Control of Collagen Stability and Heterotrimer Specificity through Repulsive Electrostatic Interactions

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Design Strategy
| A: | Ac-(Pro-Hyp-Gly)7(Asp-Asp-Gly)3-NH2 | C-terminal |
| B: | Ac-(Pro-Hyp-Gly)3(Asp-Asp-Gly)3(Pro-Hyp-Gly)4-NH2 | Middle |
| C: | Ac-(Asp-Asp-Gly)3 (Pro-Hyp-Gly)7-NH2 | N-terminal |
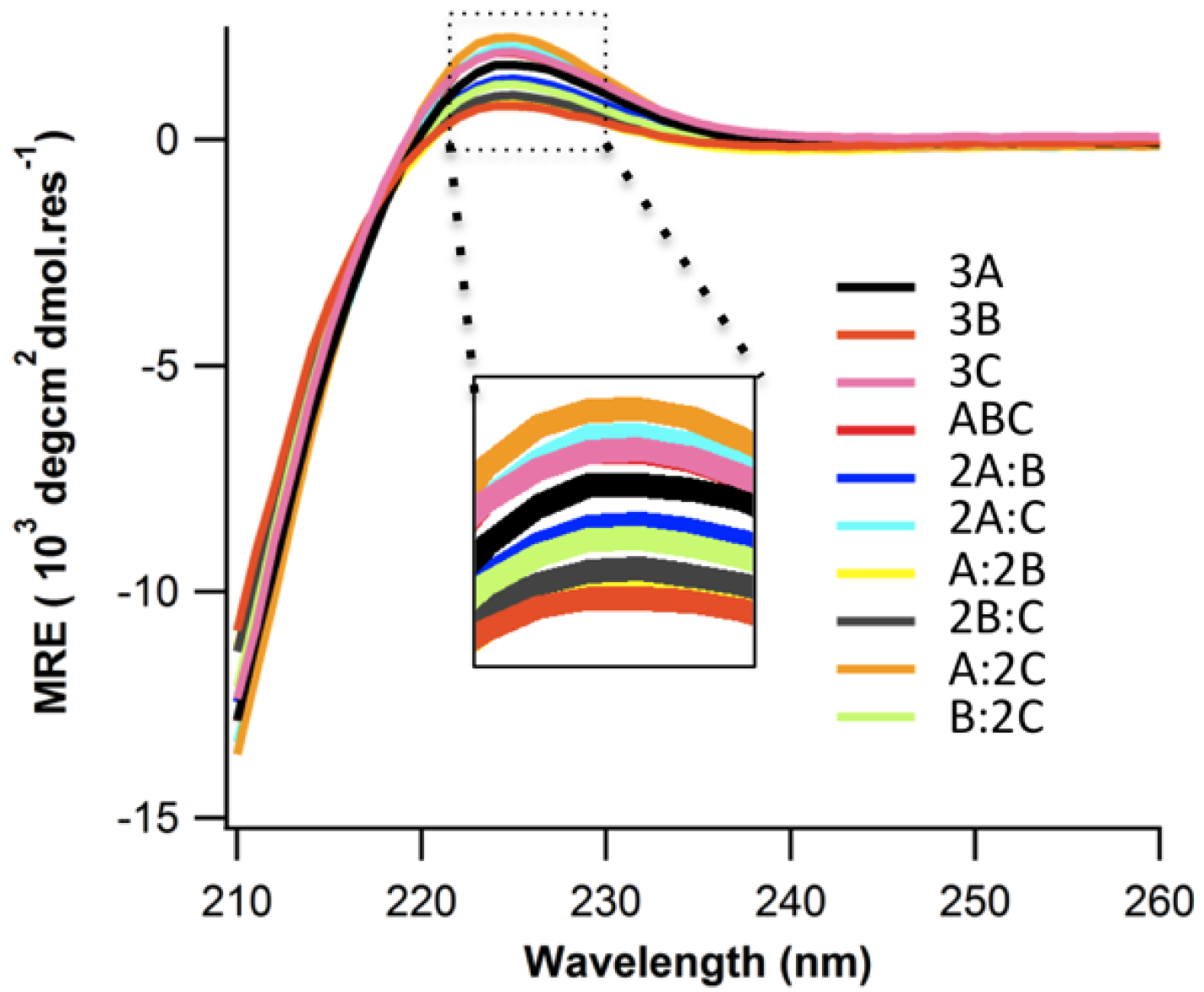
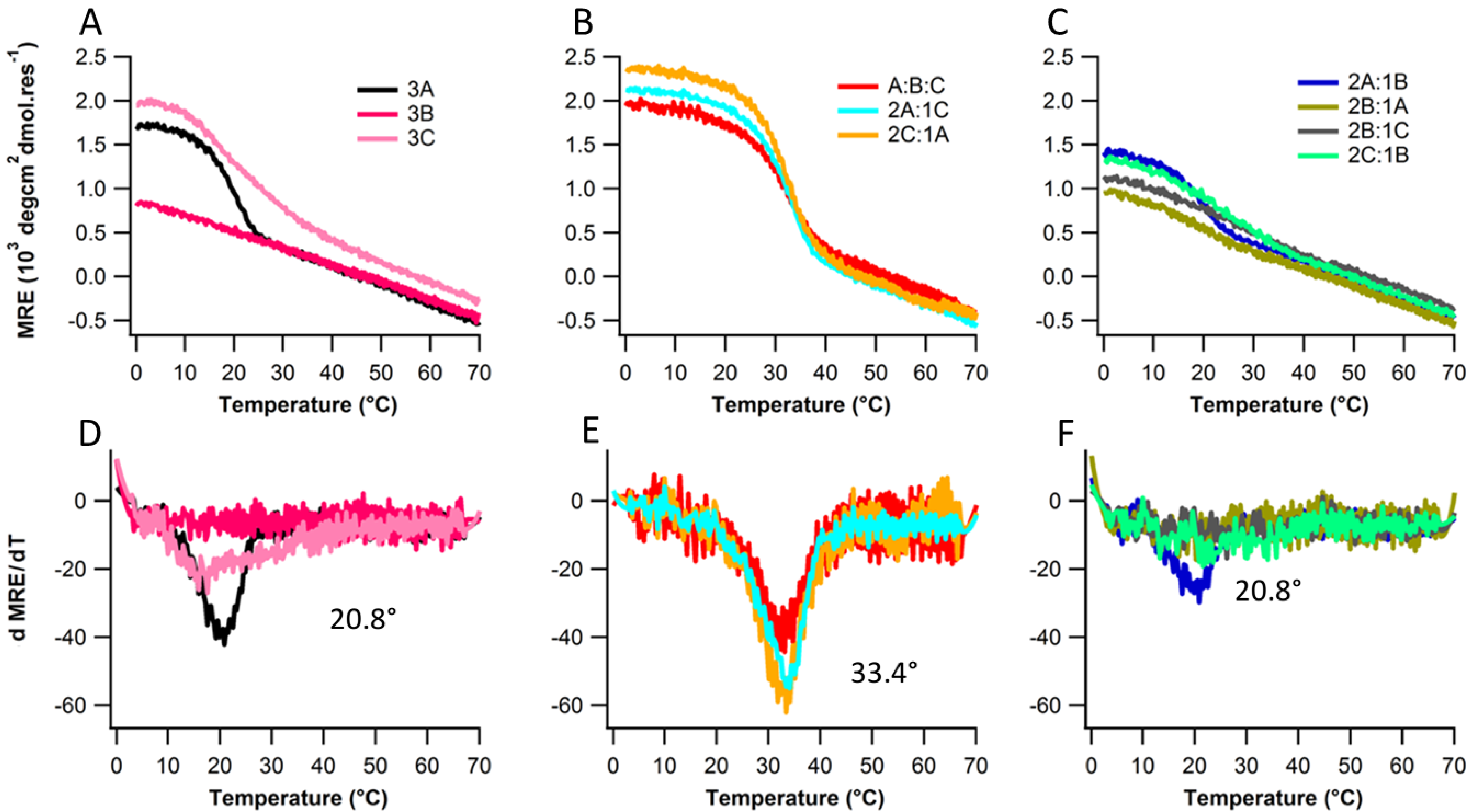
2.2. Structure and Stability
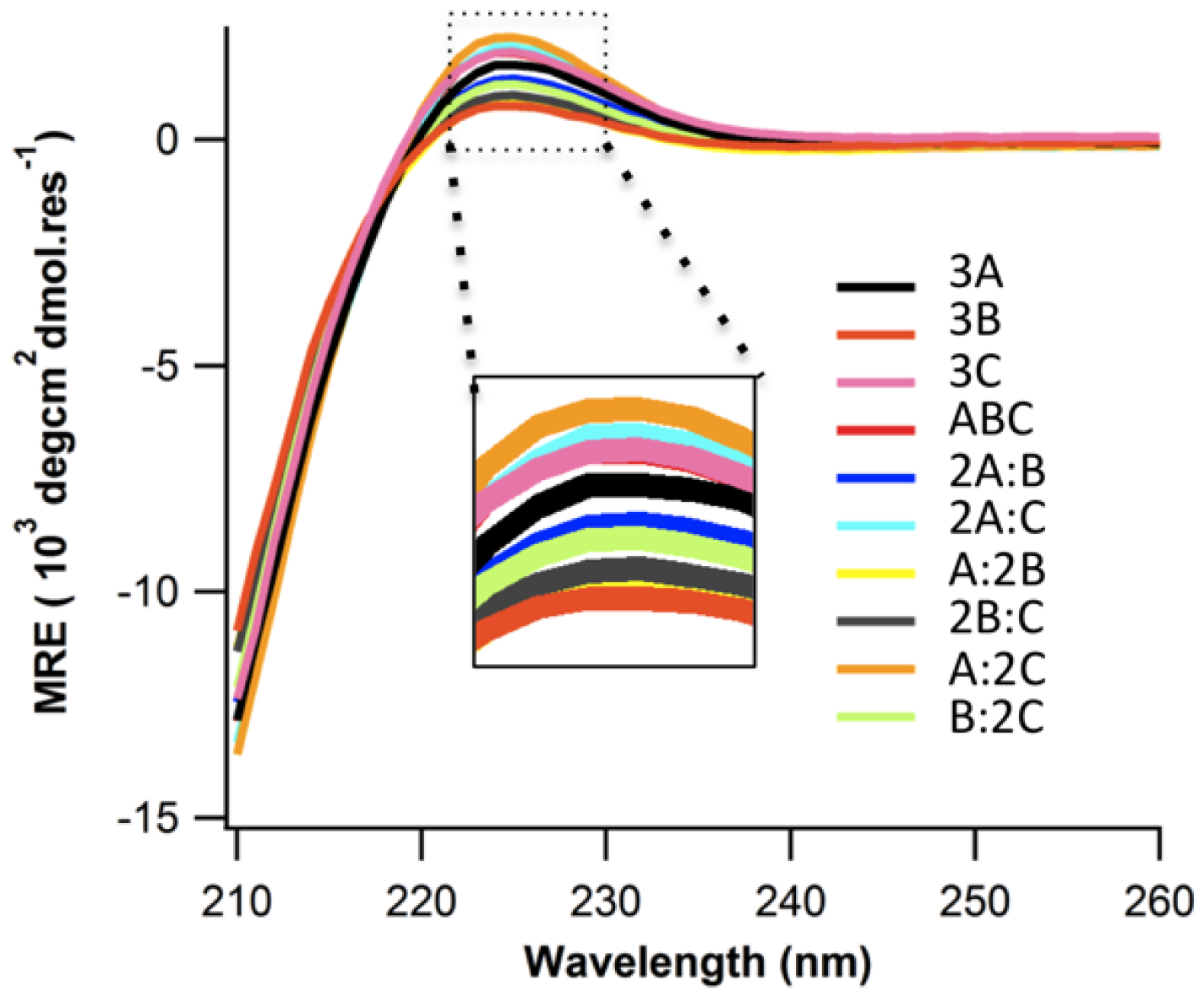
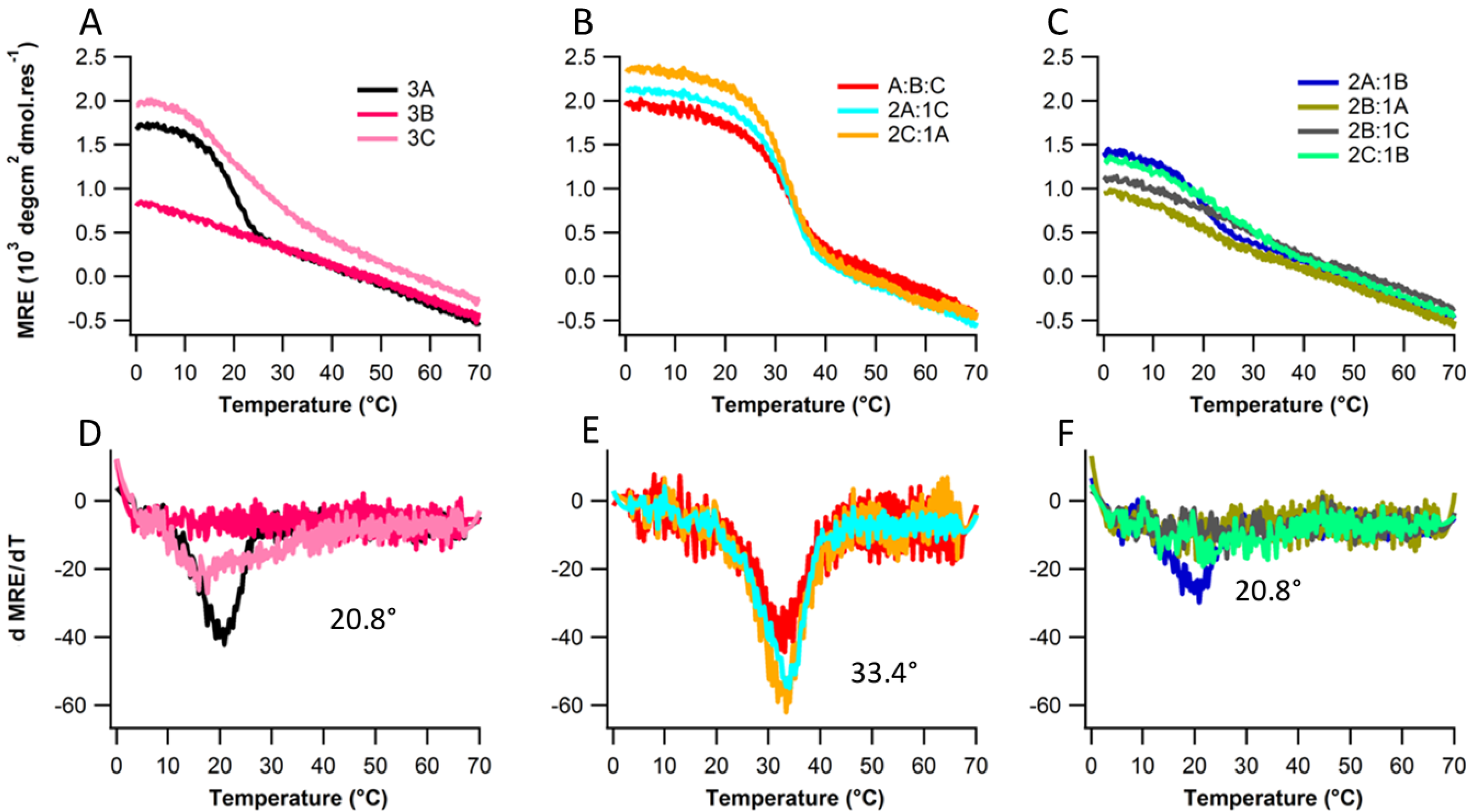
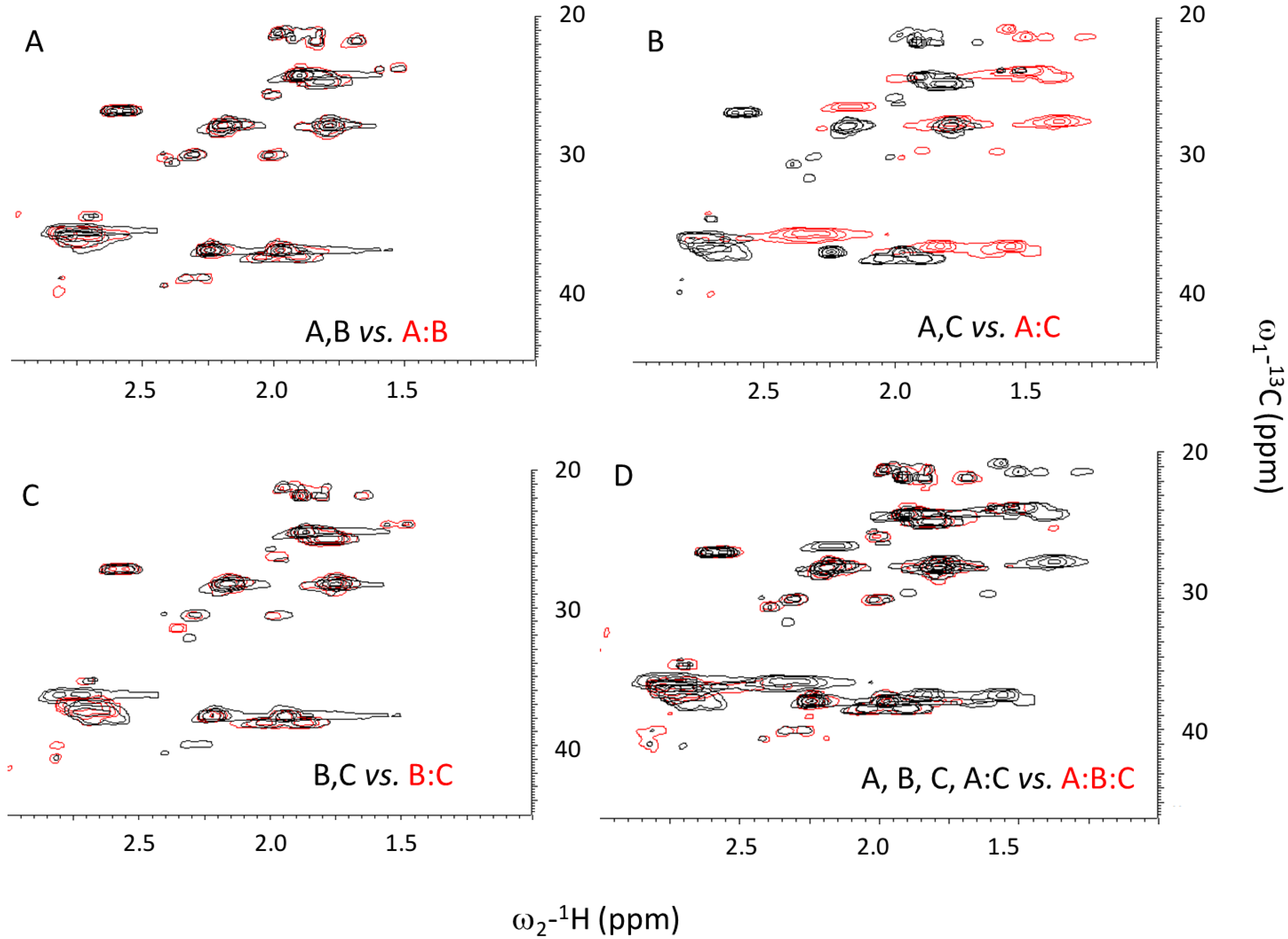
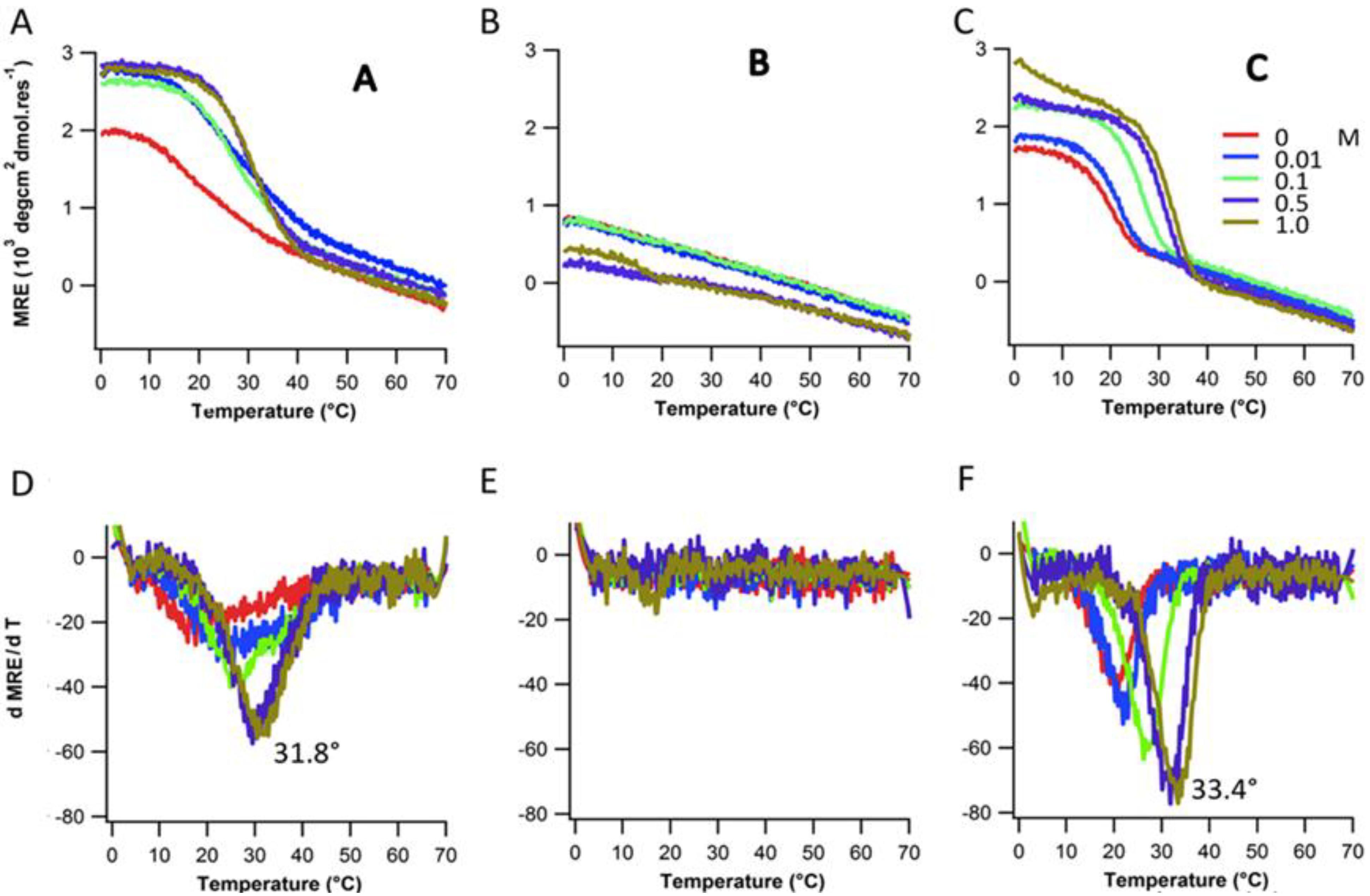
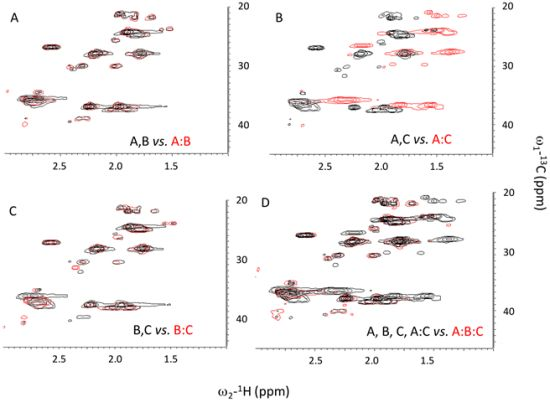
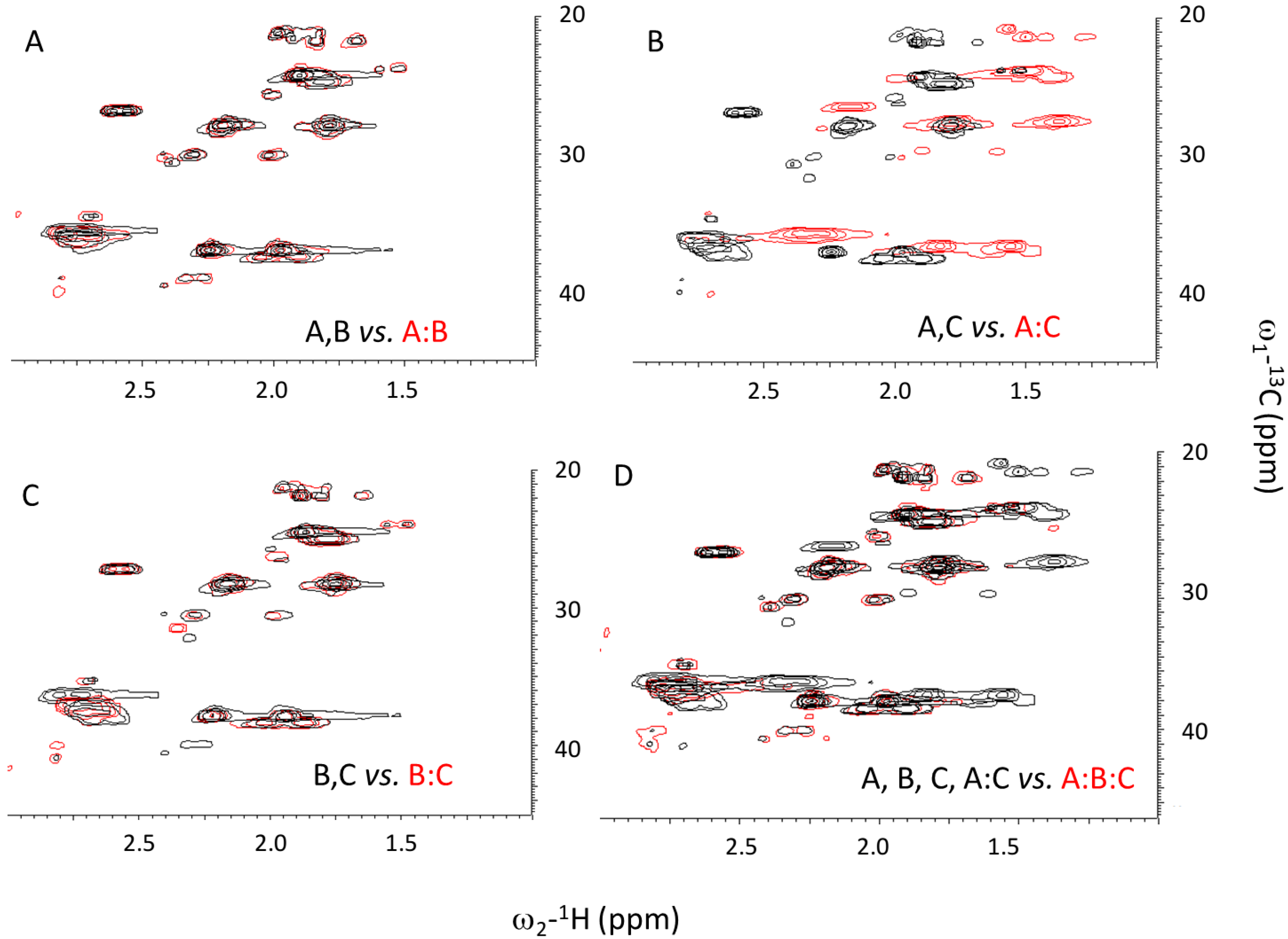
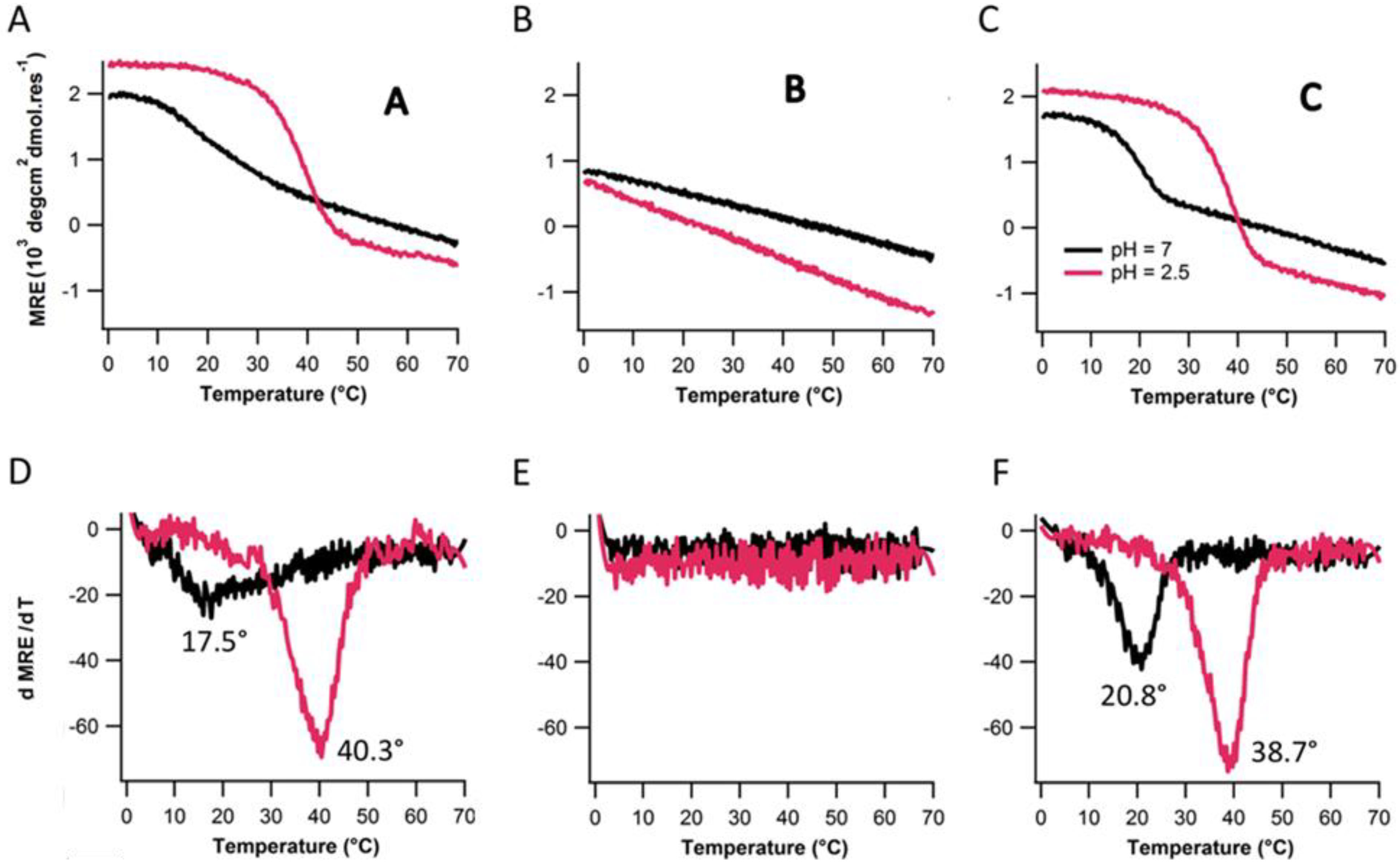
2.3. Ionic Strength Dependence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | Tm(°C) with varying NaCl Conc. | |||||
