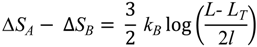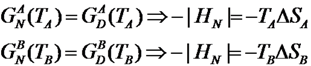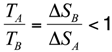1. Introduction
To survive in a permanently cold environment, psychrophilic microorganisms synthesize proteins that are resistant to cold-induced denaturation and misfolding [
1]. On the other hand, the native states of these proteins are often only marginally stable, or even unstable in temperate environments. This case clearly illustrates how, in order to understand at the molecular level the principles that regulate the adaptation of microorganisms to different thermal environments, it is crucial to identify the general physical principles that shape the structure of the proteins’ free-energy landscapes. From this perspective, it is particularly useful to compare the folding thermodynamics of homologous proteins produced by species which are evolutionarily closely related, yet ecologically separated, such as are species living, one, in polar waters and, the other one, in temperate waters. In this way, it appears to be easier, at least in principle, to identify specific structural features that are more directly responsible for the thermodynamic stability of the protein native state. In this work, we performed a theoretical analysis of protein unfolding/refolding thermodynamics, that complements a recent analysis of circular dichroism (CD) spectra [
2] of two closely homologous families of water-borne signaling proteins (known as pheromones), which regulate the vegetative (mitotic) growth and sexual mating [
3] in two ecologically separated
Euplotes species,
E. raikovi (mesophilic) living in temperate waters and
E. nobilii (psychrophilic) living in polar (Antarctic and Arctic) waters [
4]. A representative set of members of these two families of pheromones and their corresponding Protein Data Bank (PDB) codes are listed in
Table 1, while the three-dimensional structures of one
E. raikovi pheromone and one
E. nobilii pheromone are shown in
Figure 1.
Table 1.
List of the En and Er pheromones investigated and corresponding PDB codes.
Table 1.
List of the En and Er pheromones investigated and corresponding PDB codes.
| | Euplotes raikovi | Euplotes nobilii |
|---|
| Name | Er-1 | Er-2 | Er-10 | En-1 | En-2 | En-6 |
| PDB code | 1erc | 1erd | 1erp | 2nsv | 2nsw | 2jms |
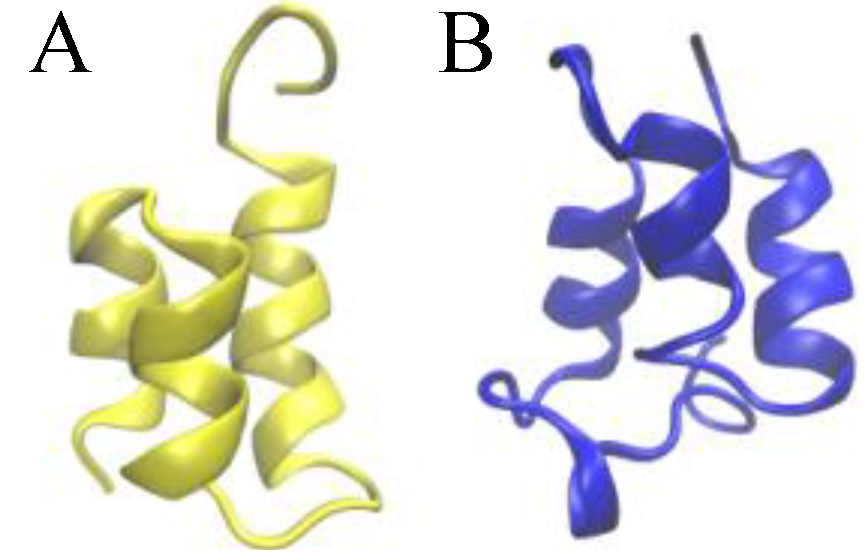
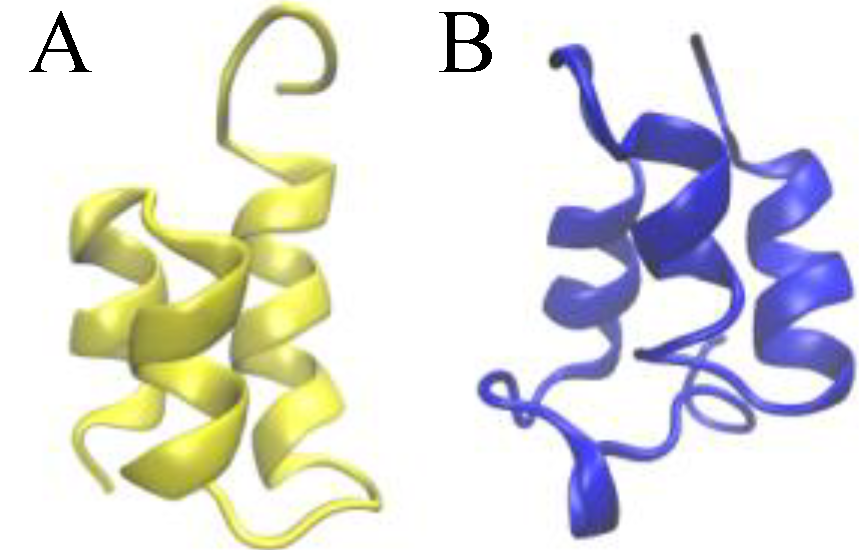
Figure 1.
Ribbon presentations of the three-dimensional native structure of the Er-1 (A) and En-1 (B) pheromones.
Figure 1.
Ribbon presentations of the three-dimensional native structure of the Er-1 (A) and En-1 (B) pheromones.
The native structure of
E. nobilii and
E. raikovi pheromones is equally characterized by densely spaced disulfide bonds (three in
E. raikovi, and four in
E. nobilii), which connect residues located in different alpha-helices (see
Figure 2). In a non-reducing environment at physiological temperatures, disulfide bonds provide unbreakable topological constraints [
5], which strongly restrict the conformational space accessible to the polypeptide chain. The impossibility of breaking a disulfide bond at room temperature and in non-reducing conditions motivated a vast research activity directed to clarify how cysteine-rich proteins realize and possibly re-arrange the network of topological constraints leading to the correct native topology, during the folding reaction [
6]. In particular, the thermodynamics for an helix-coil transition inside protein loops induced by pairs of disulfide bonds was extensively investigated, starting from the pioneering work of Scheraga and Poland, in the mid 1960s. It was realized that the presence of such covalent bonds modifies the melting curve, leading to a higher melting temperature [
7,
8], as expected from general polymer physics considerations [
9]. In particular, it was observed that disulfide bonds between Cys residues that are sequentially distant have a larger impact on thermodynamics than those that are sequentially close.
