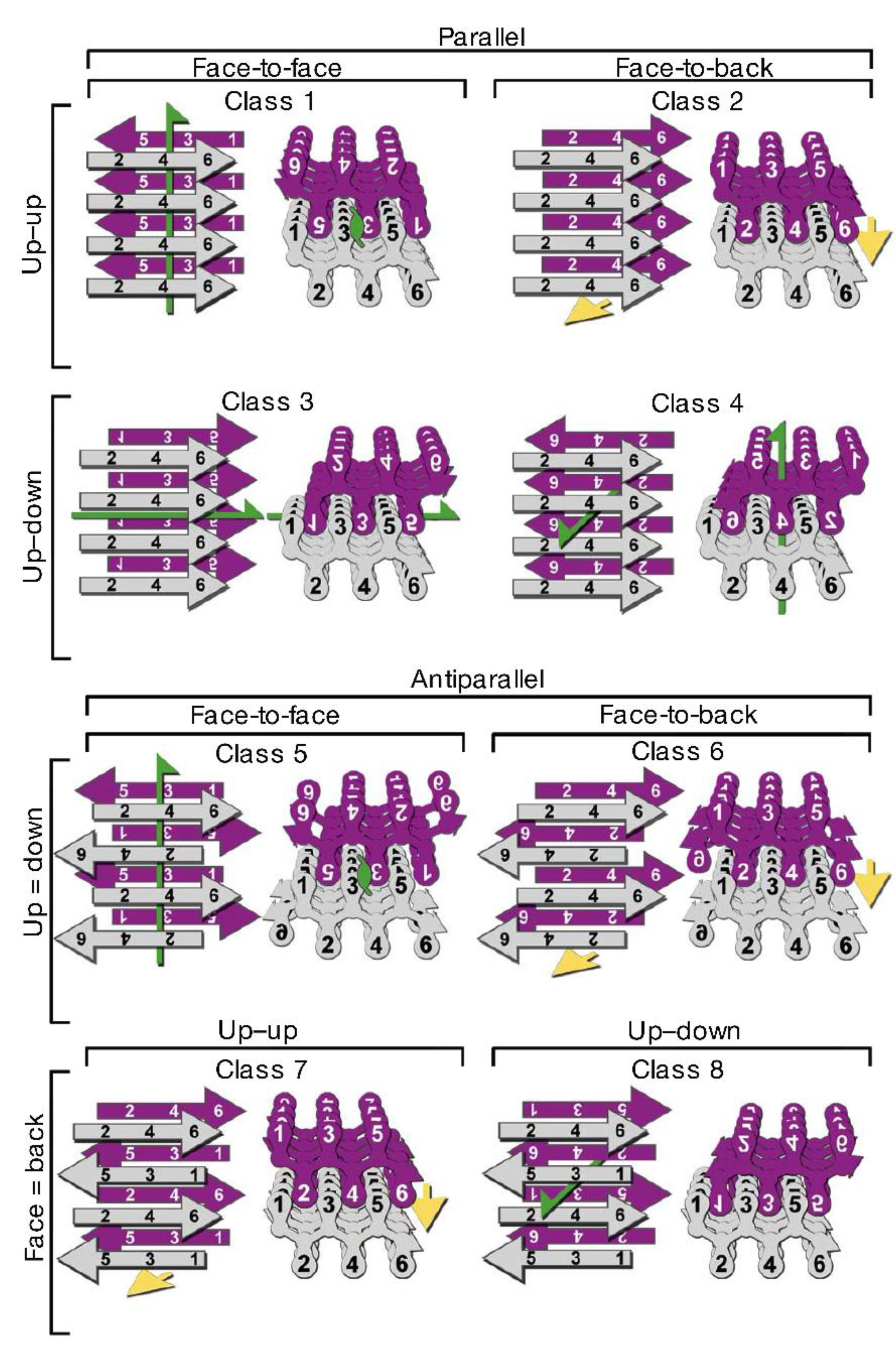
3.3. Structural Properties of Oligomers
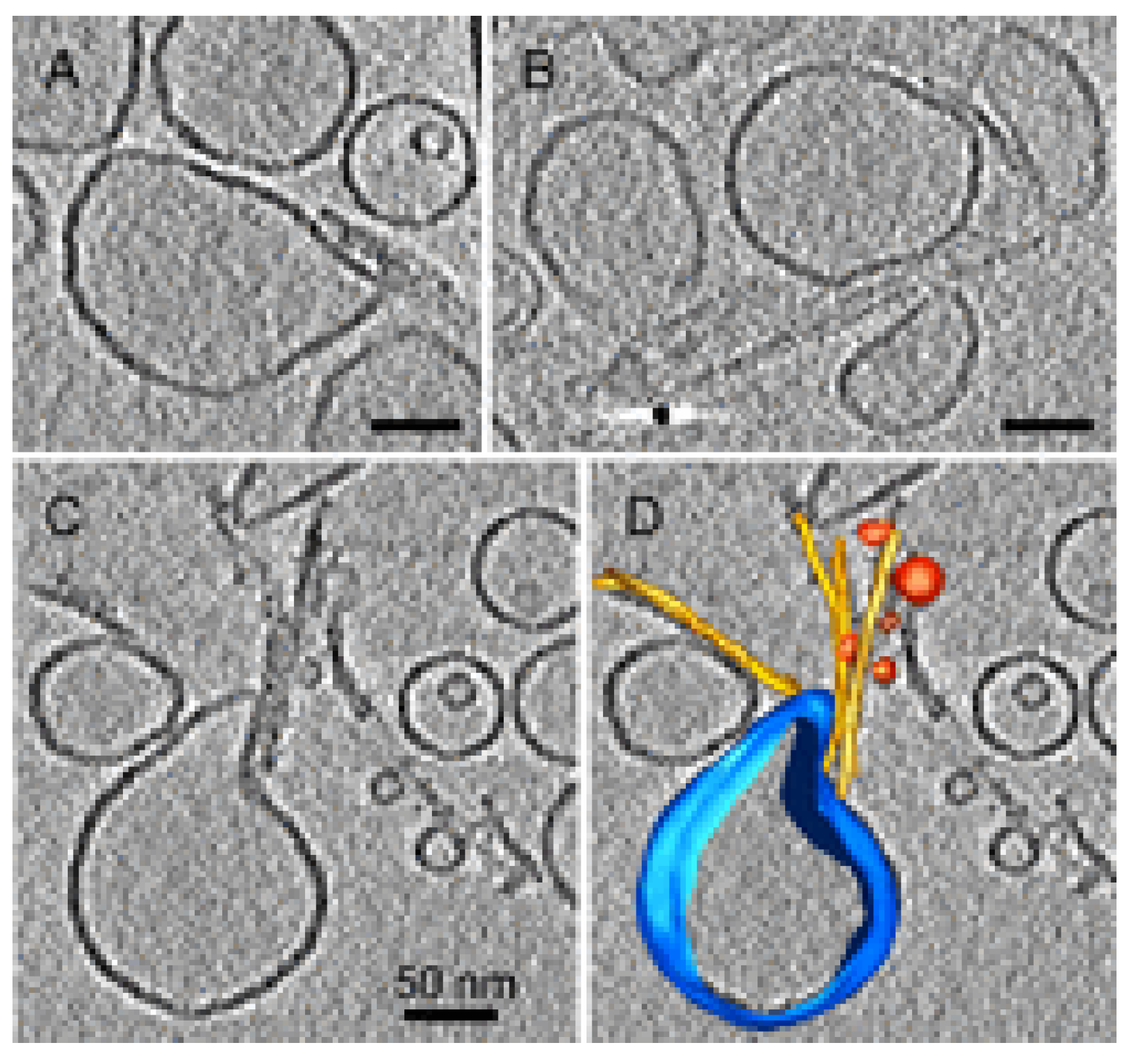
4. Amyloid Aggregates Disrupt Membrane Integrity
4.1. Formation of Ionic Pores
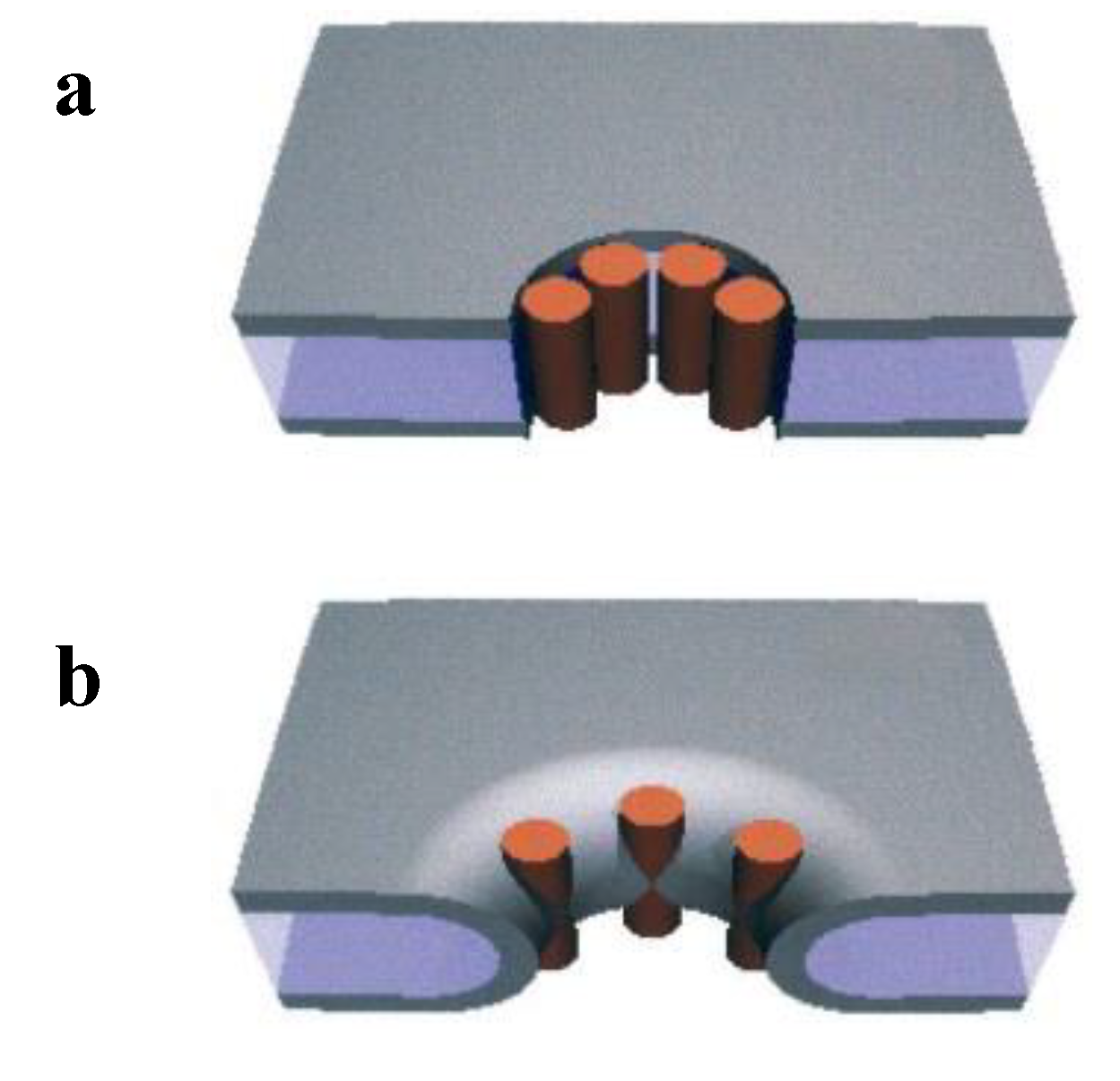
4.2. Tension-Induced Poration Mechanism
4.3. Disassembly of the Lipid Bilayer: Role of Oligomers and Fibrils
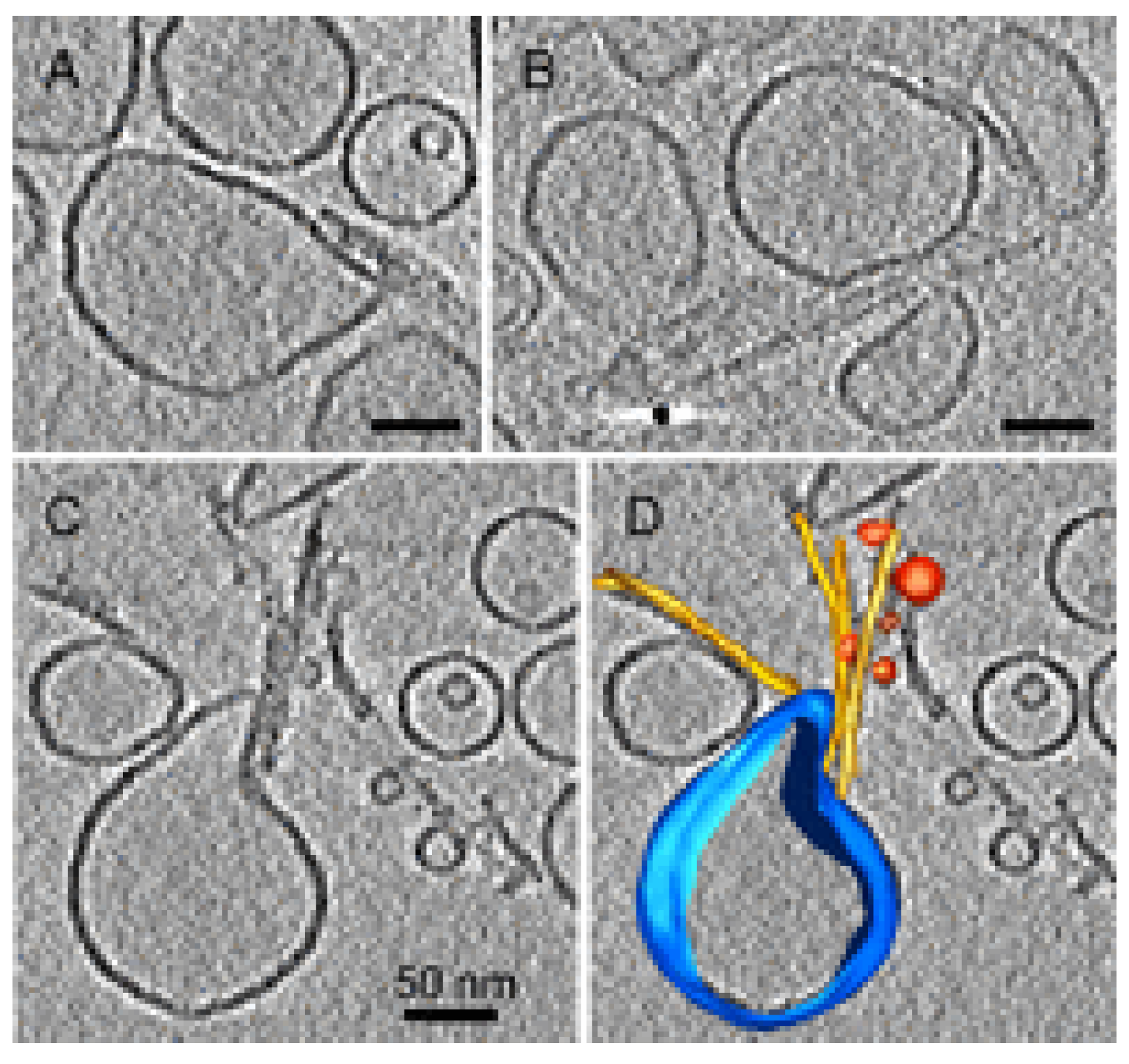
4.4. Oligomer Interaction with Specific Cell Structures
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.R.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef]
- Olsen, J.S.; DiMaio, J.T.; Doran, T.M.; Brown, C.; Nilsson, B.L.; Dewhurst, S. Seminal plasma accelerates semen-derived enhancer of viral infection (SEVI) fibril formation by the prostatic acid phosphatase (PAP248–286) peptide. J. Biol. Chem. 2012, 287, 11842–11849. [Google Scholar]
- Kelly, J.W. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 1998, 8, 101–106. [Google Scholar] [CrossRef]
- Rochet, J.; Lansbury, P.T., Jr. Amyloid fibrillogenesis: Themes and variations. Curr. Opin. Struct. Biol. 2000, 10, 60–68. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009, 5, 15–22. [Google Scholar] [CrossRef]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. BBA - Proteins Proteomics 2013, 1834, 932–951. [Google Scholar]
- Zhang, T.; Faraggi, E.; Li, Z.; Zhou, Y. Intrinsically semi-disordered state and its role in induced folding and protein aggregation. Cell Biochem. Biophys. 2013, 67, 1193–1205. [Google Scholar] [CrossRef]
- Zhao, H.; Tuominen, E.K.J.; Kinnunen, P.K.J. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry 2004, 43, 10302–10307. [Google Scholar] [CrossRef]
- Butterfield, S.; Lashuel, H. Amyloidogenic protein–membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. 2010, 49, 5628–5654. [Google Scholar] [CrossRef]
- Relini, A.; Marano, N.; Gliozzi, A. Probing the interplay between amyloidogenic proteins and membranes using lipid monolayers and bilayers. Adv. Colloid Interface Sci. 2013. in press. Available online: http://dx.doi.org/10.1016/j.cis.2013.10.015 (access on 24 December 2013).