|---|---|---|---|---|---|---|
| 0 mM | 10 mM | 100 mM | 500 mM | 1,000 mM | ||
| Ac-(POG)7(DDG)3-NH2 | 20.8 | 21.8 | 26.2 | 31.8 | 33.4 | |
| Ac-(POG)3(DDG)3(POG)4-NH2 | - | - | - | - | 16.9 | |
| Ac-(DDG)3(POG)7-NH2 | 17.5 | 22.1 | 24.8 | 29.4 | 30.5 | |
2.4. Assembly at Low pH
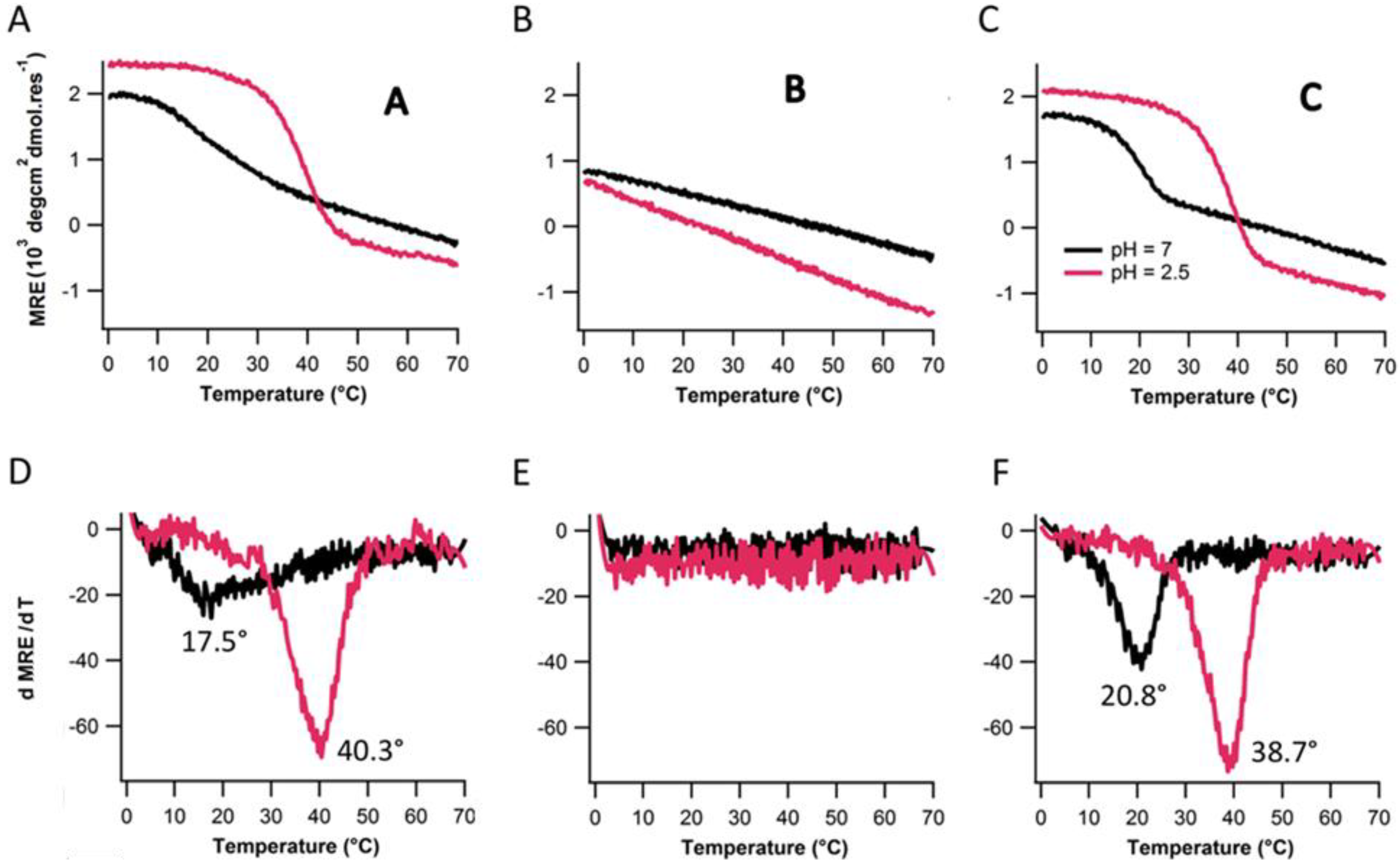
3. Experimental
3.1. Peptide Synthesis and Sequences
- A: Ac-(Pro-Hyp-Gly)7(Asp-Asp-Gly)3-NH2
- B: Ac-(Pro-Hyp-Gly)3(Asp-Asp-Gly)3(Pro-Hyp-Gly)4-NH2
- C: Ac-(Asp-Asp-Gly)3 (Pro-Hyp-Gly)7-NH2
3.2. Sample Preparation
3.3. Circular Dichroism (CD)
3.4. Nuclear Magnetic Resonance (NMR)
4. Conclusions

Acknowledgments
Conflicts of Interest
References
- Di Lullo, G.A.; Sweeney, S.M.; Korkko, J.; Ala-Kokko, L.; San Antonio, J.D. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J. Biol. Chem. 2002, 277, 4223–4231. [Google Scholar]