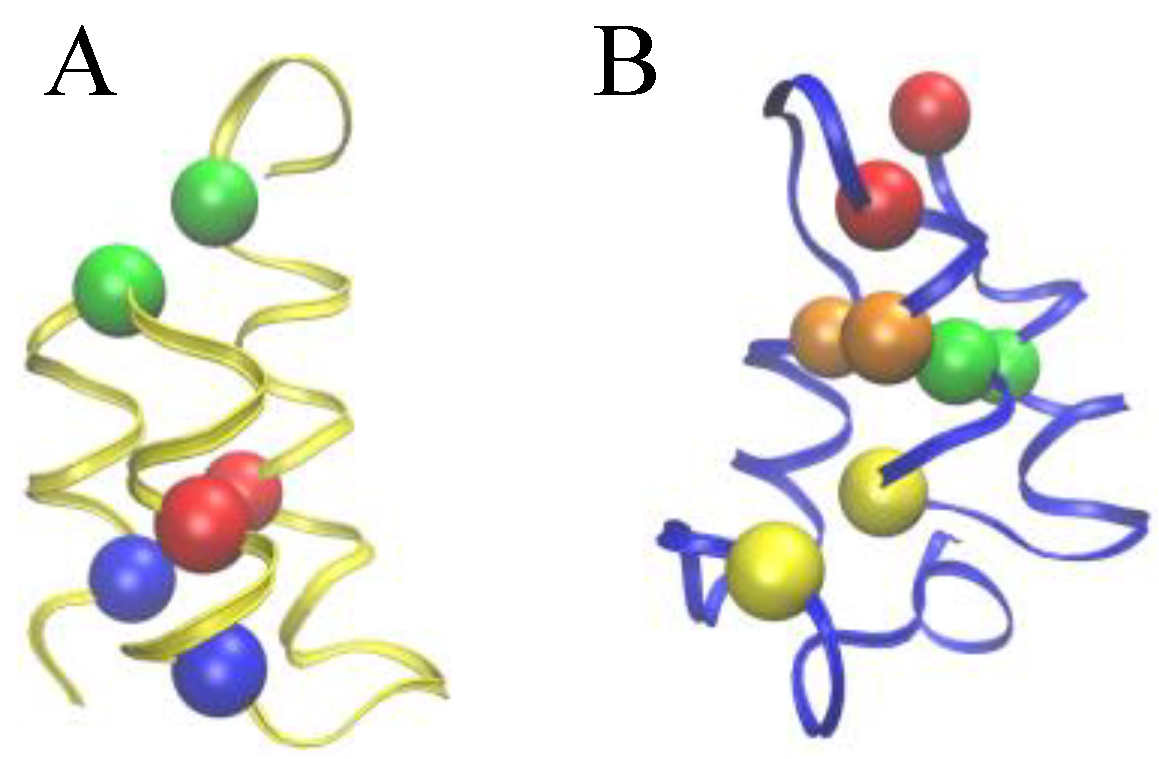
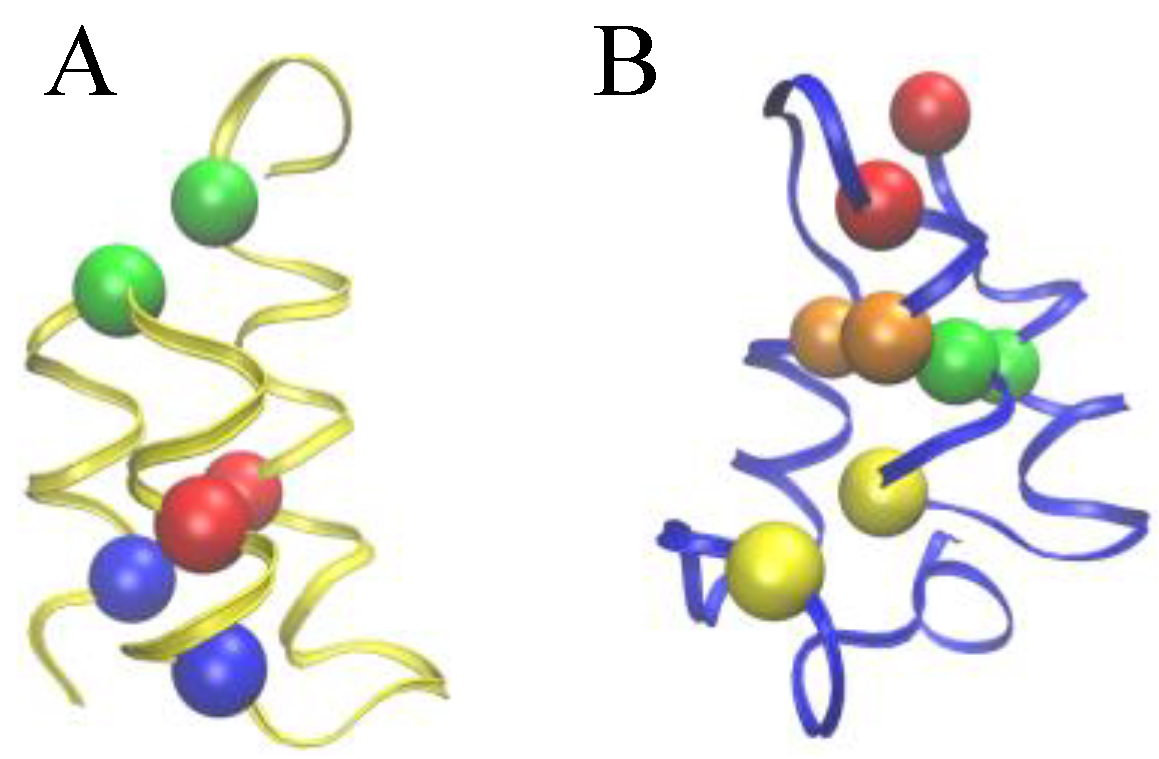
Figure 2.
Comparison of the Cys-Cys bond patterns in the native Er-1 (A) and En-1 (B) pheromone structures. Spheres of identical color indicate cysteine residues paired together into a disulfide bond.
Figure 2.
Comparison of the Cys-Cys bond patterns in the native Er-1 (A) and En-1 (B) pheromone structures. Spheres of identical color indicate cysteine residues paired together into a disulfide bond.
The folding kinetics of cysteine-rich proteins was investigated in detail by Camacho and Thirumalai in the mid 1990s using both coarse-grained models [
8,
10], and phenomenological statistical models [
11]. One striking conclusion of those studies was that, despite the fact that the disulfide bonds greatly restrict the available configuration space, it can lead to slower folding kinetics. In addition, a phenomenological scheme (the so-called proximity rule) was proposed to describe the dynamics of formation and rearrangement of disulfide bonds, during the folding reaction. From the experimental side, techniques have been developed to trap disulfide-bonded intermediates along the folding processes [
12,
13,
14,
15,
16]. Another approach to experimentally assess the role of disulfide bonds is to unfold a protein while keeping them intact [
17]. Disulfide cross-linking techniques have also been used in combination with mutagenic experiments in order to gain information on the folding pathways in a larger class of proteins, by substituting specific residues with cysteines [
18].
In this paper, we investigate the implication of these studies on the thermodynamics of cysteine-rich proteins on the molecular adaptation of the
E. nobili and
E. raikovi pheromones. In a recent paper [
2], it has been reported that, despite their high degree of structural similarity (homology), the
E. nobilii pheromones (designated E
n-1, E
n-2 and E
n-6) and
E. raikovi pheromones (designated E
r-1, E
r-2, E
r-10, E
r-11, E
r-22 and E
r-23) are characterized by very different unfolding/refolding thermodynamics. By comparing the temperature-dependent CD spectra of these pheromones it was found that while the
E. nobilii psychrophilic pheromones undergo an unfolding transition in temperature ranges from 55 °C to 70 °C, the mesophilic
E. raikovi pheromones essentially remain in their native conformation up to 100 °C.
Although this difference is not surprising from an evolutionary perspective, because these pheromones are produced by organisms adapted to very different thermal environments, it is remarkable from a biophysical perspective. Indeed, a major implication of the energy landscape theory of protein folding [
19] is that the folding kinetics and thermodynamics is shaped mostly by the interactions which are present in the native state and, hence, by the native tertiary structure and topology. The smoothness and funnel-like approximation of the folding landscape, which is the basis of the energy landscape theory, is supported by the successfulness of the topology-based,
i.e., native-centric, Gō-type or structure-based models, the terms that are used interchangeably in literature [
20]. On the other hand, the primary amino acid sequence and, in general, non-native interactions are expected to play a subsidiary role (an exception, however, may be represented by proteins with knotted native topology [
21,
22]). In particular, a minimally-frustrated and native-centric picture of protein folding predicts that homologous proteins have closely comparable unfolding temperatures.
The observed thermodynamics of the
E. nobilii and
E. raikovi pheromones challenges the native-centric view, as it appears to be dominated by effects associated to the protein primary structure and non-native interactions. In line with this new concept, it has been observed that the physicochemical properties of the polypeptide chain of cold-adapted
E. nobilii pheromones are characterized by a reduced hydrophobicity and improved backbone flexibility [
23]. Similarly, the stability of mesophilic polypeptides has been argued to be due to the presence of hydrophobic clusters [
24].
In this work, we challenge this explanation by showing that the body of experimental data on the thermal unfolding of the E. nobilii and E. raikovi pheromones can be understood in terms of physical effects entirely associated to the protein native structure and topology, i.e., without relying on solvent-induced effects and/or non-native interactions. In particular, we show that the unfolding temperatures of these cysteine-rich proteins are strongly influenced by the precise topology of the disulfide bonds in the protein three-dimensional native structure.
These conclusions are reached by analyzing the results of an extensive Monte Carlo (MC) simulations of the conformational space sampled by the E. nobilii and E. raikovi pheromones, performed at different temperatures, within a purely native-centric Gō-type model implemented on the coarse-grained (CG) level. In this approach, attractive non-bonded interactions are assigned only to the pairs of amino-acids that are in contact in the native state. Further, because sulfur bridges impose topological constraints that cannot be broken in the wide temperature range of interest (in the absence of chemical denaturants), we have modeled them appropriately in order to assure they remain unbreakable at room temperature. Indeed, the strength of disulfide bond is about 60 kcal/mol, i.e., of the order of 10 RT, at the temperature of T = 300 K. We also emphasize that this model is completely blind to any physical effect associated to the primary structure of the considered polypeptide chains.
Despite its simplicity, this model reproduces well the observed differences in the unfolding temperatures between the members of the two pheromone families. In particular, in our simulations, the E. raikovi pheromones remain native over the entire temperature range that has been considered, while the E. nobilii pheromones unfold at a (nominal) temperature which ranges from 40 °C to 60 °C. These results strongly suggest that the thermodynamics of the two pheromone families is driven by the structural properties of the protein native states, rather than by physical effects associated to the specificities of the amino-acid sequences. Using an analytically solvable statistical model, we argue that the much stronger relative stability of E. raikovi pheromons with respect to E. nobilii pheromones is due to the higher non-locality of the Cys-Cys bonds.
These conclusions are also corroborated by the behavior of the fractional helicity (mole fraction of helical backbone within the peptide or protein) of the E. nobilii En-1 and E. raikovi Er-1 pheromones at different temperatures. We find that our theoretical predictions agree well with the corresponding experimental results for the main secondary structural motifs, which were estimated by deconvoluting the whole experimental CD spectra using the DICHROWEB web interface.