- Relini, A.; Cavalleri, O.; Rolandi, R.; Gliozzi, A. The two-fold aspect of the interplay of amyloidogenic proteins with lipid membranes. Chem. Phys. Lipids 2009, 158, 1–9. [Google Scholar] [CrossRef]
- Stefani, M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 2010, 277, 4602–4613. [Google Scholar] [CrossRef]
- Evangelisti, E.; Cecchi, C.; Cascella, R.; Sgromo, C.; Becatti, M.; Dobson, C.M.; Chiti, F.; Stefani, M. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J. Cell Sci. 2012, 125, 2416–2427. [Google Scholar] [CrossRef]
- Kelly, B.L.; Ferreira, A. β-amyloid-induced dynamin 1 degradation is mediated by n-methyl-d-aspartate receptors in hippocampal neurons. J. Biol. Chem. 2006, 281, 28079–28089. [Google Scholar] [CrossRef]
- Pellistri, F.; Bucciantini, M.; Invernizzi, G.; Gatta, E.; Penco, A.; Frana, A.M.; Nosi, D.; Relini, A.; Regonesi, M.E.; Gliozzi, A.; et al. Different ataxin-3 amyloid aggregates induce intracellular Ca2+ deregulation by different mechanisms in cerebellar granule cells. BBA-Mol. Cell Res. 2013, 1833, 3155–3165. [Google Scholar]
- Kim, T.; Vidal, G.S.; Djurisic, M.; William, C.M.; Birnbaum, M.E.; Garcia, K.C.; Hyman, B.T.; Shatz, C.J. Human lilrB2 is a ß-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an alzheimer’s model. Science 2013, 341, 1399–1404. [Google Scholar] [CrossRef]
- Engel, M.F.M.; Khemtémourian, L.; Kleijer, C.C.; Meeldijk, H.J.D.; Jacobs, J.; Verkleij, A.J.; de Kruijff, B.; Killian, J.A.; Höppener, J.W.M. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar] [CrossRef]
- Campioni, S.; Mossuto, M.F.; Torrassa, S.; Calloni, G.; de Laureto, P.P.; Relini, A.; Fontana, A.; Chiti, F. Conformational properties of the aggregation precursor state of HypF-N. J. Mol. Biol. 2008, 379, 554–567. [Google Scholar]
- Calloni, G.; Lendel, C.; Campioni, S.; Giannini, S.; Gliozzi, A.; Relini, A.; Vendruscolo, M.; Dobson, C.M.; Salvatella, X.; Chiti, F. Structure and dynamics of a partially folded protein are decoupled from its mechanism of aggregation. J. Am. Chem. Soc. 2008, 130, 13040–13050. [Google Scholar] [CrossRef]
- Bouchard, M.; Zurdo, J.; Nettleton, E.J.; Dobson, C.M.; Robinson, C.V. Formation of insulin amyloid fibrils followed by FTIR simultaneously with CD and electron microscopy. Protein Sci. 2000, 9, 1960–1967. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Christensen, G.; Otzen, D.E. Modulation of S6 fibrillation by unfolding rates and gatekeeper residues. J. Mol. Biol. 2004, 341, 575–588. [Google Scholar] [CrossRef]
- Plakoutsi, G.; Taddei, N.; Stefani, M.; Chiti, F. Aggregation of the acylphosphatase from sulfolobus solfataricus: The folded and patially unfolded states can both be precursors for amyloid formation. J. Biol. Chem. 2004, 279, 14111–14119. [Google Scholar] [CrossRef]
- Plakoutsi, G.; Bemporad, F.; Monti, M.; Pagnozzi, D.; Pucci, P.; Chiti, F. Exploring the mechanism of formation of native-like and precursor amyloid oligomers for the native acylphosphatase from sulfolobus solfataricus. Structure 2006, 14, 993–1001. [Google Scholar] [CrossRef] [Green Version]
- Danielsson, J.; Jarvet, J.; Damberg, P.; Gräslund, A. Translational diffusion measured by PFG-NMR on full length and fragments of the Alzheimer Aβ(1–40) peptide. Determination of hydrodynamic radii of random coil peptides of varying length. Magn. Reson. Chem. 2002, 40, S89–S97. [Google Scholar] [CrossRef]
- Ma, K.; Clancy, E.L.; Zhang, Y.; Ray, D.G.; Wollenberg, K.; Zagorski, M.G. Residue-specific pka measurements of the β-peptide and mechanism of ph-induced amyloid formation. J. Am. Chem. Soc. 1999, 121, 8698–8706. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Rosenman, D.J.; Connors, C.R.; Chen, W.; Wang, C.; García, A.E. Aβ monomers transiently sample oligomer and fibril-like configurations: Ensemble characterization using a combined MD/NMR approach. J. Mol. Biol. 2013, 425, 3338–3359. [Google Scholar] [CrossRef]
- Knapman, T.W.; Valette, N.M.; Warriner, S.L.; Ashcroft, A.E. Ion mobility spectrometry-mass spectrometry of intrinsically unfolded proteins: Trying to put order into disorder. Curr. Anal. Chem. 2013, 9, 181–191. [Google Scholar]
- Qiao, Q.; Bowman, G.R.; Huang, X. Dynamics of an intrinsically disordered protein reveal metastable conformations that potentially seed aggregation. J. Am. Chem. Soc. 2013, 135, 16092–16101. [Google Scholar] [CrossRef]
- Jain, N.; Bhattacharya, M.; Mukhopadhyay, S. Chain collapse of an amyloidogenic intrinsically disordered protein. Biophys. J. 2011, 101, 1720–1729. [Google Scholar] [CrossRef]
- Kayed, R.; Bernhagen, J.; Greenfield, N.; Sweimeh, K.; Brunner, H.; Voelter, W.; Kapurniotu, A. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J. Mol. Biol. 1999, 287, 781–796. [Google Scholar] [CrossRef]
- Pawar, A.P.; DuBay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, C. Aβ42 is more rigid than Aβ40 at the C terminus: Implications for Aβ aggregation and toxicity. J. Mol. Biol. 2006, 364, 853–862. [Google Scholar] [CrossRef]
- Chen, Y.; Glabe, C.G. Distinct early folding and aggregation properties of alzheimer amyloid-β peptides Aβ40 and Aβ42: STABLE TRIMER OR TETRAMER FORMATION BY Aβ42. J. Biol. Chem. 2006, 281, 24414–24422. [Google Scholar] [CrossRef]
- Roychaudhuri, R.; Yang, M.; Deshpande, A.; Cole, G.M.; Frautschy, S.; Lomakin, A.; Benedek, G.B.; Teplow, D.B. C-terminal turn stability determines assembly differences between Aβ40 and Aβ42. J. Mol. Biol. 2013, 425, 292–308. [Google Scholar] [CrossRef]
- De Simone, A.; Dhulesia, A.; Soldi, G.; Vendruscolo, M.; Hsu, S.D.; Chiti, F.; Dobson, C.M. Experimental free energy surfaces reveal the mechanisms of maintenance of protein solubility. Proc. Natl. Acad. Sci. USA 2011, 108, 21057–21062. [Google Scholar] [CrossRef]
- Morozova-Roche, L.A.; Zurdo, J.; Spencer, A.; Noppe, W.; Receveur, V.; Archer, D.B.; Joniau, M.; Dobson, C.M. Amyloid fibril formation and seeding by wild-type human lysozyme and its disease-related mutational variants. J. Struct. Biol. 2000, 130, 339–351. [Google Scholar] [CrossRef]
- Fazili, N.; Naeem, A. In vitro hyperglycemic condition facilitated the aggregation of lysozyme via the passage through a molten globule state. Cell Biochem. Biophys. 2013, 66, 265–275. [Google Scholar]
- Marcon, G.; Plakoutsi, G.; Canale, C.; Relini, A.; Taddei, N.; Dobson, C.M.; Ramponi, G.; Chiti, F. Amyloid formation from HypF-N under conditions in which the protein is initially in its native state. J. Mol. Biol. 2005, 347, 323–335. [Google Scholar] [CrossRef]
- Jahn, T.R.; Parker, M.J.; Homans, S.W.; Radford, S.E. Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nat. Struct. Mol. Biol. 2006, 13, 195–201. [Google Scholar] [CrossRef]
- Rennella, E.; Cutuil, T.; Schanda, P.; Ayala, I.; Gabel, F.; Forge, V.; Corazza, A.; Esposito, G.; Brutscher, B. Oligomeric states along the folding pathways of β 2-microglobulin: Kinetics, thermodynamics, and structure. J. Mol. Biol. 2013, 425, 2722–2736. [Google Scholar] [CrossRef]
- Eichner, T.; Radford, S.E. A diversity of assembly mechanisms of a generic amyloid fold. Mol. Cell 2011, 43, 8–18. [Google Scholar] [CrossRef]
- Arnaudov, L.; de Vries, R. Thermally induced fibrillar aggregation of hen egg white lysozyme. Biophys. J. 2005, 88, 515–526. [Google Scholar] [CrossRef]
- Krebs, M.R.; Wilkins, D.K.; Chung, E.W.; Pitkeathly, M.C.; Chamberlain, A.K.; Zurdo, J.; Robinson, C.V.; Dobson, C.M. Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the β-domain. J. Mol. Biol. 2000, 300, 541–549. [Google Scholar] [CrossRef]
- Ow, S.; Dunstan, D.E. The effect of concentration, temperature and stirring on hen egg white lysozyme amyloid formation. Soft Matter 2013, 9, 9692–9701. [Google Scholar] [CrossRef]
- Nielsen, L.; Khurana, R.; Coats, A.; Frokjaer, S.; Brange, J.; Vyas, S.; Uversky, V.; Fink, A. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 2001, 40, 6036–6046. [Google Scholar] [CrossRef]
- Babenko, V.; Piejko, M.; Wojcik, S.; Mak, P.; Dzwolak, W. Vortex-induced amyloid superstructures of insulin and its component A and B chains. Langmuir 2013, 29, 5271–5278. [Google Scholar] [CrossRef]