- Kadler, K.E.; Holmes, D.F.; Trotter, J.A.; Chapman, J.A. Collagen fibril formation. Biochem. J. 1996, 316, 1–11. [Google Scholar]
- Ramachandran, G.N.; Kartha, G. Structure of collagen. Nature 1955, 176, 593–595. [Google Scholar] [CrossRef]
- Rich, A.; Crick, F.H. The molecular structure of collagen. J. Mol. Biol. 1961, 3, 483–506. [Google Scholar] [CrossRef]
- Salem, G.; Traub, W. Conformational implications of amino acid sequence regularities in collagen. FEBS Lett. 1975, 51, 94–99. [Google Scholar] [CrossRef]
- Ramshaw, J.A.; Shah, N.K.; Brodsky, B. Gly-X-Y tripeptide frequencies in collagen: A context for host-guest triple-helical peptides. J. Struct. Biol. 1998, 122, 86–91. [Google Scholar]
- Mason, J.M.; Hagemann, U.B.; Arndt, K.M. Role of hydrophobic and electrostatic interactions in coiled coil stability and specificity. Biochemistry 2009, 48, 10380–10388. [Google Scholar]
- O’Shea, E.K.; Lumb, K.J.; Kim, P.S. Peptide “Velcro”: Design of a heterodimeric coiled coil. Curr. Biol. 1993, 3, 658–667. [Google Scholar] [CrossRef]
- Nautiyal, S.; Woolfson, D.N.; King, D.S.; Alber, T. A designed heterotrimeric coiled coil. Biochemistry 1995, 34, 11645–11651. [Google Scholar]
- Kohn, W.D.; Monera, O.D.; Kay, C.M.; Hodges, R.S. The effects of interhelical electrostatic repulsions between glutamic acid residues in controlling the dimerization and stability of two-stranded alpha-helical coiled-coils. J. Biol. Chem. 1995, 270, 25495–25506. [Google Scholar]
- Kohn, W.D.; Kay, C.M.; Hodges, R.S. Protein destabilization by electrostatic repulsions in the two-stranded alpha-helical coiled-coil/leucine zipper. Protein Sci. 1995, 4, 237–250. [Google Scholar]
