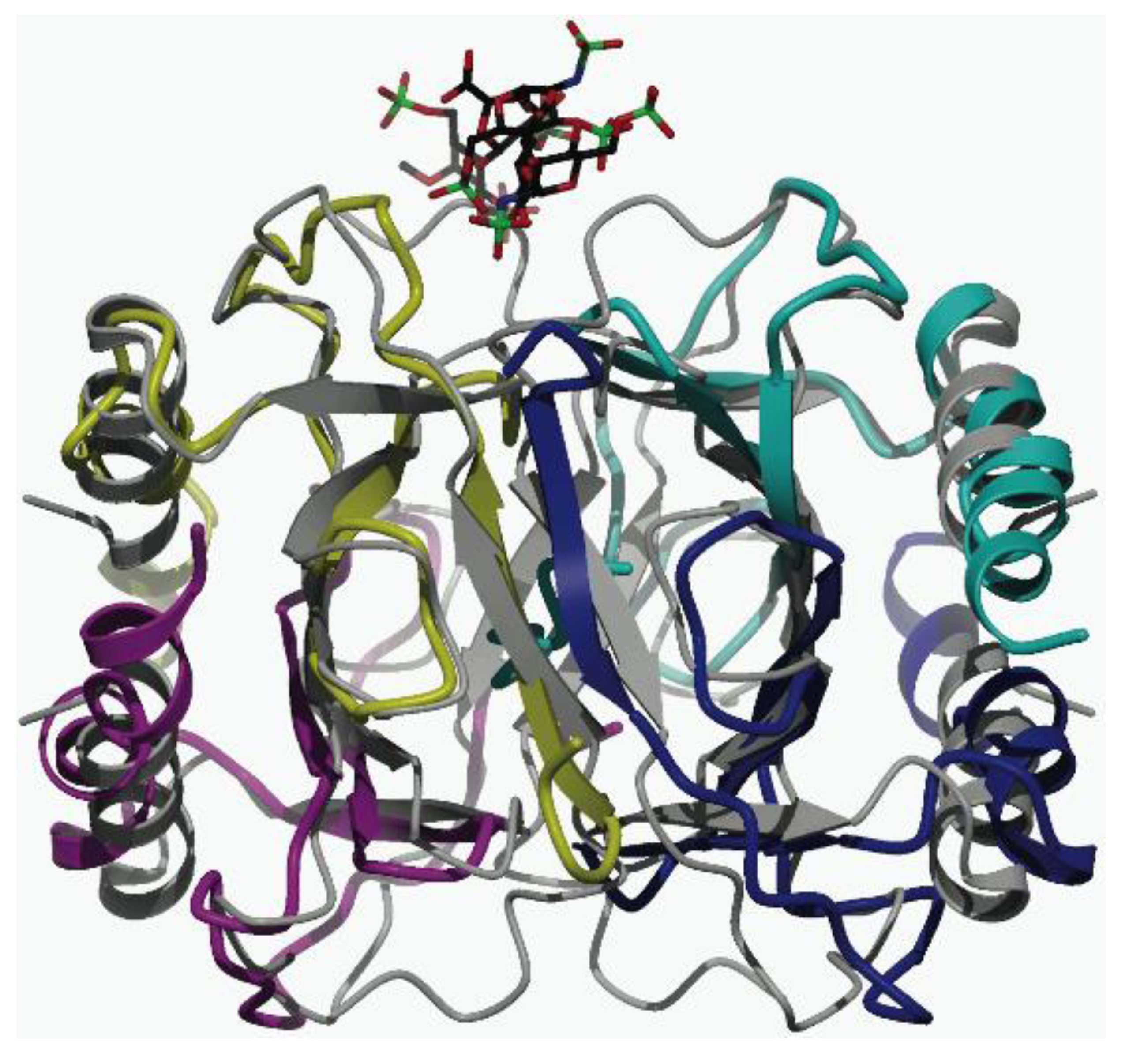
Structural Evidence for the Tetrameric Assembly