- Sluzky, V.; Tamada, J.A.; Klibanov, A.M.; Langer, R. Kinetics of insulin aggregation in aqueous solutions upon agitation in the presence of hydrophobic surfaces. Proc. Natl. Acad. Sci. USA 1991, 88, 9377–9381. [Google Scholar] [CrossRef]
- Bekard, I.B.; Dunstan, D.E. Shear-induced deformation of bovine insulin in couette flow. J. Phys. Chem. B 2009, 113, 8453–8457. [Google Scholar] [CrossRef]
- Mangione, P.P.; Esposito, G.; Relini, A.; Raimondi, S.; Porcari, R.; Giorgetti, S.; Corazza, A.; Fogolari, F.; Penco, A.; Goto, Y.; et al. Structure, folding dynamics, and amyloidogenesis of D76N ß2-microglobulin: Roles of shear flow, hydrophobic surfaces, and a-crystallin. J. Biol. Chem. 2013, 288, 30917–30930. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Lee, S.; Liu, Y.; Lim, M.H. Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem. Biol. 2013, 8, 856–865. [Google Scholar] [CrossRef]
- Hoernke, M.; Koksch, B.; Brezesinski, G. Influence of the hydrophobic interface and transition metal ions on the conformation of amyloidogenic model peptides. Biophys. Chem. 2010, 150, 64–72. [Google Scholar] [CrossRef]
- Leal, S.S.; Cardoso, I.; Valentine, J.S.; Gomes, C.M. Calcium ions promote superoxide dismutase 1 (SOD1) aggregation into non-fibrillar amyloid: A link to toxic effects of calcium overload in amyotrophic lateral sclerOSIS (ALS)? J. Biol. Chem. 2013, 288, 25219–25228. [Google Scholar] [CrossRef]
- Diaz-Espinoza, R.; Mukherjee, A.; Soto, C. Kosmotropic anions promote conversion of recombinant prion protein into a PrPSc-like misfolded form. PLoS One 2012, 7, e31678. [Google Scholar] [CrossRef]
- Rubin, J.; Khosravi, H.; Bruce, K.L.; Lydon, M.E.; Behrens, S.H.; Chernoff, Y.O.; Bommarius, A.S. Ion-specific effects on prion nucleation and strain formation. J. Biol. Chem. 2013, 288, 30300–30308. [Google Scholar]
- Munishkina, L.; Phelan, C.; Uversky, V.; Fink, A. Conformational behavior and aggregation of alpha-synuclein in organic solvents: Modeling the effects of membranes. Biochemistry 2003, 42, 2720–2730. [Google Scholar] [CrossRef]
- Alonso Vilatela, M.E.; López-López, M.; Yescas-Gómez, P. Genetics of Alzheimer’s disease. Arch. Med. Res. 2012, 43, 622–631. [Google Scholar] [CrossRef]
- Canet, D.; Sunde, M.; Last, A.M.; Miranker, A.; Spencer, A.; Robinson, C.V.; Dobson, C.M. Mechanistic studies of the folding of human lysozyme and the origin of amyloidogenic behavior in its disease-related variants. Biochemistry 1999, 38, 6419–6427. [Google Scholar] [CrossRef]
- Valleix, S.; Gillmore, J.D.; Bridoux, F.; Mangione, P.P.; Dogan, A.; Nedelec, B.; Boimard, M.; Touchard, G.; Goujon, J.; Lacombe, C.; et al. Hereditary systemic amyloidosis due to Asp76Asn variant β2-microglobulin. N. Engl. J. Med. 2012, 366, 2276–2283. [Google Scholar] [CrossRef]
- Bellotti, V.; Stoppini, M.; Mangione, P.; Sunde, M.; Robinson, C.; Asti, L.; Brancaccio, D.; Ferri, G. β2-microglobulin can be refolded into a native state from ex vivo amyloid fibrils. Eur. J. Biochem. 1998, 258, 61–67. [Google Scholar]
- Esposito, G.; Michelutti, R.; Verdone, G.; Viglino, P.; Hernández, H.; Robinson, C.V.; Amoresano, A.; Dal Piaz, F.; Monti, M.; Pucci, P.; et al. Removal of the N-terminal hexapeptide from human β2-microglobulin facilitates protein aggregation and fibril formation. Protein Sci. 2000, 9, 831–845. [Google Scholar] [CrossRef]
- Eichner, T.; Kalverda, A.P.; Thompson, G.S.; Homans, S.W.; Radford, S.E. Conformational conversion during amyloid formation at atomic resolution. Mol. Cell 2011, 41, 161–172. [Google Scholar] [CrossRef]
- Piazza, R.; Pierno, M.; Iacopini, S.; Mangione, P.; Esposito, G.; Bellotti, V. Micro-heterogeneity and aggregation in β2-microglobulin solutions: Effects of temperature, pH, and conformational variant addition. Eur. Biophys. J. 2006, 35, 439–445. [Google Scholar] [CrossRef]
- Calamai, M.; Taddei, N.; Stefani, M.; Ramponi, G.; Chiti, F. Relative influence of hydrophobicity and net charge in the aggregation of two homologous proteins. Biochemistry 2003, 42, 15078–15083. [Google Scholar] [CrossRef]
- Chiti, F.; Calamai, M.; Taddei, N.; Stefani, M.; Ramponi, G.; Dobson, C.M. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. 4), 16419–16426. [Google Scholar]
- Chiti, F.; Taddei, N.; Baroni, F.; Capanni, C.; Stefani, M.; Ramponi, G.; Dobson, C.M. Kinetic partitioning of protein folding and aggregation. Nat. Struct. Biol. 2002, 9, 137–143. [Google Scholar] [CrossRef]
- Kim, W.; Hecht, M.H. Sequence determinants of enhanced amyloidogenicity of alzheimer Aβ42 peptide relative to Aβ40. J. Biol. Chem. 2005, 280, 35069–35076. [Google Scholar] [CrossRef]
- Cummings, C.J.; Zoghbi, H.Y. Fourteen and counting: Unraveling trinucleotide repeat diseases. Hum. Mol. Genet. 2000, 9, 909–916. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Orr, H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [Google Scholar] [CrossRef]
- Perutz, M.F.; Johnson, T.; Suzuki, M.; Finch, J.T. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 1994, 91, 5355–5358. [Google Scholar] [CrossRef]
- Chen, S.; Berthelier, V.; Hamilton, J.B.; O’Nuallai, B.; Wetzel, R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry 2002, 41, 7391–7399. [Google Scholar] [CrossRef]
- Shehi, E.; Fusi, P.; Secundo, F.; Pozzuolo, S.; Bairati, A.; Tortora, P. Temperature-dependent, irreversible formation of amyloid fibrils by a soluble human ataxin-3 carrying a moderately expanded polyglutamine stretch (Q36). Biochemistry 2003, 42, 14626–14632. [Google Scholar] [CrossRef]
- Shen, L.; Adachi, T.; Vanden Bout, D.; Zhu, X.-Y. A mobile precursor determines amyloid-β peptide fibril formation at interfaces. J. Am. Chem. Soc. 2012, 134, 14172–14178. [Google Scholar] [CrossRef]
- Krasowska, M.; Zawala, J.; Malysa, K. Air at hydrophobic surfaces and kinetics of three phase contact formation. Adv. Colloid Interface Sci. 2009, 147–148, 155–169. [Google Scholar] [CrossRef]
- Hoernke, M.; Falenski, J.A.; Schwieger, C.; Koksch, B.; Brezesinski, G. Triggers for β-Sheet formation at the hydrophobic-hydrophilic interface: High concentration, in-plane orientational order, and metal ion complexation. Langmuir 2011, 27, 14218–14231. [Google Scholar] [CrossRef]
- Nault, L.; Guo, P.; Jain, B.; Brechet, Y.; Bruckert, F.; Weidenhaupt, M. Human insulin adsorption kinetics, conformational changes and amyloidal aggregate formation on hydrophobic surfaces. Acta Biomater. 2013, 9, 5070–5079. [Google Scholar] [CrossRef]
- Apicella, A.; Soncini, M.; Deriu, MA; Natalello, A.; Bonanomi, M.; Dellasega, D.; Tortora, P.; Regonesi, M.E; Casari, C.S. A hydrophobic gold surface triggers misfolding and aggregation of the amyloidogenic Josephin domain in monomeric form, while leaving the oligomers unaffected. PLoS One 2013, 8, e58794. [Google Scholar]
- Zhu, M.; Souillac, P.; Ionescu-Zanetti, C.; Carter, S.; Fink, A. Surface-catalyzed amyloid fibril formation. J. Biol. Chem. 2002, 277, 50914–50922. [Google Scholar] [CrossRef]
- Meng, X.; Fink, A.L.; Uversky, V.N. The effect of membranes on the in vitro fibrillation of an amyloidogenic light-chain variable-domain SMA. J. Mol. Biol. 2008, 381, 989–999. [Google Scholar] [CrossRef]
- Bellotti, V.; Chiti, F. Amyloidogenesis in its biological environment: Challenging a fundamental issue in protein misfolding diseases. Curr. Opin. Struct. Biol. 2008, 18, 771–779. [Google Scholar] [CrossRef]
- Homma, N.; Gejyo, F.; Isemura, M.; Arakawa, M. Collagen-binding affinity of β-2-microglobulin, a preprotein of hemodialysis-associated amyloidosis. Nephron 1989, 53, 37–40. [Google Scholar] [CrossRef]
- Giorgetti, S.; Rossi, A.; Mangione, P.; Raimondi, S.; Marini, S.; Stoppini, M.; Corazza, A.; Viglino, P.; Esposito, G.; Cetta, G.; et al. β2-Microglobulin isoforms display an heterogeneous affinity for type I collagen. Protein Sci. 2005, 14, 696–702. [Google Scholar] [CrossRef]
- Relini, A.; Canale, C.; de Stefano, S.; Rolandi, R.; Giorgetti, S.; Stoppini, M.; Rossi, A.; Fogolari, F.; Corazza, A.; Esposito, G.; et al. Collagen plays an active role in the aggregation of β2-microglobulin under physiopathological conditions of dialysis-related amyloidosis. J. Biol. Chem. 2006, 281, 16521–16529. [Google Scholar] [CrossRef]