2. Results and Discussion
To quantify the amount of unfolding of secondary structures in the two pheromone families, we used the raw CD data reported in Geralt
et al. [
2] to evaluate the temperature dependence of the fractional helicity
fH for two representative polypeptide chains,
i.e., E
n-1 and E
r-1. The temperature dependent CD measurements of E
n-1 and E
r-1 pheromones (expressed as the difference in the molar extinction coefficients; for more details see Experimental Section) are shown in
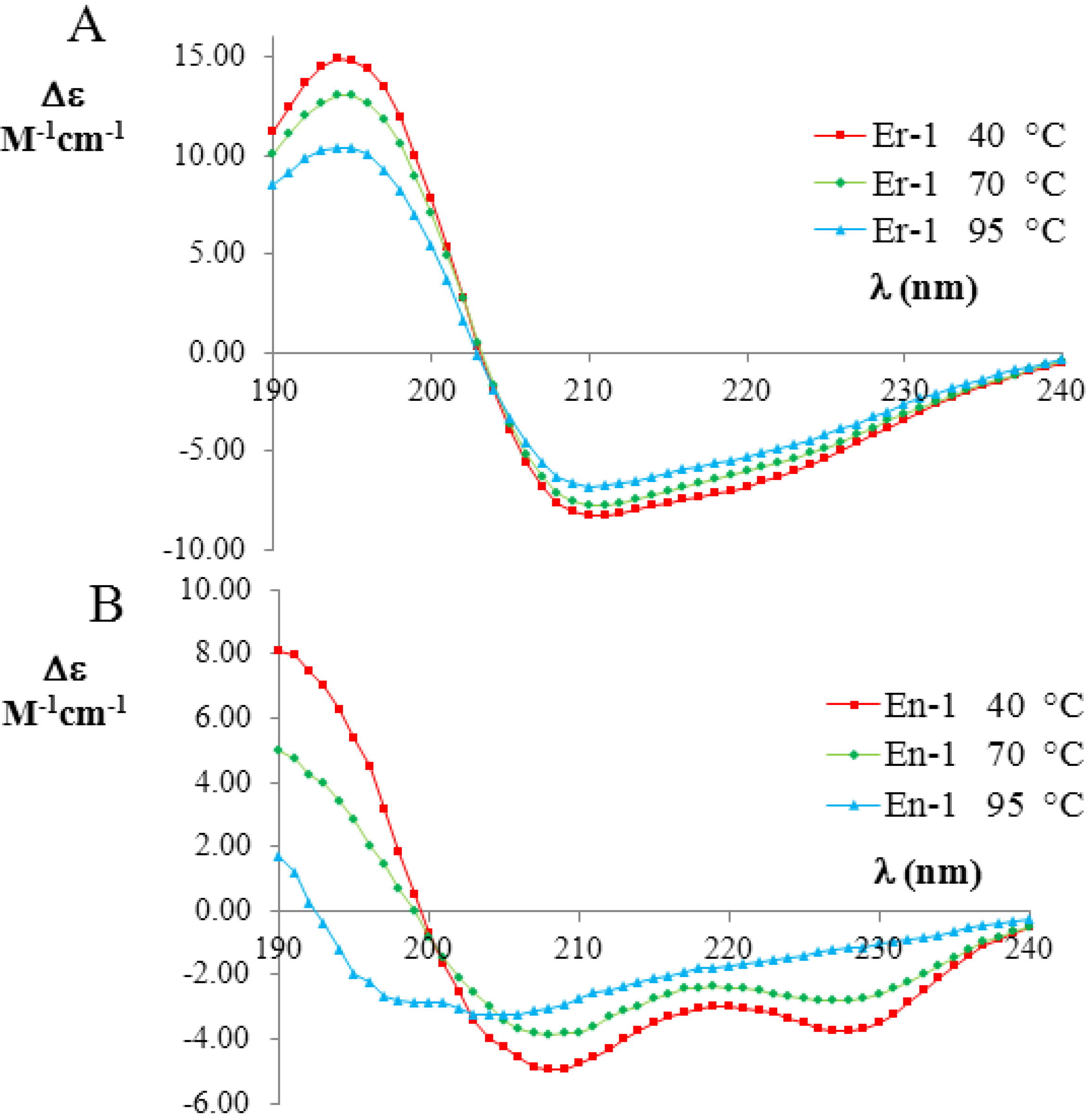
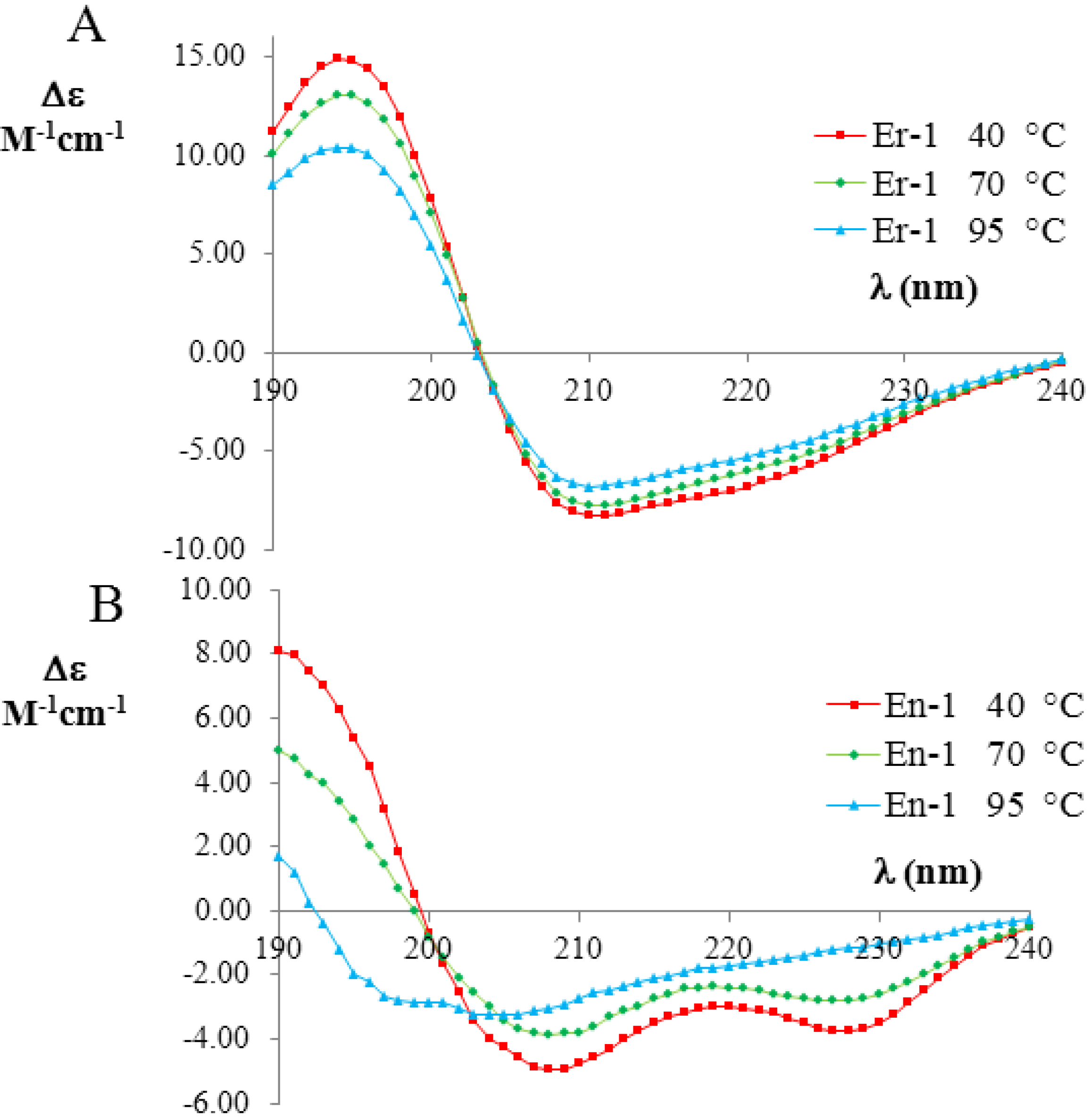
Figure 3A,B, respectively. The far-UV CD spectra of E
r-1 showed a strong negative Cotton effect at 208 nm and 222 nm, thus indicating that the
α-helix conformations provide a relevant contribution to the overall secondary structure of this protein. The deconvolution of the E
r-1 CD spectra obtained at 40 °C and 70 °C from the DICHROWEB application, indicated a high and similar helix content
fH = (70 ± 8)%, in a good agreement with E
r-1 NMR [
25] and X-ray analysis [
26,
27,
28] in which the contribution to helical structures was estimated to be 62% and 67%, respectively. Moreover, from the deconvolution of the E
r-1 CD spectrum at 95 °C, the fraction of residues involved in
α-helical structures was found to remain high,
i.e., (53 ± 3)%. Overall, within the wide temperature range 20–100 °C, the
fH parameter decreases less than 25%.