of Chemokine CCL11 and the Glycosaminoglycan Arixtra™

Abstract
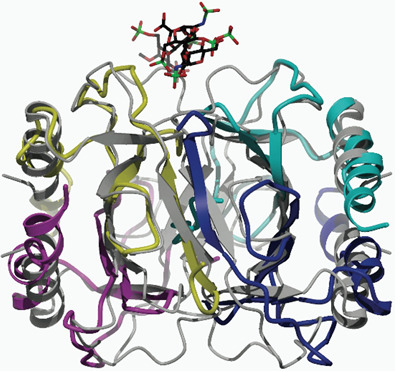
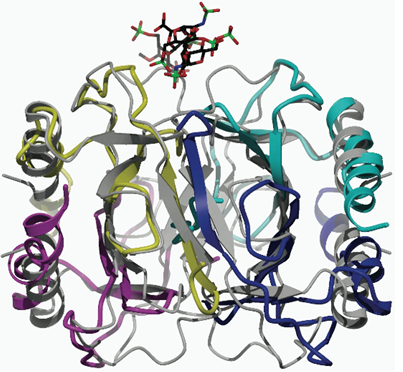
:
1. Introduction
2. Results and Discussion
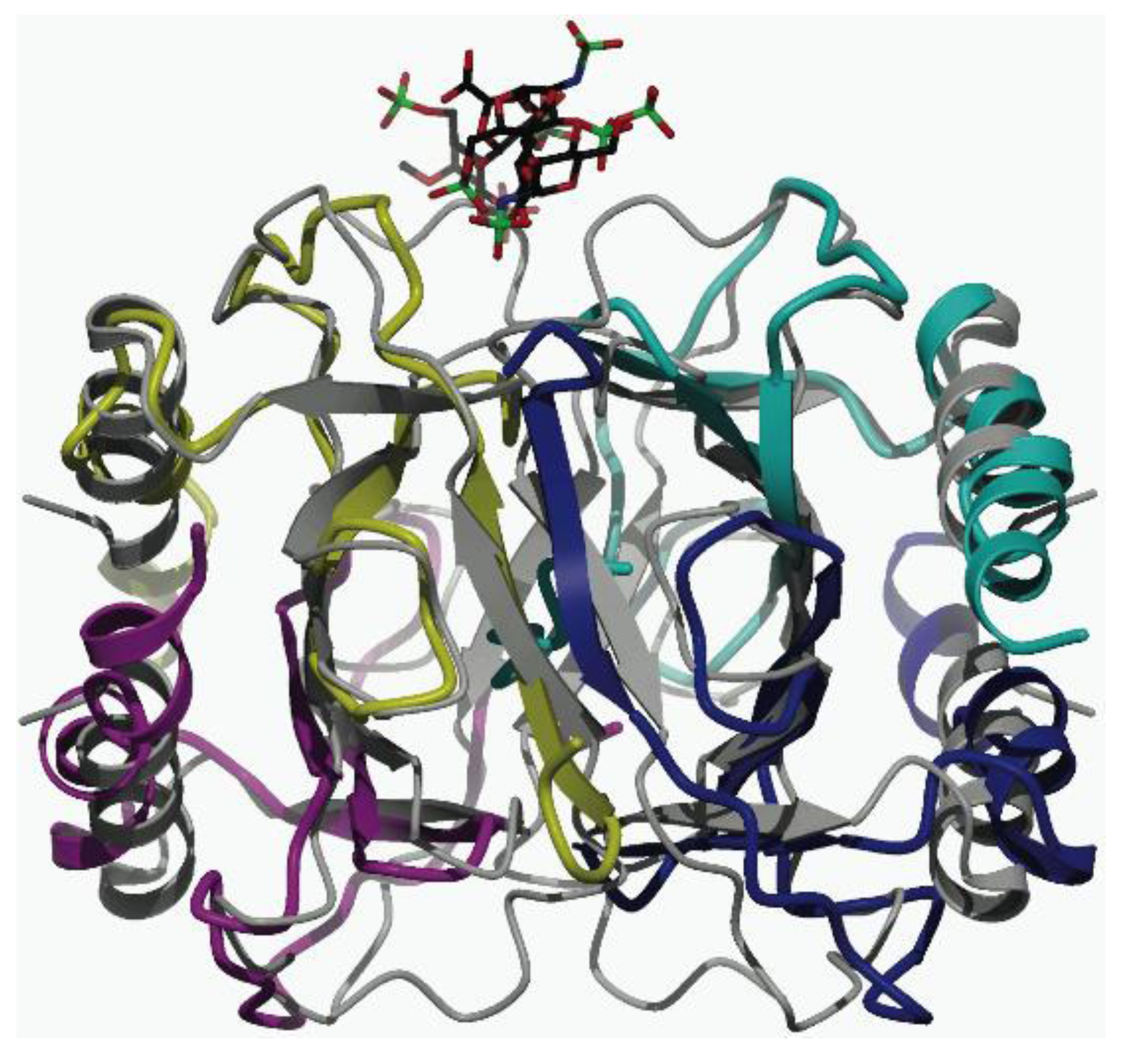
2.1. Binding of CCL11 to Arixtra