- Bertoletti, L.; Regazzoni, L.; Aldini, G.; Colombo, R.; Abballe, F.; Caccialanza, G.; de Lorenzi, E. Separation and characterisation of β2-microglobulin folding conformers by ion-exchange liquid chromatography and ion-exchange liquid chromatography-mass spectrometry. Anal. Chim. Acta 2013, 771, 108–114. [Google Scholar] [CrossRef]
- Myers, S.L.; Jones, S.; Jahn, T.R.; Morten, I.J.; Tennent, G.A.; Hewitt, E.W.; Radford, S.E. A systematic study of the effect of physiological factors on β2-microglobulin amyloid formation at neutral pH. Biochemistry 2006, 45, 2311–2321. [Google Scholar] [CrossRef]
- Harris, D.L.; King, E.; Ramsland, P.A.; Edmundson, A.B. Binding of nascent collagen by amyloidogenic light chains and amyloid fibrillogenesis in monolayers of human fibrocytes. J. Mol. Recognit. 2000, 13, 198–212. [Google Scholar] [CrossRef]
- Borysik, A.J.; Morten, I.J.; Radford, S.E.; Hewitt, E.W. Specific glycosaminoglycans promote unseeded amyloid formation from β2-microglobulin under physiological conditions. Kidney Int. 2007, 72, 174–181. [Google Scholar] [CrossRef]
- Yamamoto, S.; Hasegawa, K.; Yamaguchi, I.; Goto, Y.; Gejyo, F.; Naiki, H. Kinetic analysis of the polymerization and depolymerization of β2-microglobulin-related amyloid fibrils in vitro. BBA - Proteins Proteomics 2005, 1753, 34–43. [Google Scholar] [CrossRef]
- Relini, A.; de Stefano, S.; Torrassa, S.; Cavalleri, O.; Rolandi, R.; Gliozzi, A.; Giorgetti, S.; Raimondi, S.; Marchese, L.; Verga, L.; et al. Heparin strongly enhances the formation of β2-microglobulin amyloid fibrils in the presence of type I collagen. J. Biol. Chem. 2008, 283, 4912–4920. [Google Scholar]
- Ancsin, J.B. Amyloidogenesis: Historical and modern observations point to heparan sulfate proteoglycans as a major culprit. Amyloid 2003, 10, 67–79. [Google Scholar] [CrossRef]
- Ariga, T.; Miyatake, T.; Yu, R.K. Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer’s disease and related disorders: Amyloidogenesis and therapeutic strategies—A review. J. Neurosci. Res. 2010, 88, 2303–2315. [Google Scholar] [CrossRef]
- Kisilevsky, R.; Ancsin, J.B.; Szarek, W.A.; Petanceska, S. Heparan sulfate as a therapeutic target in amyloidogenesis: Prospects and possible complications. Amyloid 2007, 14, 21–32. [Google Scholar] [CrossRef]
- Castillo, G.M.; Cummings, J.A.; Yang, W.; Judge, M.E.; Sheardown, M.J.; Rimvall, K.; Hansen, J.B.; Snow, A.D. Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan’s enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes 1998, 47, 612–620. [Google Scholar] [CrossRef]
- Potter-Perigo, S.; Hull, R.L.; Tsoi, C.; Braun, K.R.; Andrikopoulos, S.; Teague, J.; Bruce Verchere, C.; Kahn, S.E.; Wight, T.N. Proteoglycans synthesized and secreted by pancreatic islet β-cells bind amylin. Arch. Biochem. Biophys. 2003, 413, 182–190. [Google Scholar] [CrossRef]
- Hull, R.L.; Peters, M.J.; Perigo, S.P.; Chan, C.K.; Wight, T.N.; Kinsella, M.G. Overall Sulfation of heparan Sulfate from pancreatic islet ß-TC3 cells increases maximal fibril formation but does not determine binding to the amyloidogenic peptide islet amyloid polypeptide. J. Biol. Chem. 2012, 287, 37154–37164. [Google Scholar]
- Castillo, G.M.; Ngo, C.; Cummings, J.; Wight, T.N.; Snow, A.D. Perlecan binds to the β-amyloid proteins (Aβ) of Alzheimer’s disease, accelerates A? Fibril formation, and maintains Aβ fibril stability. J. Neurochem. 1997, 69, 2452–2465. [Google Scholar]
- Cotman, S.L.; Halfter, W.; Cole, G.J. Agrin binds to β-amyloid (Aβ), accelerates Aβ fibril formation, and is localized to Aβ deposits in Alzheimer’s disease brain. Mol. Cell. Neurosci. 2000, 15, 183–198. [Google Scholar] [CrossRef]
- Castillo, G.M.; Lukito, W.; Peskind, E.; Raskind, M.; Kirschner, D.A.; Yee, A.G.; Snow, A.D. Laminin inhibition of β-amyloid protein (Aβ) fibrillogenesis and identification of an Aβ binding site localized to the globular domain repeats on the laminin a chain. J. Neurosci. Res. 2000, 62, 451–462. [Google Scholar] [CrossRef]
- Drouet, B.; Pinçon-Raymond, M.; Chambaz, J.; Pillot, T. Laminin 1 attenuates ß-amyloid peptide Aß(1–40) neurotoxicity of cultured fetal rat cortical neurons. J. Neurochem. 1999, 73, 742–749. [Google Scholar]
- Kiuchi, Y.; Isobe, Y.; Fukushima, K. Entactin-induced inhibition of human amyloid ß-protein fibril formation in vitro. Neurosci. Lett. 2001, 305, 119–122. [Google Scholar] [CrossRef]
- Kiuchi, Y.; Isobe, Y.; Fukushima, K. Type IV collagen prevents amyloid ß-protein fibril formation. Life Sci. 2002, 70, 1555–1564. [Google Scholar] [CrossRef]
- Hasegawa, K.; Ozawa, D.; Ookoshi, T.; Naiki, H. Surface-bound basement membrane components accelerate amyloid-β peptide nucleation in air-free wells: An in vitro model of cerebral amyloid angiopathy. BBA - Proteins Proteomics 2013, 1834, 1624–1631. [Google Scholar] [CrossRef]
- Gorbenko, G.P.; Kinnunen, P.K. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids 2006, 141, 72–82. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar]
- Jo, E.; McLaurin, J.; Yip, C.M.; St. George-Hyslop, P.; Fraser, P.E. Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000, 275, 34328–34334. [Google Scholar] [CrossRef]
- Ferreon, A.C.M.; Gambin, Y.; Lemke, E.A.; Deniz, A.A. Interplay of a-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 2009, 106, 5645–5650. [Google Scholar] [CrossRef]
- Apostolidou, M.; Jayasinghe, S.A.; Langen, R. Structure of α-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J. Biol. Chem. 2008, 283, 17205–17210. [Google Scholar] [CrossRef]
- Jayasinghe, S.A.; Langen, R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 2005, 44, 12113–12119. [Google Scholar]
- Knight, J.D.; Hebda, J.A.; Miranker, A.D. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry 2006, 45, 9496–9508. [Google Scholar] [CrossRef]
- Lee, C.; Sun, Y.; Huang, H.W. How type II diabetes-related islet amyloid polypeptide damages lipid bilayers. Biophys. J. 2012, 102, 1059–1068. [Google Scholar]
- Jia, Y.; Qian, Z.; Zhang, Y.; Wei, G. Adsorption and orientation of human islet amyloid polypeptide (hIAPP) monomer at anionic lipid bilayers: Implications for membrane-mediated aggregation. Int. J. Mol. Sci. 2013, 14, 6241–6258. [Google Scholar] [CrossRef]
- Lee, C.; Sun, Y.; Huang, H.W. Membrane-mediated peptide conformation change from alpha-monomers to β-aggregates. Biophys. J. 2010, 98, 2236–2245. [Google Scholar] [CrossRef]
- Schauerte, J.A.; Wong, P.T.; Wisser, K.C.; Ding, H.; Steel, D.G.; Gafni, A. Simultaneous single-molecule fluorescence and conductivity studies reveal distinct classes of Aβ species on lipid bilayers. Biochemistry 2010, 49, 3031–3039. [Google Scholar] [CrossRef]
- Ding, H.; Schauerte, J.; Steel, D.; Gafni, A. β-Amyloid (1-40) peptide interactions with supported phospholipid membranes: A single-molecule study. Biophys. J. 2012, 103, 1500–1509. [Google Scholar] [CrossRef]
- Sani, M.; Gehman, J.D.; Separovic, F. Lipid matrix plays a role in Aβ fibril kinetics and morphology. FEBS Lett. 2011, 585, 749–754. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. The driving force of α-synuclein insertion and amyloid channel formation in the plasma membrane of neural cells: Key role of ganglioside- and cholesterol-binding domains. Adv. Exp. Med. Biol. 2013, 991, 15–26. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: Common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med. 2010, 12, e27. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N.; Garmy, N. Cholesterol accelerates the binding of Alzheimer’s β-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front. Physiol. 2013, 4, 120. [Google Scholar]
- Mao, Y.; Shang, Z.; Imai, Y.; Hoshino, T.; Tero, R.; Tanaka, M.; Yamamoto, N.; Yanagisawa, K.; Urisu, T. Surface-induced phase separation of a sphingomyelin/cholesterol/ganglioside GM1-planar bilayer on mica surfaces and microdomain molecular conformation that accelerates Aβ oligomerization. Biochim. Biophys. Acta-Biomembr. 2010, 1798, 1090–1099. [Google Scholar] [CrossRef]