Figure 3.
Far-UV circular dichroism (CD) spectra (expressed in
Δε units) of the mesophilic, E
r-1 (
A), and psychrophilic, E
n-1 (
B), pheromones (20 mM, pH 6) at three selected temperatures (red 40 °C; green 70 °C; and blue 90 °C). The raw CD data were kindly supplied by M. Geralt and reported in [
2].
Figure 3.
Far-UV circular dichroism (CD) spectra (expressed in
Δε units) of the mesophilic, E
r-1 (
A), and psychrophilic, E
n-1 (
B), pheromones (20 mM, pH 6) at three selected temperatures (red 40 °C; green 70 °C; and blue 90 °C). The raw CD data were kindly supplied by M. Geralt and reported in [
2].
In contrast, the deconvolution of the corresponding E
n-1 CD spectra in the same temperature range lead to a strikingly different result. At 40 °C, the helical contribution was measured to be in rough agreement
fH = (57 ± 8)% with the value coming from NMR analysis, where it was estimated to be 46% [
23,
29]. However, the
fH values measured from the deconvolution of the CD spectra at 70 °C and 95 °C were markedly lower ((41 ± 14)% and (16 ± 7)%, respectively) than at 40 °C, implying that there is a significant loss (by a factor greater than three) in
α-helix structures.
Let us now compare this body of experimental results with the results of the MC simulations performed in the native-centric CG model described in the Experimental Section. To this end, it is instructive to first consider the temperature dependence of the average value of the fluctuations in the fraction of native contacts,
i.e.,
where < >
T denotes the thermal average at the temperature
T,
N is the total number of native contacts,
Q is the fraction of native contacts that are present in a given conformation,
ε0 = 2 kcal/mol is a typical contact energy and
kB is the Boltzmann constant. To this regard it is worth recalling that, in the present simplified model, non-bonded attractive interactions are assigned exclusively to pairs of amino-acids that are in native contact. Hence, the definition in Equation (1) basically coincides with a rescaled specific heat, since it includes only the contributions from the statistical fluctuations of the native interactions, which are directly correlated with the unfolding transition, and hence drive the characteristic peak in the specific heat