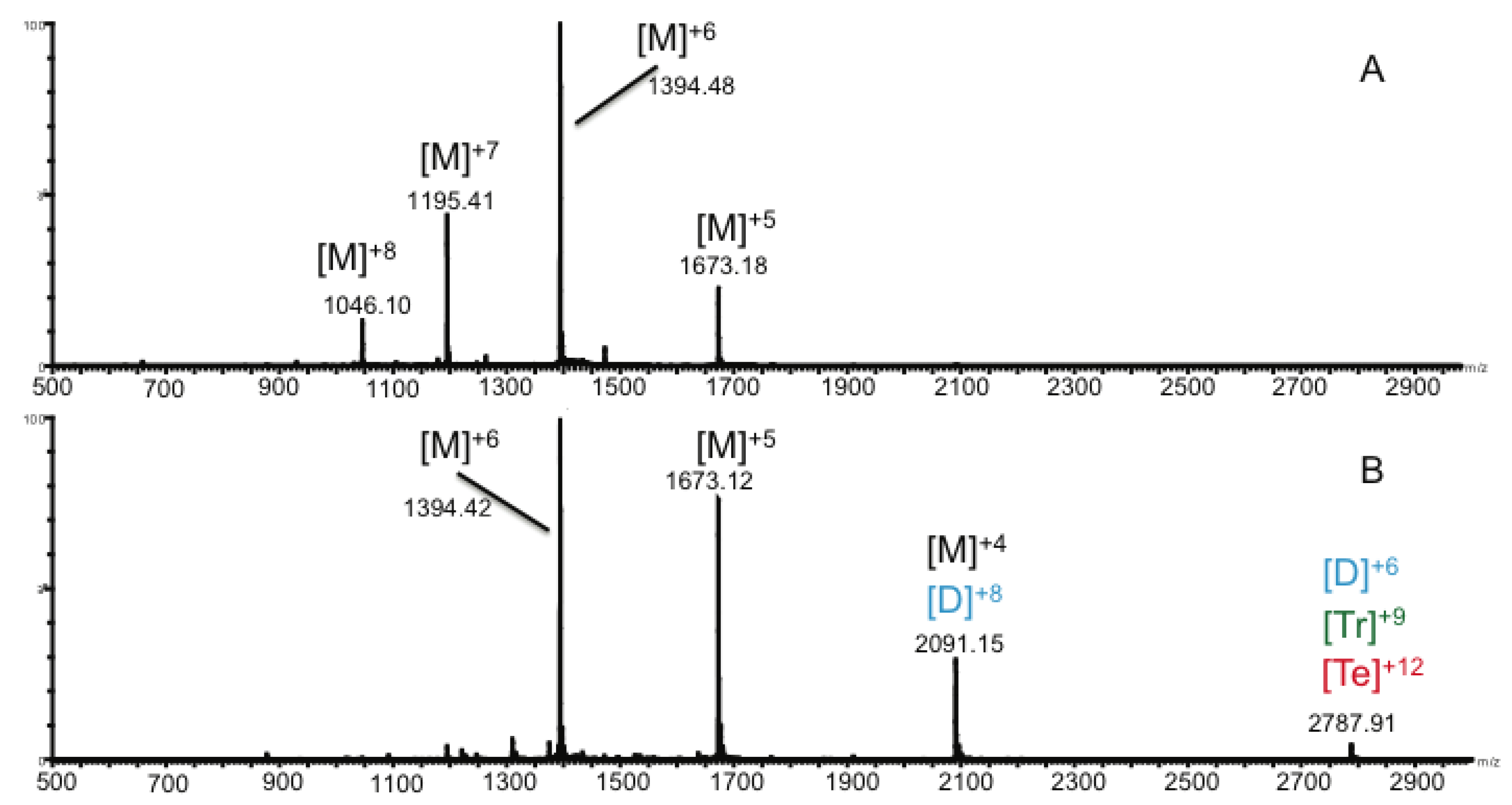
2.2. Ion Mobility Mass Spectrometry of CCL11 Monomer