- Mikhalyov, I.; Olofsson, A.; Grobner, G.; Johansson, L.B.-A. Designed fluorescent probes reveal interactions between amyloid-β(1–40) peptides and G(M1) gangliosides in micelles and lipid vesicles. Biophys. J. 2010, 99, 1510–1519. [Google Scholar]
- Matsuzaki, K.; Kato, K.; Yanagisawa, K. Aβ polymerization through interaction with membrane gangliosides. BBA-Mol. Cell Biol. Lipids 2010, 1801, 868–877. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef]
- Apetri, M.M.; Maiti, N.C.; Zagorski, M.G.; Carey, P.R.; Anderson, V.E. Secondary structure of α-synuclein oligomers: Characterization by Raman and atomic force microscopy. J. Mol. Biol. 2006, 355, 63–71. [Google Scholar] [CrossRef]
- Hill, S.E.; Robinson, J.; Matthews, G.; Muschol, M. Amyloid protofibrils of lysozyme nucleate and grow via oligomer fusion. Biophys. J. 2009, 96, 3781–3790. [Google Scholar] [CrossRef]
- Modler, A.J.; Gast, K.; Lutsch, G.; Damaschun, G. Assembly of amyloid protofibrils via critical oligomers—A novel pathway of amyloid formation. J. Mol. Biol. 2003, 325, 135–148. [Google Scholar] [CrossRef]
- Kumar, S.; Udgaonkar, J.B. Conformational conversion may precede or follow aggregate elongation on alternative pathways of amyloid protofibril formation. J. Mol. Biol. 2009, 385, 1266–1276. [Google Scholar] [CrossRef]
- Kheterpal, I.; Chen, M.; Cook, K.D.; Wetzel, R. Structural differences in Aβ amyloid protofibrils and fibrils mapped by hydrogen exchange—Mass spectrometry with on-line proteolytic fragmentation. J. Mol. Biol. 2006, 361, 785–795. [Google Scholar]
- Scheidt, H.A.; Morgado, I.; Rothemund, S.; Huster, D.; Fändrich, M. Solid-state NMR spectroscopic investigation of Aβ protofibrils: Implication of a β-sheet remodeling upon maturation into terminal amyloid fibrils. Angew. Chem. Int. Ed. 2011, 50, 2837–2840. [Google Scholar] [CrossRef]
- Scheidt, H.A.; Morgado, I.; Huster, D. Solid-state NMR reveals a close structural relationship between amyloid-ß protofibrils and oligomers. J.Biol.Chem. 2012, 287, 22822–22826. [Google Scholar] [CrossRef]
- Petty, S.A.; Decatur, S.M. Intersheet rearrangement of polypeptides during nucleation of ß-sheet aggregates. Proc. Natl. Acad. Sci. USA 2005, 102, 14272–14277. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Lin, Y.; Yagi, H.; Lee, Y.H.; Kitayama, H.; Sakurai, K.; So, M.; Ogi, H.; Naiki, H.; Goto, Y. Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation. Proc. Natl. Acad. Sci. USA 2012, 109, 14446–14451. [Google Scholar] [CrossRef]
- Morris, A.M.; Watzky, M.A.; Finke, R.G. Protein aggregation kinetics, mechanism, and curve-fitting: A review of the literature. BBA - Proteins Proteomics 2009, 1794, 375–397. [Google Scholar] [CrossRef]
- Knowles, T.P.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef]
- Andersen, C.B.; Yagi, H.; Manno, M.; Martorana, V.; Ban, T.; Christiansen, G.; Otzen, D.E.; Goto, Y.; Rischel, C. Branching in amyloid fibril growth. Biophys. J. 2009, 96, 1529–1536. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-ß42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef]
- Ban, T.; Hamada, D.; Hasegawa, K.; Naiki, H.; Goto, Y. Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. J. Biol. Chem. 2003, 278, 16462–16465. [Google Scholar]
- Heldt, C.L.; Zhang, S.; Belfort, G. Asymmetric amyloid fibril elongation: A new perspective on a symmetric world. Proteins: Struct. Funct. Bioinforma. 2011, 79, 92–98. [Google Scholar] [CrossRef]
- Collins, S.R.; Douglass, A.; Vale, R.D.; Weissman, J.S. Mechanism of prion propagation: Amyloid growth occurs by monomer addition. PLoS Biol. 2004, 2, e321. [Google Scholar] [CrossRef] [Green Version]
- Ferkinghoff-Borg, J.; Fonslet, J.; Andersen, C.B.; Krishna, S.; Pigolotti, S.; Yagi, H.; Goto, Y.; Otzen, D.; Jensen, M.H. Stop-and-go kinetics in amyloid fibrillation. Phys. Rev. E 2010, 82, 010901. [Google Scholar] [CrossRef]
- Kellermayer, M.S.Z.; Karsai, Á.; Benke, M.; Soós, K.; Penke, B. Stepwise dynamics of epitaxially growing single amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 141–144. [Google Scholar] [CrossRef]
- Gosal, W.S.; Morten, I.J.; Hewitt, E.W.; Smith, D.A.; Thomson, N.H.; Radford, S.E. Competing pathways determine fibril morphology in the self-assembly of β2-microglobulin into amyloid. J. Mol. Biol. 2005, 351, 850–864. [Google Scholar] [CrossRef]
- Ramachandran, G.; Udgaonkar, J.B. Understanding the kinetic roles of the inducer heparin and of rod-like protofibrils during amyloid fibril formation by tau protein. J. Biol. Chem. 2011, 286, 38948–38959. [Google Scholar] [CrossRef]
- Zakharov, V.V.; Mosevitsky, M.I. Oligomeric structure of brain abundant proteins GAP-43 and BASP1. J. Struct. Biol. 2010, 170, 470–483. [Google Scholar] [CrossRef]
- Walsh, D.M.; Lomakin, A.; Benedek, G.B.; Condron, M.M.; Teplow, D.B. Amyloid ß-protein fibrillogenesis: Detection of a protofibrillar intermediate. J. Biol. Chem. 1997, 272, 22364–22372. [Google Scholar]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical Amyloid. Biochemistry 2000, 39, 2552–2563. [Google Scholar] [CrossRef]
- Relini, A.; Torrassa, S.; Ferrando, R.; Rolandi, R.; Campioni, S.; Chiti, F.; Gliozzi, A. Detection of populations of amyloid-like protofibrils with different physical properties. Biophys. J. 2010, 98, 1277–1284. [Google Scholar]
- Juárez, J.; Taboada, P.; Mosquera, V. Existence of different structural intermediates on the fibrillation pathway of human serum albumin. Biophys. J. 2009, 96, 2353–2370. [Google Scholar] [CrossRef]
- Pires, R.H.; Saraiva, M.J.; Damas, A.M.; Kellermayer, M.S.Z. Structure and assembly-disassembly properties of wild-type transthyretin amyloid protofibrils observed with atomic force microscopy. J. Mol. Recognit. 2011, 24, 467–476. [Google Scholar] [CrossRef]
- Relini, A.; Torrassa, S.; Rolandi, R.; Gliozzi, A.; Rosano, C.; Canale, C.; Bolognesi, M.; Plakoutsi, G.; Bucciantini, M.; Chiti, F.; et al. Monitoring the process of HypF fibrillization and liposome permeabilization by protofibrils. J. Mol. Biol. 2004, 338, 943–957. [Google Scholar] [CrossRef]
- Ding, T.; Lee, S.; Rochet, J.; Lansbury, P. Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 2002, 41, 10209–10217. [Google Scholar] [CrossRef]
- Malisauskas, M.; Zamotin, V.; Jass, J.; Noppe, W.; Dobson, C.; Morozova-Roche, L. Amyloid protofilaments from the calcium-binding protein equine lysozyme: Formation of ring and linear structures depends on pH and metal ion concentration. J. Mol. Biol. 2003, 330, 879–890. [Google Scholar] [CrossRef]
- Zhu, M.; Han, S.; Zhou, F.; Carter, S.A.; Fink, A.L. Annular oligomeric amyloid intermediates observed by in situ atomic force microscopy. J. Biol. Chem. 2004, 279, 24452–24459. [Google Scholar] [CrossRef]
- Quist, A.; Doudevski, L.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar]
- Lashuel, H.A.; Lansbury, P.T., Jr. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 2006, 39, 167–201. [Google Scholar] [CrossRef]
- Lagerstedt, J.O.; Cavigiolio, G.; Roberts, L.M.; Hong, H.; Jin, L.; FitzGerald, P.G.; Oda, M.N.; Voss, J.C. Mapping the structural transition in an amyloidogenic apolipoprotein A-I. Biochemistry 2007, 46, 9693–9699. [Google Scholar] [CrossRef]
- Ghodke, S.; Nielsen, S.B.; Christiansen, G.; Hjuler, H.A.; Flink, J.; Otzen, D. Mapping out the multistage fibrillation of glucagon. FEBS J. 2012, 279, 752–765. [Google Scholar] [CrossRef]
- Kayed, R.; Pensalfini, A.; Margol, L.; Sokolov, Y.; Sarsoza, F.; Head, E.; Hall, J.; Glabe, C. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J. Biol. Chem. 2009, 284, 4230–4237. [Google Scholar]
- Lasagna-Reeves, C.A.; Kayed, R. Astrocytes contain amyloid-β annular protofibrils in Alzheimer's disease brains. FEBS Lett. 2011, 585, 3052–3057. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Glabe, C.G.; Kayed, R. Amyloid-ß annular protofibrils evade fibrillar fate in Alzheimer disease brain. J. Biol. Chem. 2011, 286, 22122–22130. [Google Scholar] [CrossRef]
- Bauer, H.H.; Aebi, U.; Häner, M.; Hermann, R.; Müller, M.; Merkle, H.P. Architecture and polymorphism of fibrillar supramolecular assemblies produced by in vitro aggregation of human calcitonin. J. Struct. Biol. 1995, 115, 1–15. [Google Scholar] [CrossRef]