versus temperature curve. On the other side, the statistical fluctuations coming from repulsive hard-core interactions are quite insensitive to the unfolding transition. Indeed, due to topological constraints imposed by sulfur bridges, the chain remains in a compact state, even at high temperature. Consequently, the hard-core interactions introduce a flat background in the specific heat curve, which makes it difficult to identify the unfolding transition temperature. In order to remove this background and emphasize the differences in the thermodynamics of the two pheromone families, we have chosen to analyze the rescaled specific heat given by Equation (1). Therefore, the unfolding transition temperature can be identified by a peak in the
χ(T) curve. We also note that since the disulfide bridges are unbreakable in the temperature range of interest, the observed peaks are not as sharp as in the case of small globular proteins of the same size with no cysteine bridges at all, e.g., protein-G [
33].
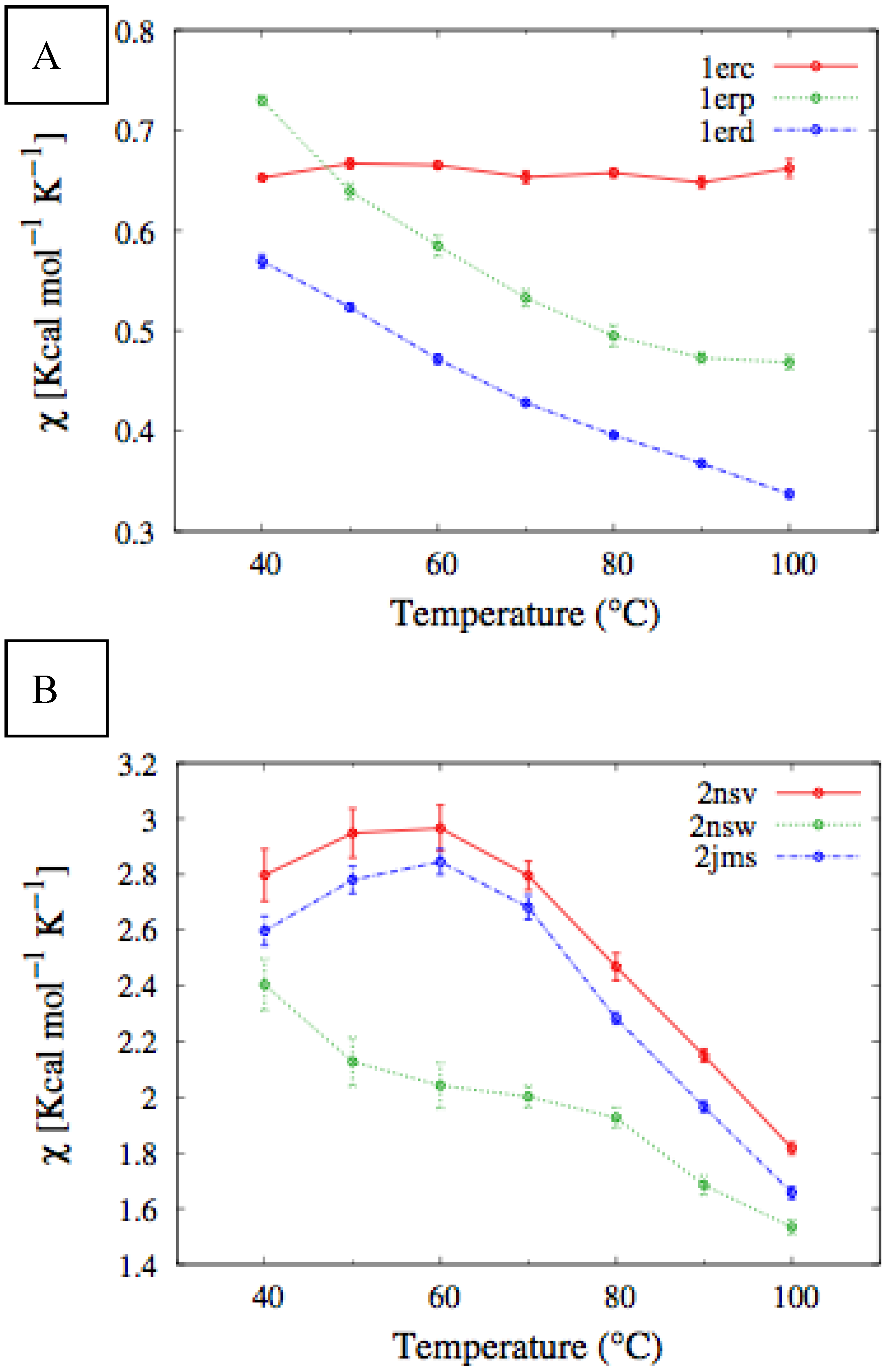
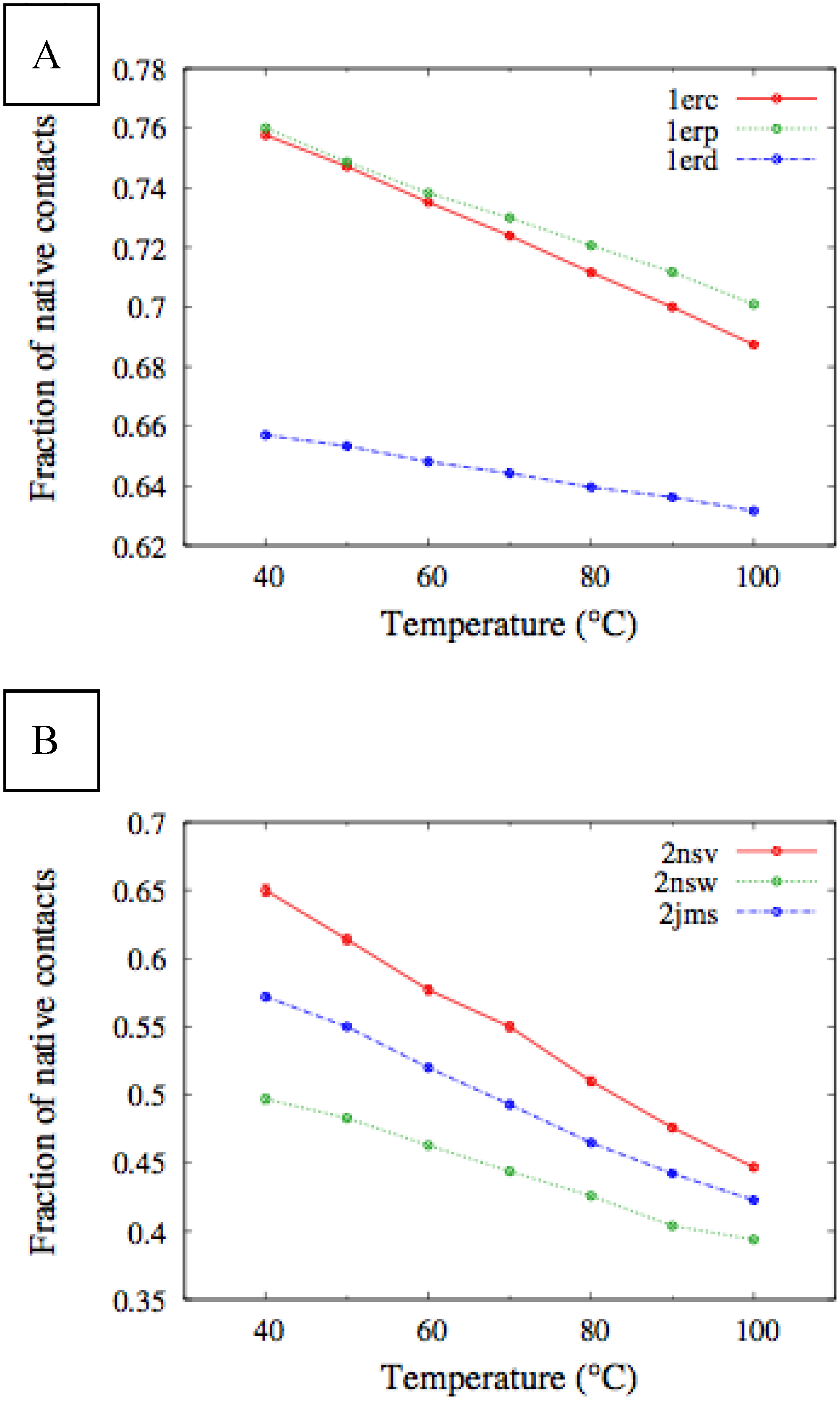
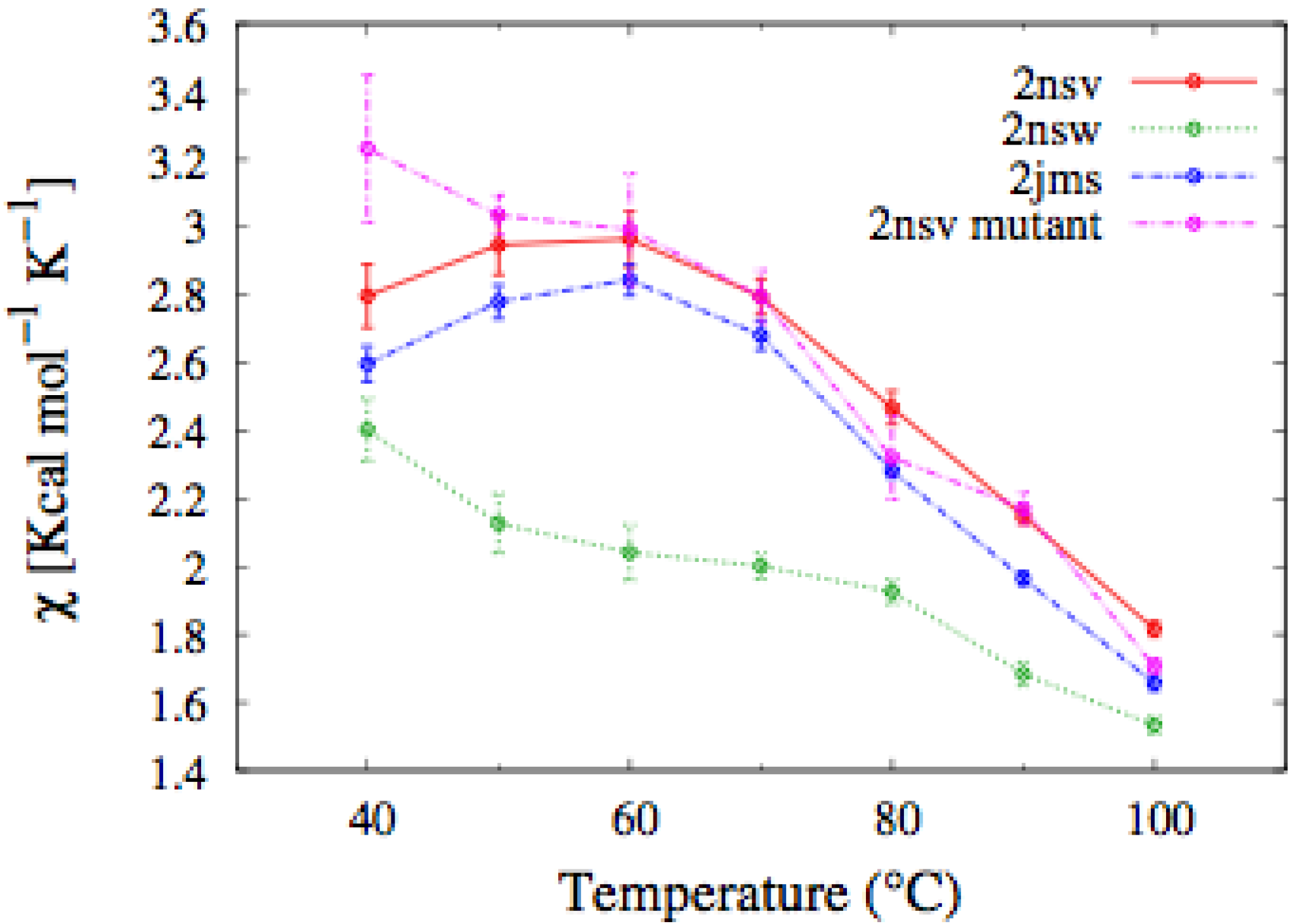
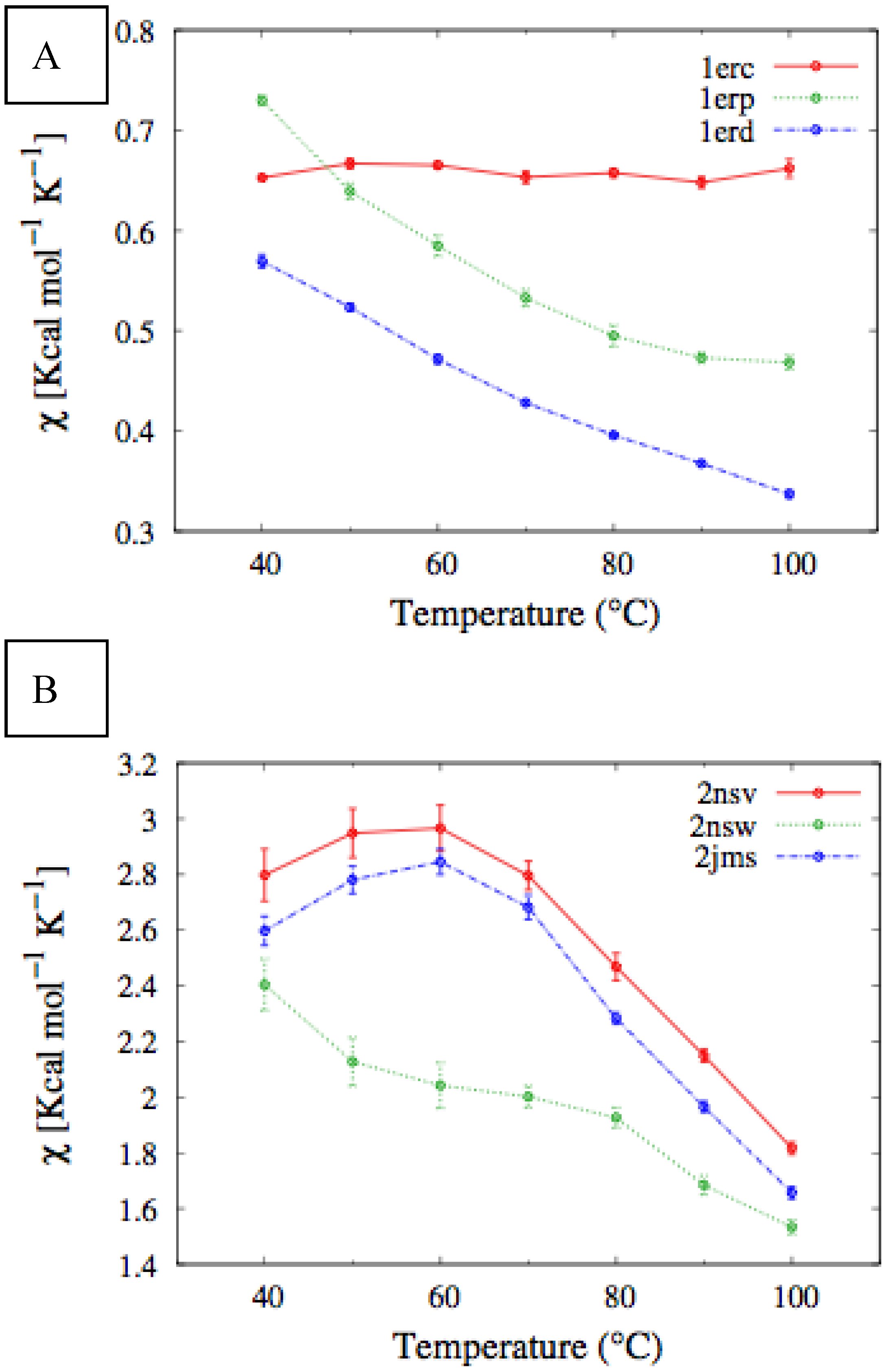
In
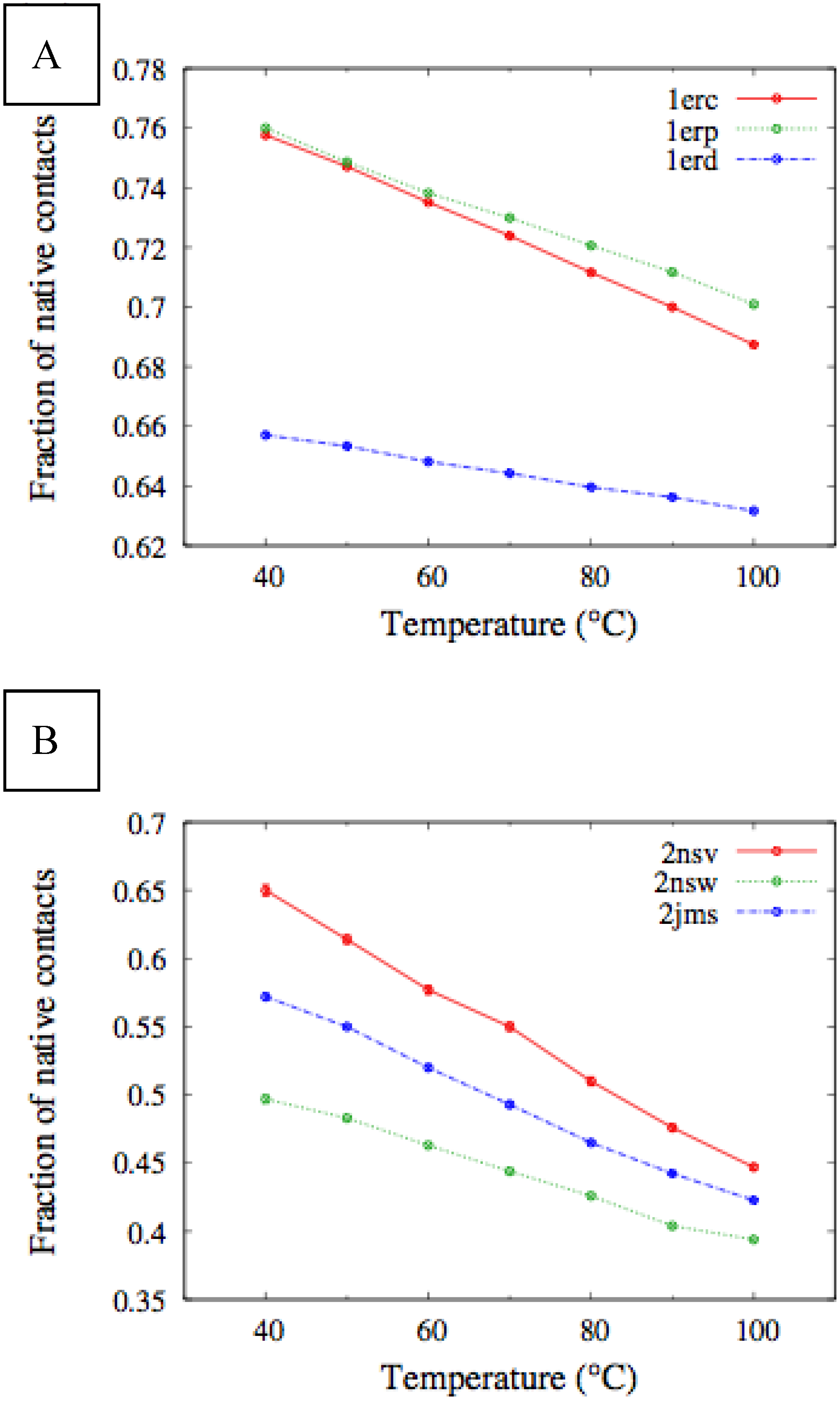
Figure 4, we report the values of
χ(T) at different temperatures for the polypeptide chains belonging to the
E. nobilii and
E. raikovi pheromone families, respectively, while in
Figure 5 we show the corresponding average fraction of native contacts. It appears that
E. nobilii pheromones are characterized by an unfolding transition in the temperature range considered. Remarkably, no unfolding peak in the fluctuation of native contacts is shown by any of the simulated
E. raikovi pheromones. In addition, we emphasize that the magnitude and the temperature dependence of the fraction of native contacts in
α-helixes computed from our MC simulations qualitatively resembles those of the helical fraction
fH, which was extracted directly from the deconvolution of the CD data. Indeed, both quantities show an overall smaller helical content, as well as steeper secondary motif suppression with temperature, in the
E. nobili pheromones.
Figure 4.
Comparison of the temperature-dependence of the fluctuation of fraction of native contacts defined in Equation (1) for different Er (A) and En (B) pheromones obtained by means of MC simulations in the CG model defined in the Experimental Section.
Figure 4.