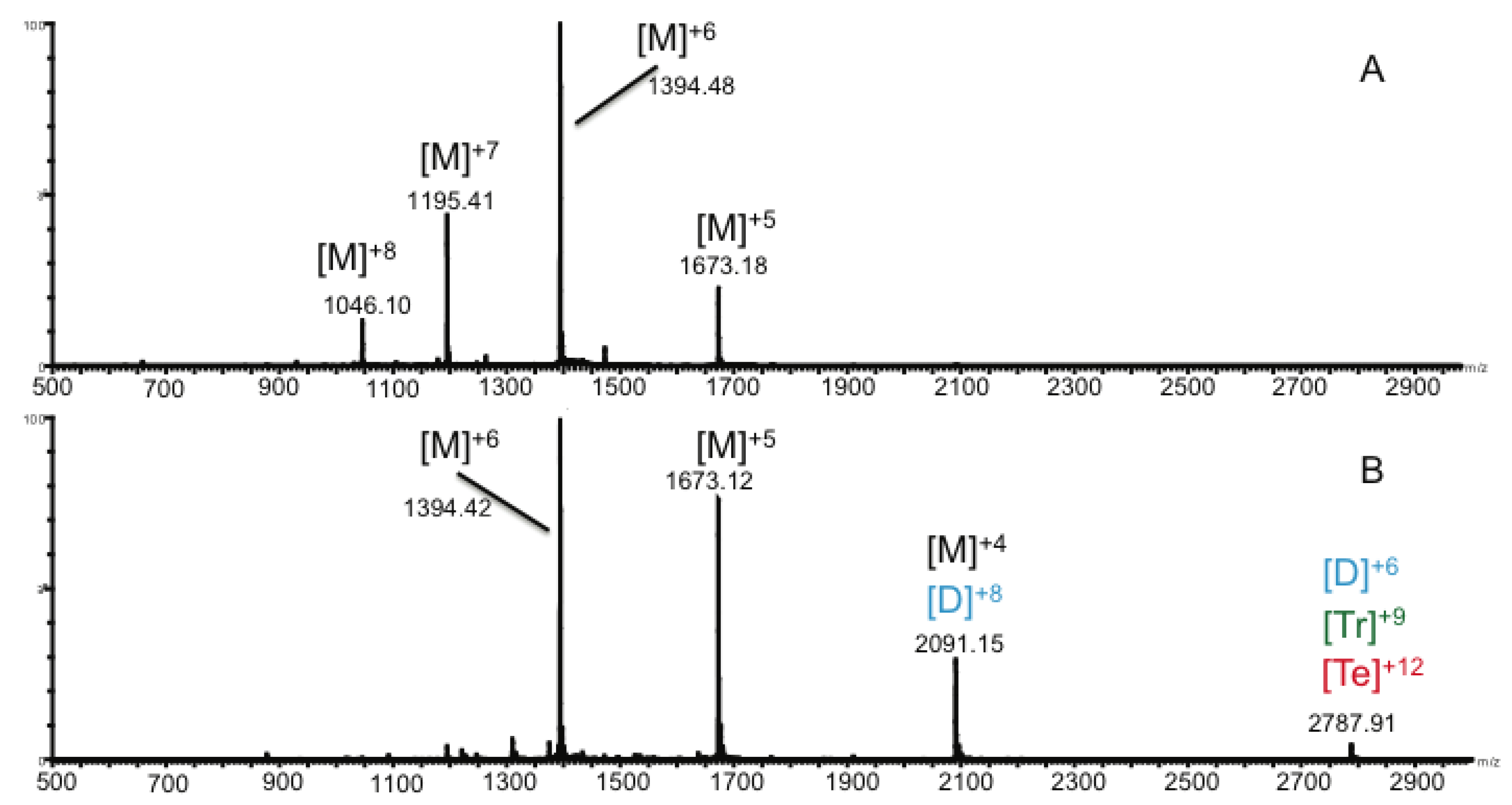
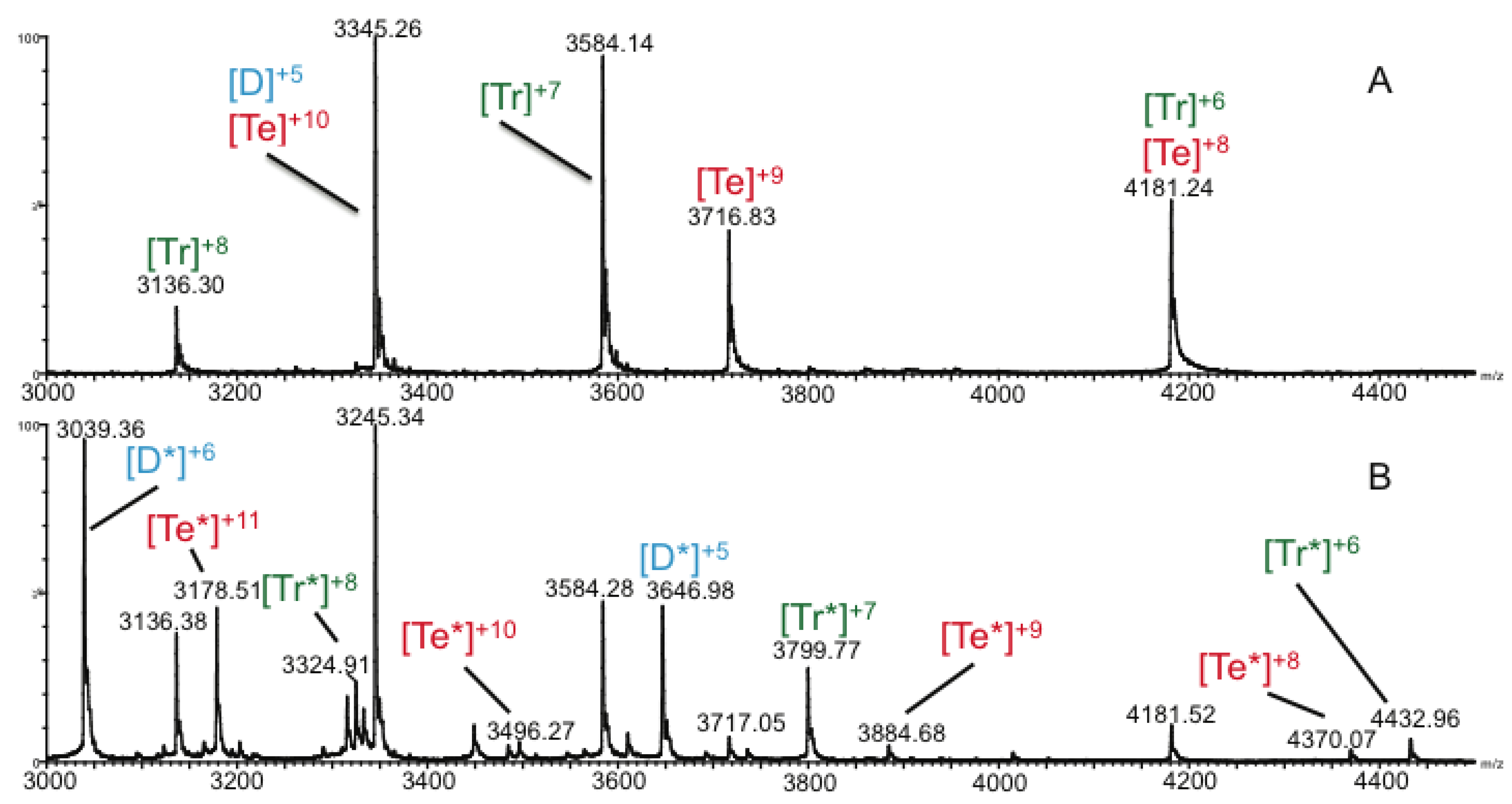