- Kohn, W.D.; Kay, C.M.; Hodges, R.S. Positional dependence of the effects of negatively charged Glu side chains on the stability of two-stranded alpha-helical coiled-coils. J. Pept. Sci. 1997, 3, 209–223. [Google Scholar] [CrossRef]
- Fallas, J.A.; Hartgerink, J.D. Computational design of self-assembling register-specific collagen heterotrimers. Nat. Commun. 2012, 3, e1087. [Google Scholar] [CrossRef]
- Fallas, J.A.; Lee, M.A.; Jalan, A.A.; Hartgerink, J.D. Rational design of single-composition ABC collagen heterotrimers. J. Am. Chem. Soc. 2012, 134, 1430–1433. [Google Scholar] [CrossRef]
- Fallas, J.A.; Dong, J.; Tao, Y.J.; Hartgerink, J.D. Structural insights into charge pair interactions in triple helical collagen-like proteins. J. Biol. Chem. 2012, 287, 8039–8047. [Google Scholar]
- Gauba, V.; Hartgerink, J.D. Surprisingly high stability of collagen ABC heterotrimer: Evaluation of side chain charge pairs. J. Am. Chem. Soc. 2007, 129, 15034–15041. [Google Scholar] [CrossRef]
- Gauba, V.; Hartgerink, J.D. Self-assembled heterotrimeric collagen triple helices directed through electrostatic interactions. J. Am. Chem. Soc. 2007, 129, 2683–2690. [Google Scholar] [CrossRef]
- Xu, F.; Zahid, S.; Silva, T.; Nanda, V. Computational design of a collagen A:B:C-type heterotrimer. J. Am. Chem. Soc. 2011, 133, 15260–15263. [Google Scholar] [CrossRef]
- Parmar, A.S.; Zahid, S.; Belure, S.V.; Young, R.; Hasan, N.; Nanda, V. Design of net-charged abc-type collagen heterotrimers. J. Struct. Biol. 2013. [Google Scholar] [CrossRef]
- Giddu, S.; Xu, F.; Nanda, V. Sequence recombination improves target specificity in a redesigned collagen peptide abc-type heterotrimer. Proteins 2013, 81, 386–393. [Google Scholar] [CrossRef]
- Xu, F.; Zhang, L.; Koder, R.L.; Nanda, V. De novo self-assembling collagen heterotrimers using explicit positive and negative design. Biochemistry 2010, 49, 2307–2316. [Google Scholar] [CrossRef]