Comparison of the temperature-dependence of the fluctuation of fraction of native contacts defined in Equation (1) for different Er (A) and En (B) pheromones obtained by means of MC simulations in the CG model defined in the Experimental Section.
Figure 5.
Temperature dependence of the fraction of native contacts for different Er (A) and En (B) pheromones, obtained by means of MC simulations in the CG model defined in the Experimental Section.
Figure 5.
Temperature dependence of the fraction of native contacts for different Er (A) and En (B) pheromones, obtained by means of MC simulations in the CG model defined in the Experimental Section.
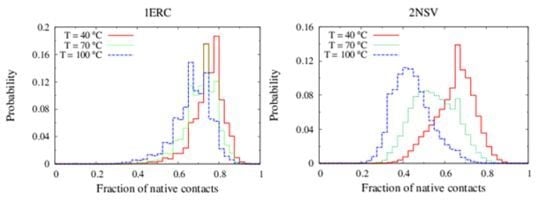
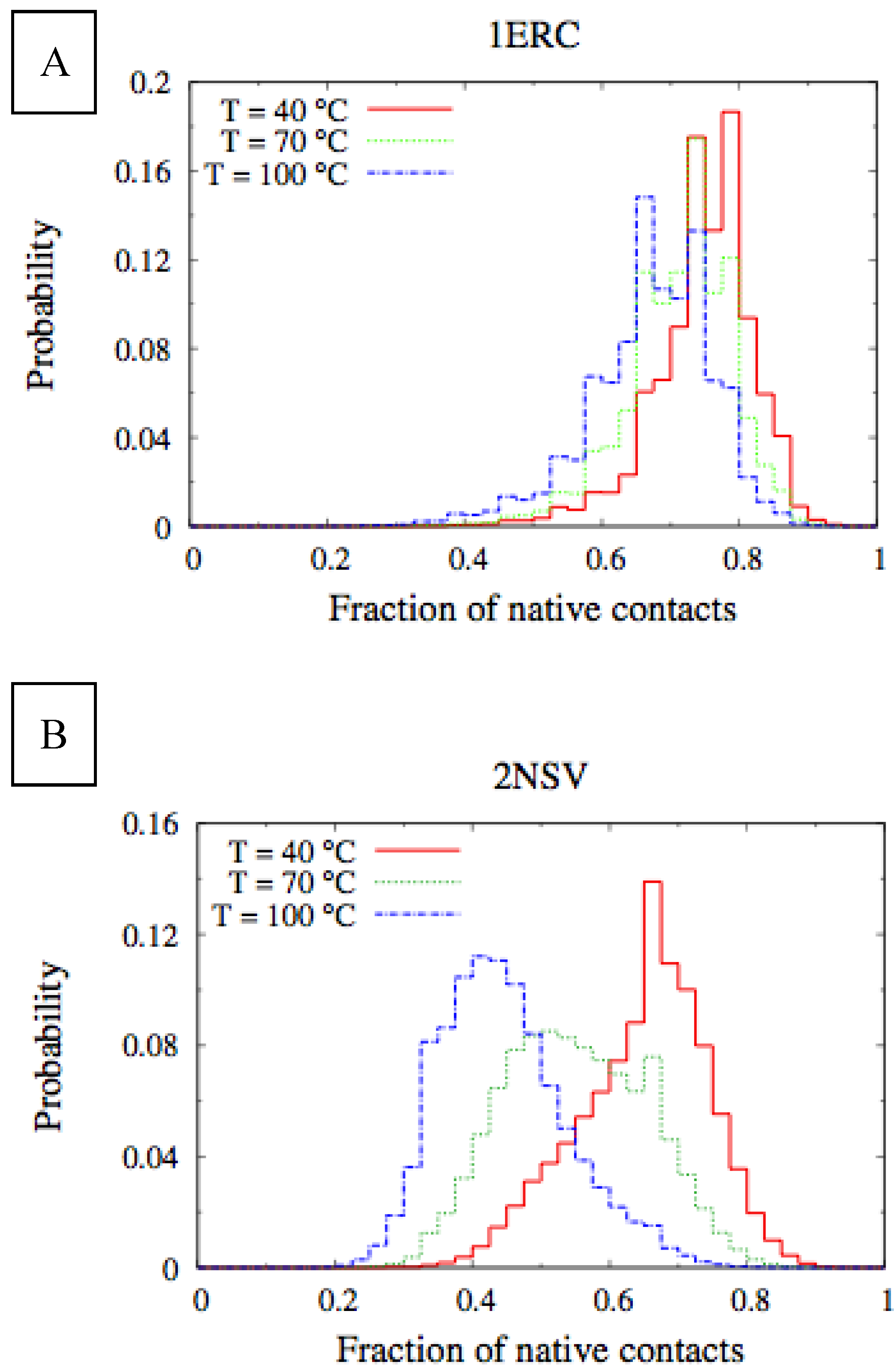
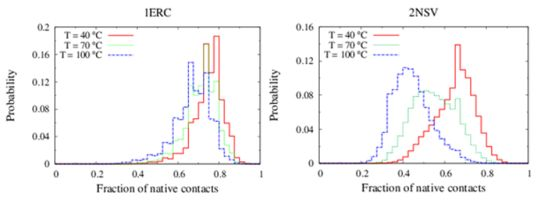
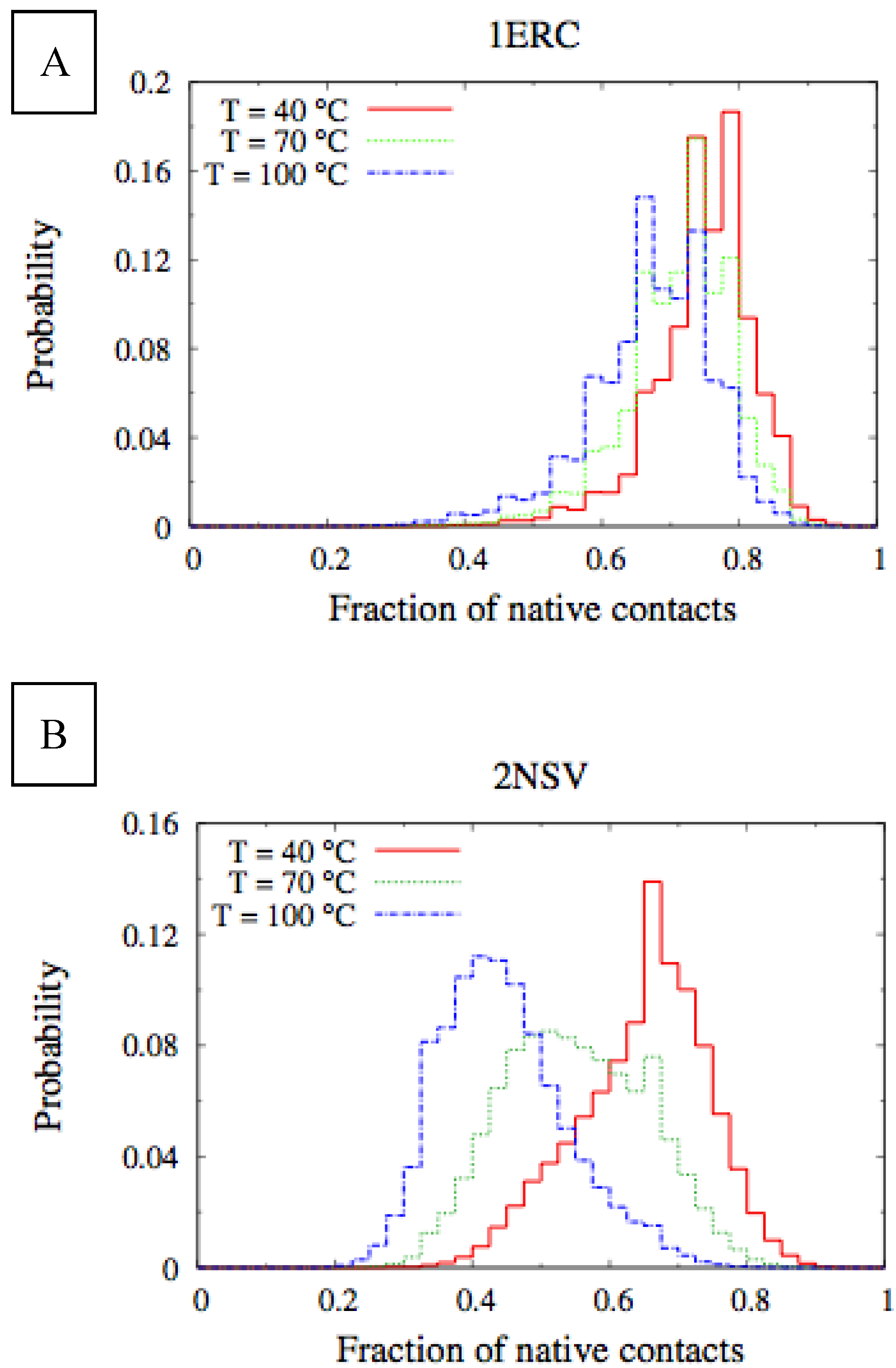
In
Figure 6, we compare the equilibrium distributions of the fraction of native contacts in two representative pheromones, namely E
r-1of
E. raikovi and E
n-1 of
E. nobilii, at different temperatures. In E
r-1, this distribution appears to be almost insensitive to temperature variations and the protein remains native even at the highest temperature considered. On the other hand, the fraction of native contacts in the E
n-1 polypeptide chain drops significantly at high temperatures. These results match even at a semi-quantitative level the distributions shown in
Table 2, which were obtained by deconvoluting the CD experimental data. In principle, the model’s parameters may be adjusted to in order to reproduce quantitatively also the experimentally observed relatively low helicity of the high-temperature experiments (close to 100 °C), e.g., by slightly decreasing the strength of the native interactions involved in the secondary structures. However, the main point of this analysis was to show that such a model allows for rationalization of the mechanism behind the qualitative difference in the thermodynamics of the two pheromone families. For this reason we choose to avoid the fine tuning of the parameters, thereby allowing for this small discrepancy.
Figure 6.
Equilibrium distributions of the fraction of native contacts at different temperatures for the Er-1 (A) and En-1 (B) pheromones.
Figure 6.
Equilibrium distributions of the fraction of native contacts at different temperatures for the Er-1 (A) and En-1 (B) pheromones.
Table 2.
Values of fractional helicity
fH at different temperatures for the E
r-1 and E