- Jiménez, J.L.; Nettleton, E.J.; Bouchard, M.; Robinson, C.V.; Dobson, C.M.; Saibil, H.R. The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. USA 2002, 99, 9196–9201. [Google Scholar] [CrossRef]
- Morgado, I.; Fändrich, M. Assembly of Alzheimer’s Aβ peptide into nanostructured amyloid fibrils. Curr. Opin. Colloid Interface Sci. 2011, 16, 508–514. [Google Scholar] [CrossRef]
- Meinhardt, J.; Sachse, C.; Hortschansky, P.; Grigorieff, N.; Fändrich, M. Aβ(1–40) fibril polymorphism implies diverse interaction patterns in amyloid fibrils. J. Mol. Biol. 2009, 386, 869–877. [Google Scholar] [CrossRef]
- Knowles, T.; Smith, J.; Craig, A.; Dobson, C.; Welland, M. Spatial persistence of angular correlations in amyloid fibrils. Phys. Rev. Lett. 2006, 96, 238301. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomic structure and hierarchical assembly of a cross-ß amyloid fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef]
- Sweers, K.K.M.; Segers-Nolten, I.; Bennink, M.L.; Subramaniam, V. Structural model for α-synuclein fibrils derived from high resolution imaging and nanomechanical studies using atomic force microscopy. Soft Matter 2012, 8, 7215–7222. [Google Scholar] [CrossRef]
- Petkova, A.; Leapman, R.; Guo, Z.; Yau, W.; Mattson, M.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef]
- Teoh, C.L.; Bekard, I.B.; Asimakis, P.; Griffin, M.D.W.; Ryan, T.M.; Dunstan, D.E.; Howlett, G.J. Shear flow induced changes in apolipoprotein C-II conformation and amyloid fibril formation. Biochemistry 2011, 50, 4046–4057. [Google Scholar] [CrossRef]
- Adamcik, J.; Mezzenga, R. Adjustable twisting periodic pitch of amyloid fibrils. Soft Matter 2011, 7, 5437–5443. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Dikov, D.; Flink, J.L.; Hjuler, H.A.; Christiansen, G.; Otzen, D.E. The changing face of glucagon fibrillation: Structural polymorphism and conformational imprinting. J. Mol. Biol. 2006, 355, 501–523. [Google Scholar] [CrossRef]
- Chatani, E.; Yagi, H.; Naiki, H.; Goto, Y. Polymorphism of β2-microglobulin amyloid fibrils manifested by ultrasonication-enhanced fibril formation in trifluoroethanol. J. Biol. Chem. 2012, 287, 22827–22837. [Google Scholar] [CrossRef]
- Dzwolak, W.; Smirnovas, V.; Jansen, R.; Winter, R. Insulin forms amyloid in a strain-dependent manner: An FT-IR spectroscopic study. Protein Sci. 2004, 13, 1927–1932. [Google Scholar] [CrossRef]
- Surmacz-Chwedoruk, W.; Nieznańska, H.; Wójcik, S.; Dzwolak, W. Cross-seeding of fibrils from two types of insulin induces new amyloid strains. Biochemistry 2012, 51, 9460–9469. [Google Scholar] [CrossRef]
- Macchi, F.; Hoffmann, S.; Carlsen, M.; Vad, B.; Imparato, A.; Rischel, C.; Otzen, D.E. Mechanical stress affects glucagon fibrillation kinetics and fibril structure. Langmuir 2011, 27, 12539–12549. [Google Scholar] [CrossRef]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.O.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef]
- Yau, J.; Sharpe, S. Structures of amyloid fibrils formed by the prion protein derived peptides PrP(244–249) and PrP(245–250). J. Struct. Biol. 2012, 180, 290–302. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, M.; Jiang, L.; Cheng, P.; Park, J.; Sawaya, M.R.; Pensalfini, A.; Gou, D.; Berk, A.J.; Glabe, C.G.; et al. Out-of-register β-β suggest a pathway to toxic amyloid aggregates. Proc. Natl. Acad. Sci. USA 2012, 109, 20913–20918. [Google Scholar] [CrossRef]
- Laganowsky, A.; Liu, C.; Sawaya, M.R.; Whitelegge, J.P.; Park, J.; Zhao, M.; Pensalfini, A.; Soriaga, A.B.; Landau, M.; Teng, P.K.; et al. Atomic view of a toxic amyloid small oligomer. Science 2012, 335, 1228–1231. [Google Scholar] [CrossRef]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar] [CrossRef]
- Glabe, C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008, 283, 29639–29643. [Google Scholar] [CrossRef]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef]
- Fändrich, M. Oligomeric intermediates in amyloid formation: Structure determination and mechanisms of toxicity. J. Mol. Biol. 2012, 421, 427–440. [Google Scholar] [CrossRef]
- Pham, J.D.; Chim, N.; Goulding, C.W.; Nowick, J.S. Structures of oligomers of a peptide from β-amyloid. J. Am. Chem. Soc. 2013, 135, 12460–12467. [Google Scholar] [CrossRef]
- Liu, C.; Sawaya, M.R.; Cheng, P.; Zheng, J.; Nowick, J.S.; Eisenberg, D. Characteristics of amyloid-related oligomers revealed by crystal structures of macrocyclic β-sheet mimics. J. Am. Chem. Soc. 2011, 133, 6736–6744. [Google Scholar]
- Streltsov, V.A.; Varghese, J.N.; Masters, C.L.; Nuttall, S.D. Crystal structure of the amyloid-β p3 fragment provides a model for oligomer formation in Alzheimer’s disease. J. Neurosci. 2011, 31, 1419–1426. [Google Scholar] [CrossRef]
- Domanska, K.; Vanderhaegen, S.; Srinivasan, V.; Pardon, E.; Dupeux, F.; Marquez, J.A.; Giorgetti, S.; Stoppini, M.; Wyns, L.; Bellotti, V.; et al. Atomic structure of a nanobody-trapped domain-swapped dimer of an amyloidogenic β2-microglobulin variant. Proc. Natl. Acad. Sci. USA 2011, 108, 1314–1319. [Google Scholar] [CrossRef]
- Chimon, S.; Shaibat, M.A.; Jones, C.R.; Calero, D.C.; Aizezi, B.; Ishii, Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat. Struct. Mol. Biol. 2007, 14, 1157–1164. [Google Scholar] [CrossRef]
- Yu, L.; Edalji, R.; Harlan, J.E.; Holzman, T.F.; Lopez, A.P.; Labkovsky, B.; Hillen, H.; Barghorn, S.; Ebert, U.; Richardson, P.L.; et al. Structural characterization of a soluble amyloid β-peptide oligomer. Biochemistry 2009, 48, 1870–1877. [Google Scholar] [CrossRef]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef]
- Suzuki, Y.; Brender, J.R.; Soper, M.T.; Krishnamoorthy, J.; Zhou, Y.; Ruotolo, B.T.; Kotov, N.A.; Ramamoorthy, A.; Marsh, E.N. Resolution of oligomeric species during the aggregation of Aβ1–40 using 19F NMR. Biochemistry 2013, 52, 1903–1912. [Google Scholar] [CrossRef]
- Giehm, L.; Svergun, D.I.; Otzen, D.E.; Vestergaard, B. Low-resolution structure of a vesicle disrupting α-synuclein oligomer that accumulates during fibrillation. Proc. Natl. Acad. Sci. USA 2011, 108, 3246–3251. [Google Scholar] [CrossRef]
- Gu, L.; Liu, C.; Guo, Z. Structural insights into Aß42 oligomers using site-directed spin labeling. J. Biol. Chem. 2013, 288, 18673–18683. [Google Scholar] [CrossRef]
- Plakoutsi, G.; Bemporad, F.; Calamai, M.; Taddei, N.; Dobson, C.M.; Chiti, F. Evidence for a mechanism of amyloid formation involving molecular reorganisation within native-like precursor aggregates. J. Mol. Biol. 2005, 351, 910–922. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef]
- Stroud, J.C.; Liu, C.; Teng, P.K.; Eisenberg, D. Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc. Natl. Acad. Sci. USA 2012, 109, 7717–7722. [Google Scholar] [CrossRef]
- Stefani, M. Structural features and cytotoxicity of amyloid oligomers: Implications in Alzheimer’s disease and other diseases with amyloid deposits. Prog. Neurobiol. 2012, 99, 226–245. [Google Scholar] [CrossRef]
- Milanesi, L.; Sheynis, T.; Xue, W.F.; Orlova, E.V.; Hellewell, A.L.; Jelinek, R.; Hewitt, E.W.; Radford, S.E.; Saibil, H.R. Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proc. Natl. Acad. Sci. USA 2012, 109, 20455–20460. [Google Scholar] [CrossRef] [Green Version]
- Pieri, L.; Madiona, K.; Bousset, L.; Melki, R. Fibrillar alpha-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 2012, 102, 2894–2905. [Google Scholar] [CrossRef]
- Arispe, N.; Pollard, H.B.; Rojas, E. Giant multilevel cation channels formed by Alzheimer disease amyloid β-protein Aβ P-(1–40) in bilayer membranes. Proc. Natl. Acad. Sci. USA 1993, 90, 10573–10577. [Google Scholar] [CrossRef]
- Yang, L.; Harroun, T.; Weiss, T.; Ding, L.; Huang, H. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; de Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Demuro, A.; Smith, M.; Parker, I. Single-channel Ca2+ imaging implicates Aβ 1-42 amyloid pores in Alzheimer’s disease pathology. J. Cell Biol. 2011, 195, 515–524. [Google Scholar] [CrossRef]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Azimova, R.; Kagan, B.L.; Nussinov, R.; Lal, R. Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer’s disease and down syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 6538–6543. [Google Scholar]