- Parmar, A.S.; Nunes, A.M.; Baum, J.; Brodsky, B. A peptide study of the relationship between the collagen triple-helix and amyloid. Biopolymers 2012, 97, 795–806. [Google Scholar] [CrossRef]
- Persikov, A.V.; Ramshaw, J.A.; Brodsky, B. Prediction of collagen stability from amino acid sequence. J. Biol. Chem. 2005, 280, 19343–19349. [Google Scholar] [CrossRef]
- Bretscher, L.E.; Jenkins, C.L.; Taylor, K.M.; DeRider, M.L.; Raines, R.T. Conformational stability of collagen relies on a stereoelectronic effect. J. Am. Chem. Soc. 2001, 123, 777–778. [Google Scholar]
- Venugopal, M.G.; Ramshaw, J.A.; Braswell, E.; Zhu, D.; Brodsky, B. Electrostatic interactions in collagen-like triple-helical peptides. Biochemistry 1994, 33, 7948–7956. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and differentiation of data by simplified least squares procedures. Anal. Chem. 1964, 36, e13. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Parmar, A.S.; Joshi, M.; Nosker, P.L.; Hasan, N.F.; Nanda, V. Control of Collagen Stability and Heterotrimer Specificity through Repulsive Electrostatic Interactions. Biomolecules 2013, 3, 986-996. https://doi.org/10.3390/biom3040986
Parmar AS, Joshi M, Nosker PL, Hasan NF, Nanda V. Control of Collagen Stability and Heterotrimer Specificity through Repulsive Electrostatic Interactions. Biomolecules. 2013; 3(4):986-996. https://doi.org/10.3390/biom3040986
Chicago/Turabian StyleParmar, Avanish S., Mihir Joshi, Patrick L. Nosker, Nida F. Hasan, and Vikas Nanda. 2013. "Control of Collagen Stability and Heterotrimer Specificity through Repulsive Electrostatic Interactions" Biomolecules 3, no. 4: 986-996. https://doi.org/10.3390/biom3040986