n-1 proteins, obtained by DICHROWEB/CONTINLL-4 analysis. The values in parenthesis are the corresponding normalized root mean square deviations. The last column reports the values of fractional helicity
fH of E
r-1 and E
n-1 as established by X-ray [
27] and NMR [
23], respectively.
Table 2.
Values of fractional helicity fH at different temperatures for the Er-1 and En-1 proteins, obtained by DICHROWEB/CONTINLL-4 analysis. The values in parenthesis are the corresponding normalized root mean square deviations. The last column reports the values of fractional helicity fH of Er-1 and En-1 as established by X-ray [27] and NMR [23], respectively.
| Pheromone | fH (CD, 40 °C) | fH (CD, 70 °C) | fH (CD, 95 °C) | fH reported |
|---|
| Er-1 | 0.72 (0.07) | 0.68 (0.08) | 0.53 (0.03) | 0.678 (X-ray) |
| En-1 | 0.57 (0.08) | 0.41 (0.14) | 0.16 (0.07) | 0.46 (NMR) |
These results overall indicate that our simple CG model, which only encodes the information related to three dimensional native structure and the location of the disulfide bonds, is able to closely reproduce the observed difference in the pheromone unfolding/refolding thermodynamics. We emphasize that no parameter in the energy function described in the Experimental Section was fit in order to produce the observed differences between the En and Er pheromones.
It is important to point out that the energy function of the CG model encodes only the information related to the protein tertiary structure and the location of the Cys-Cys bonds. Therefore, the remarkable agreement that we find with the experimental data suggests that the specificities of the chemical composition of the polypeptide chain and the non-native interactions do not significantly affect the thermodynamics of these systems. This finding contrasts with the explanation of the enhanced thermal stability of the E
n pheromones based on the existence of a strongly hydrophobic cluster, which was proposed [
23].
In general, it is difficult to justify the observed large effects on the unfolding temperatures in terms of the relatively small differences in the three-dimensional crystal structures of the different pheromones. In view of the results of the theoretical studies on the thermodynamics of cysteine-rich proteins [
7,
8,
10,
11], we argue that the observed differences are instead generated by the different topological constraints that are imposed by the specific location of the disulfide bonds.