- Prangkio, P.; Yusko, E.C.; Sept, D.; Yang, J.; Mayer, M. Multivariate analyses of amyloid-β oligomer populations indicate a connection between pore formation and cytotoxicity. PLoS One 2012, 7, e47261. [Google Scholar]
- Ludtke, S.; He, K.; Heller, W.; Harroun, T.; Yang, L.; Huang, H. Membrane pores induced by magainin. Biochemistry 1996, 35, 13723–13728. [Google Scholar] [CrossRef]
- Huang, H.; Chen, F.; Lee, M. Molecular mechanism of peptide-induced pores in membranes. Phys. Rev. Lett. 2004, 92, 198304. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef]
- Tamba, Y.; Ariyama, H.; Levadny, V.; Yamazaki, M. Kinetic pathway of antimicrobial peptide magainin 2-induced pore formation in lipid membranes. J. Phys. Chem. B 2010, 114, 12018–12026. [Google Scholar] [CrossRef]
- Gregory, S.M.; Pokorny, A.; Almeida, P.F.F. Magainin 2 revisited: A test of the quantitative model for the all-or-none permeabilization of phospholipid vesicles. Biophys. J. 2009, 96, 116–131. [Google Scholar] [CrossRef]
- Last, N.B.; Schlamadinger, D.E.; Miranker, A.D. A common landscape for membrane-active peptides. Protein Sci. 2013, 22, 870–882. [Google Scholar] [CrossRef]
- Dittmer, J.; Thogersen, L.; Underhaug, J.; Bertelsen, K.; Vosegaard, T.; Pedersen, J.M.; Schiott, B.; Tajkhorshid, E.; Skrydstrup, T.; Nielsen, N.C. Incorporation of antimicrobial peptides into membranes: A combined liquid-state NMR and molecular dynamics study of alamethicin in DMPC/DHPC bicelles. J. Phys. Chem. B 2009, 113, 6928–6937. [Google Scholar] [CrossRef]
- Last, N.B.; Miranker, A.D. Common mechanism unites membrane poration by amyloid and antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2013, 110, 6382–6387. [Google Scholar] [CrossRef]
- Gregory, S.M.; Cavenaugh, A.; Journigan, V.; Pokorny, A.; Almeida, P.F.F. A quantitative model for the all-or-none permeabilization of phospholipid vesicles by the antimicrobial peptide cecropin A. Biophys. J. 2008, 94, 1667–1680. [Google Scholar] [CrossRef]
- Lee, M.; Hung, W.; Chen, F.; Huang, H.W. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 5087–5092. [Google Scholar] [CrossRef]
- Last, N.B.; Rhoades, E.; Miranker, A.D. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proc. Natl. Acad. Sci. USA 2011, 108, 9460–9465. [Google Scholar] [CrossRef]
- Canale, C.; Torrassa, S.; Rispoli, P.; Relini, A.; Rolandi, R.; Bucciantini, M.; Stefani, M.; Gliozzi, A. Natively folded HypF-N and its early amyloid aggregates interact with phospholipid monolayers and destabilize supported phospholipid bilayers. Biophys. J. 2006, 91, 4575–4588. [Google Scholar] [CrossRef]
- Valincius, G.; Heinrich, F.; Budvytyte, R.; Vanderah, D.J.; McGillivray, D.J.; Sokolov, Y.; Hall, J.E.; Losche, M. Soluble amyloid β-oligomers affect dielectric membrane properties by bilayer insertion and domain formation: Implications for cell toxicity. Biophys. J. 2008, 95, 4845–4861. [Google Scholar]
- Sparr, E.; Engel, M.; Sakharov, D.; Sprong, M.; Jacobs, J.; de Kruijff, B.; Hoppener, J.; Killian, J. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004, 577, 117–120. [Google Scholar] [CrossRef]
- Bucciantini, M.; Nosi, D.; Forzan, M.; Russo, E.; Calamai, M.; Pieri, L.; Formigli, L.; Quercioli, F.; Soria, S.; Pavone, F.; et al. Toxic effects of amyloid fibrils on cell membranes: The importance of ganglioside GM1. FASEB J. 2012, 26, 818–831. [Google Scholar] [CrossRef]
- Brender, J.R.; Heyl, D.L.; Samisetti, S.; Kotler, S.A.; Osborne, J.M.; Pesaru, R.R.; Ramamoorthy, A. Membrane disordering is not sufficient for membrane permeabilization by islet amyloid polypeptide: Studies of IAPP(20–29) fragments. Phys. Chem. Chem. Phys. 2013, 15, 8908–8915. [Google Scholar]
- Engel, M.; Yigittop, H.; Elgersma, R.; Rijkers, D.; Liskamp, R.; de Kruijff, B.; Hoppener, J.; Killian, J. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J. Mol. Biol. 2006, 356, 783–789. [Google Scholar] [CrossRef]
- Brender, J.R.; Hartman, K.; Reid, K.R.; Kennedy, R.T.; Ramamoorthy, A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry 2008, 47, 12680–12688. [Google Scholar] [CrossRef]
- Brender, J.R.; Lee, E.L.; Cavitt, M.A.; Gafni, A.; Steel, D.G.; Ramamoorthy, A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J. Am. Chem. Soc. 2008, 130, 6424–6429. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef]
- Snyder, E.; Nong, Y.; Almeida, C.; Paul, S.; Moran, T.; Choi, E.; Nairn, A.; Salter, M.; Lombroso, P.; Gouras, G.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Kowalewski, J.M.; Renner, M.; Bousset, L.; Koulakoff, A.; Melki, R.; Giaume, C.; Triller, A. β-amyloid and ATP-induced diffusional trapping of astrocyte and neuronal metabotropic glutamate type-5 receptors. Glia 2013, 61, 1673–1686. [Google Scholar] [CrossRef]
- Pellistri, F.; Bucciantini, M.; Relini, A.; Nosi, D.; Gliozzi, A.; Robello, M.; Stefani, M. Nonspecific interaction of prefibrillar amyloid aggregates with glutamatergic receptors results in Ca2+ increase in primary neuronal cells. J. Biol. Chem. 2008, 283, 29950–29960. [Google Scholar] [CrossRef]
- Tatini, F.; Pugliese, A.M.; Traini, C.; Niccoli, S.; Maraula, G.; Dami, T.E.; Mannini, B.; Scartabelli, T.; Pedata, F.; Casamenti, F.; et al. Amyloid-β oligomer synaptotoxicity is mimicked by oligomers of the model protein HypF-N. Neurobiol. Aging 2013, 34, 2100–2109. [Google Scholar] [CrossRef]
- Deshpande, A.; Mina, E.; Glabe, C.; Busciglio, J. Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J. Neurosci. 2006, 26, 6011–6018. [Google Scholar] [CrossRef]
- Yates, E.A.; Owens, S.L.; Lynch, M.F.; Cucco, E.M.; Umbaugh, C.S.; Legleiter, J. Specific domains of Aβ facilitate aggregation on and association with lipid bilayers. J. Mol. Biol. 2013, 425, 1915–1933. [Google Scholar]
- Diociaiuti, M.; Gaudiano, M.C.; Malchiodi-Albedi, F. The slowly aggregating salmon calcitonin: A useful tool for the study of the amyloid oligomers structure and activity. Int. J. Mol. Sci. 2011, 12, 9277–9295. [Google Scholar] [CrossRef]
- Sousa, M.M.; Du Yan, S.; Fernandes, R.; Guimarães, A.; Stern, D.; Saraiva, M.J. Familial amyloid polyneuropathy: Receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J. Neurosci. 2001, 21, 7576–7586. [Google Scholar]
- Monteiro, F.A.; Sousa, M.M.; Cardoso, I.; do Amaral, J.B.; Guimarães, A.; Saraiva, M.J. Activation of ERK1/2 MAP kinases in familial amyloidotic polyneuropathy. J. Neurochem. 2006, 97, 151–161. [Google Scholar] [CrossRef]
- Du Yan, S.; Zhu, H.; Fu, J.; Yan, S.F.; Roher, A.; Tourtellotte, W.W.; Rajavashisth, T.; Chen, X.; Godman, G.C.; Stern, D.; et al. Amyloid-β peptide–receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: A proinflammatory pathway in Alzheimer dproduct. Proc. Natl. Acad. Sci. USA 1997, 94, 5296–5301. [Google Scholar] [CrossRef]
- Chaney, M.O.; Stine, W.B.; Kokjohn, T.A.; Kuo, Y.; Esh, C.; Rahman, A.; Luehrs, D.C.; Schmidt, A.M.; Stern, D.; Yan, S.D.; et al. RAGE and amyloid β interactions: Atomic force microscopy and molecular modeling. BBA - Mol. Basis Dis. 2005, 1741, 199–205. [Google Scholar] [CrossRef]
- Mapelli, L.; Canale, C.; Pesci, D.; Averaimo, S.; Guizzardi, F.; Fortunati, V.; Falasca, L.; Piacentini, M.; Gliozzi, A.; Relini, A.; et al. Toxic effects of expanded ataxin-1 involve mechanical instability of the nuclear membrane. BBA - Mol. Basis Dis. 2012, 1822, 906–917. [Google Scholar] [CrossRef] [Green Version]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Relini, A.; Marano, N.; Gliozzi, A. Misfolding of Amyloidogenic Proteins and Their Interactions with Membranes. Biomolecules 2014, 4, 20-55. https://doi.org/10.3390/biom4010020
Relini A, Marano N, Gliozzi A. Misfolding of Amyloidogenic Proteins and Their Interactions with Membranes. Biomolecules. 2014; 4(1):20-55. https://doi.org/10.3390/biom4010020
Chicago/Turabian StyleRelini, Annalisa, Nadia Marano, and Alessandra Gliozzi. 2014. "Misfolding of Amyloidogenic Proteins and Their Interactions with Membranes" Biomolecules 4, no. 1: 20-55. https://doi.org/10.3390/biom4010020