To substantiate this hypothesis, we analyzed the localization of the pheromone hydrogen-bonded and disulfide-bonded native contacts, and compared the contact order (
CO) of the pheromone native structures in accordance with the parameters provided by [
30] where,
In this formula,
N is the total number of contacts,
L is the total number of amino acids in the protein, and
ΔSij is the sequence separation between contacting residues
i and
j. The
CO values of all the
E. nobilii and
E. raikovi pheromones are reported in
Table 3. It appears that all the polypeptide chains have
CO values close to 0.3, which is a typical value for
α-helix proteins. Hence, the localization of generic tertiary contacts cannot be at the origin of the observed large difference in the unfolding temperature.
Table 3.
The contact order calculated using all native contacts and the contact order calculated only for Cys-Cys native contacts are reported for the investigated pheromones.
Table 3.
The contact order calculated using all native contacts and the contact order calculated only for Cys-Cys native contacts are reported for the investigated pheromones.
| | Euplotes raikovi | Euplotes nobilii |
|---|
| Name | Er-1 | Er-2 | Er-10 | En-1 | En-2 | En-6 |
| COtotal | 0.19 | 0.19 | 0.19 | 0.22 | 0.19 | 0.21 |
| COCys-Cys | 0.46 | 0.43 | 0.48 | 0.34 | 0.32 | 0.32 |
On the other hand, a significant dissimilarity between the
E. nobilii and
E. raikovi pheromones emerges when we compare a
CO variant in which the sum runs only over disulfide native contacts:
In this formula, the indexes
i and
j run over Cys residues only and
NCys denotes the total number of sulfur bridges. The values of this observable for all pheromones are reported in
Table 3. It appears that the
E. raikovi pheromones have
COCys-Cys values close to 0.45, while the
E. nobilii pheromones have
COCys-Cys close to 0.33. This difference implies that the disulfide bonds in the mesophilic
E. raikovi pheromones are systematically less local than in the psychrophilic
E. nobilii pheromones. In order to illustrate how a lower locality of the disulfide bonds can imply a higher unfolding temperature, in the appendix we discuss a minimalistic statistical toy model, which can be solved analytically.
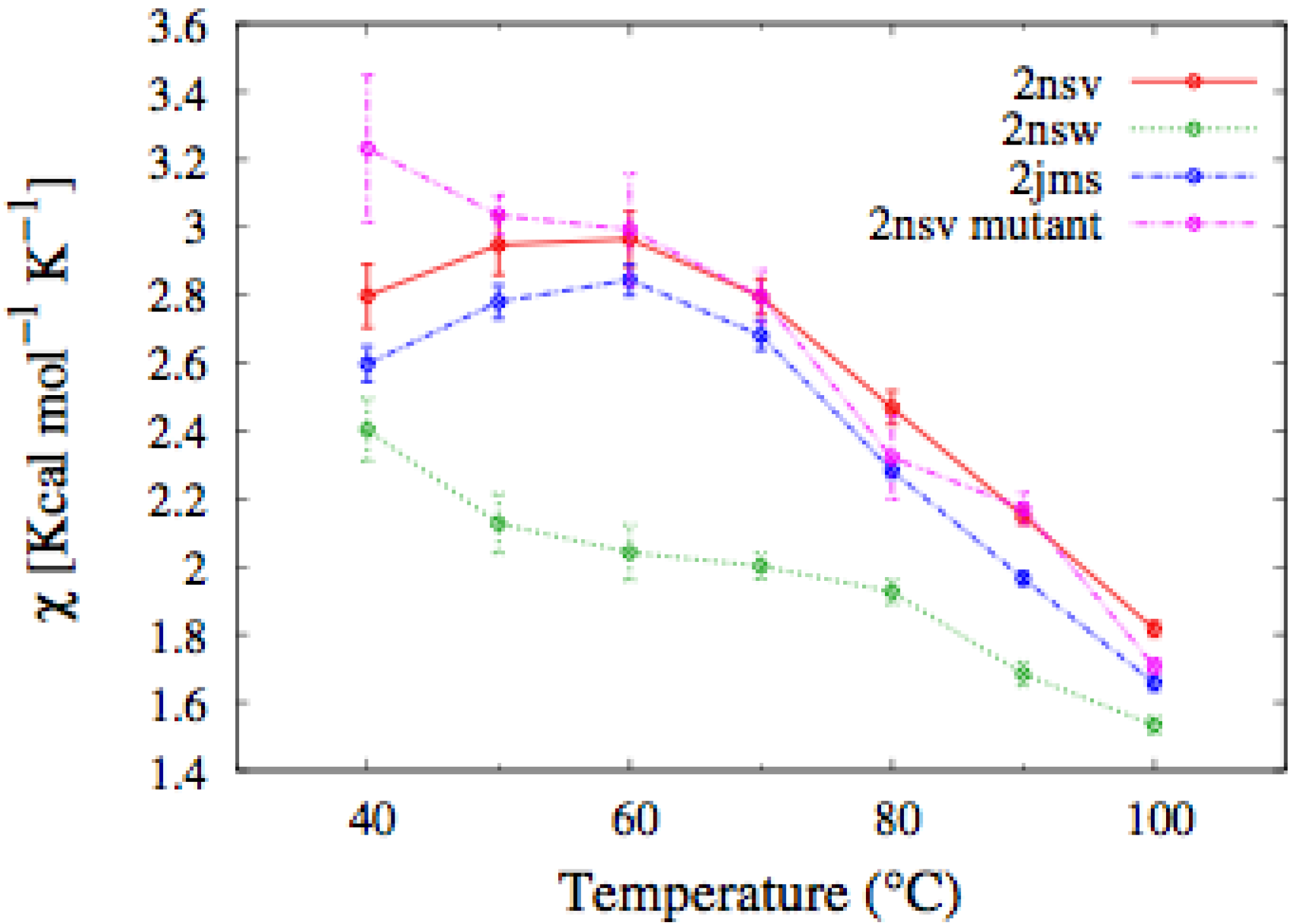
To allow for a further validation of our theoretical explanation of the CD data, we lastly addressed the question of the minimal set of mutations capable of increasing the unfolding temperature of
E. nobilii pheromones up to the values typical of
E. raikovi pheromones. The basic rationale was to reposition the Cys-Cys bonds in the native structure of a representative
E. nobilii pheromone in a way to attain the values of the contact order of cysteine-bonds that are typical of the other, that of
E. raikovi pheromone family. In particular, we designed a mutant of the
E. nobilii E
n-1 protein in which two of the Cys-Cys bonds have been translated from positions 11–37 to 15–37 and from 30–52 to 27–52, and Cys residues in the positions 23 and 33 have been replaced by Gly residues. In this way, the disulfide bonds reproduce the same disulfide bond patterns of the
E. raikovi E
r-1 polypeptide chain (see the right panel of
Figure 2). With these modifications, the new mutant protein, despite it contains one disulfide bond less, has an increased value of the contact order of sulfur bridges, and thus is expected to obey a higher thermal stability, that typical of
E. raikovi pheromones. The primary sequences of the wild-type and mutant E
n-1 chains are the following:
By assuming that such a small mutation does not affect at all the three dimensional structure of the E
n-1 polypeptide chain, we simulated the thermodynamics of the new mutant protein in the native-centric CG model. The results are presented in
Figure 7. As expected, the E
n-1 mutant protein showed a qualitatively similar thermodynamics in respect of that of the
E. raikovii pheromones. In particular, the temperature dependence of the average fluctuation of the fraction of native contacts, defined in Equation 1, for the mutant does not display a peak signaling the unfolding transition, which is present in the curve relative to the wilde type protein. It would be very interesting in the future to assess this simulation by direct CD measurements of this mutant protein. In concluding this section, we deserve a comment to the hypothesis, proposed by Geralt
et al. [
2], that the differences in the thermodynamic properties between the
E. nobilii and
E. raikovi pheromones may be due to a substantially unstructured N-terminal extension distinctive of
E. nobilii pheromones. This hypothesis does not appear to be supported by the above observations on the enhanced stability of the E
n-1 mutant protein, that includes an extension of 13 amino-acids at the
N-terminal with respect to the E
r-1 pheromone. In addition, a simulation of a second mutant protein, in which the 13 residues at the
N-terminal were removed and the remaining residues are the same as in the first mutant, showed the thermodynamics of the E
r-type (data not showed).
Figure 7.
Temperature dependence of the fluctuation in the number of native contacts defined in Equation (1), for the En pheromones and the mutant of the En-1 chain. In the mutant, the characteristic peak signaling the unfolding transition disappears.
Figure 7.
Temperature dependence of the fluctuation in the number of native contacts defined in Equation (1), for the En pheromones and the mutant of the En-1 chain. In the mutant, the characteristic peak signaling the unfolding transition disappears.


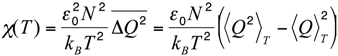
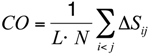
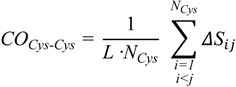
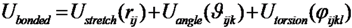



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}