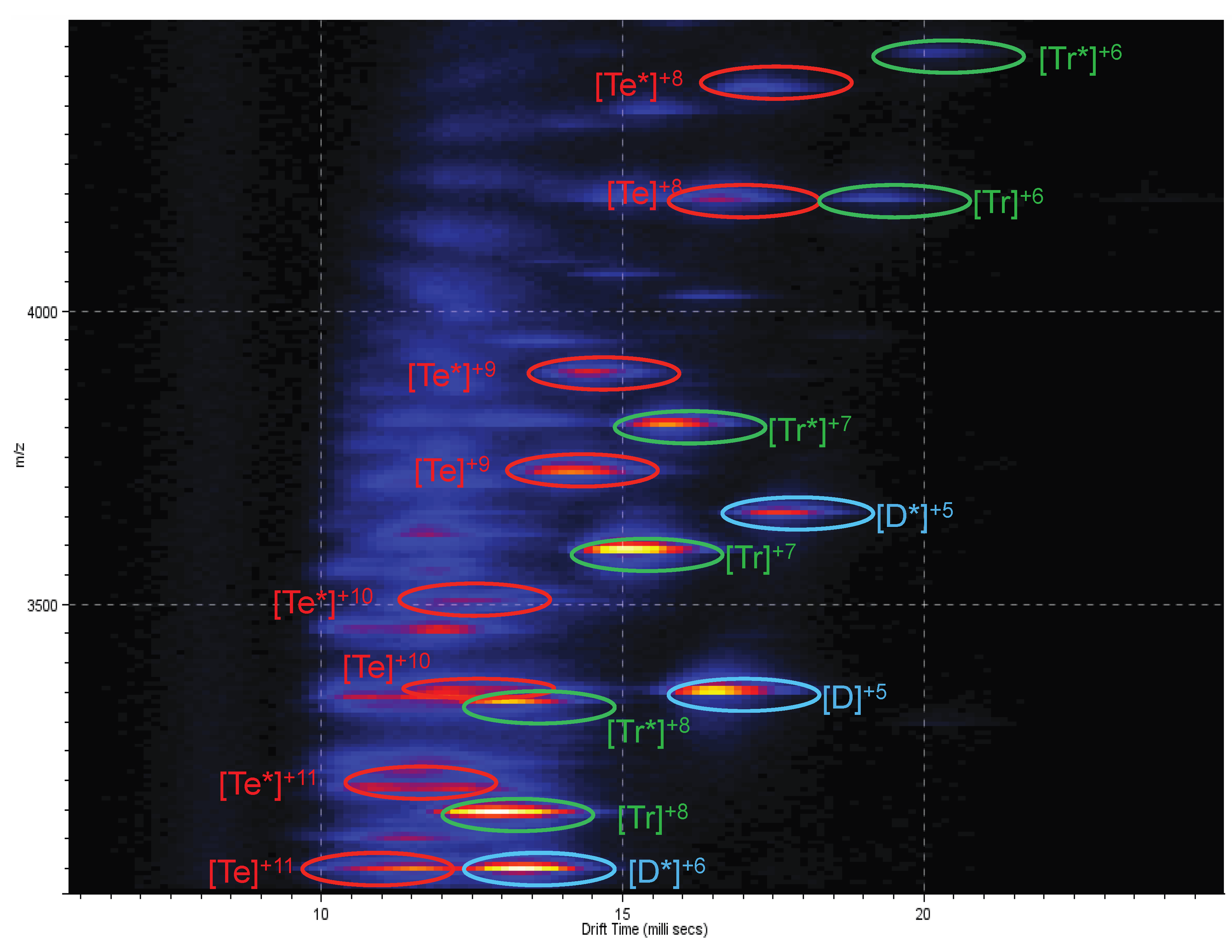
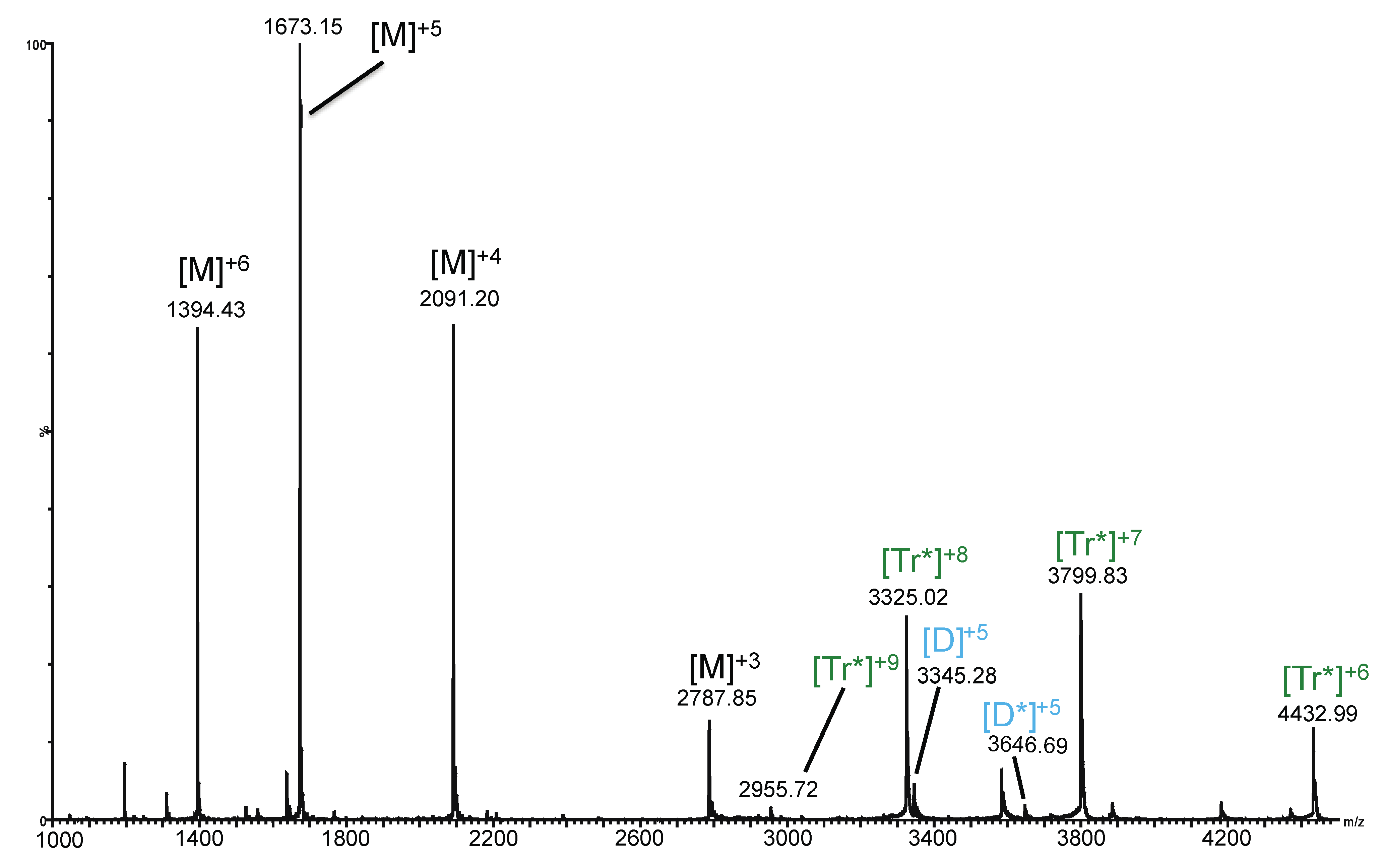
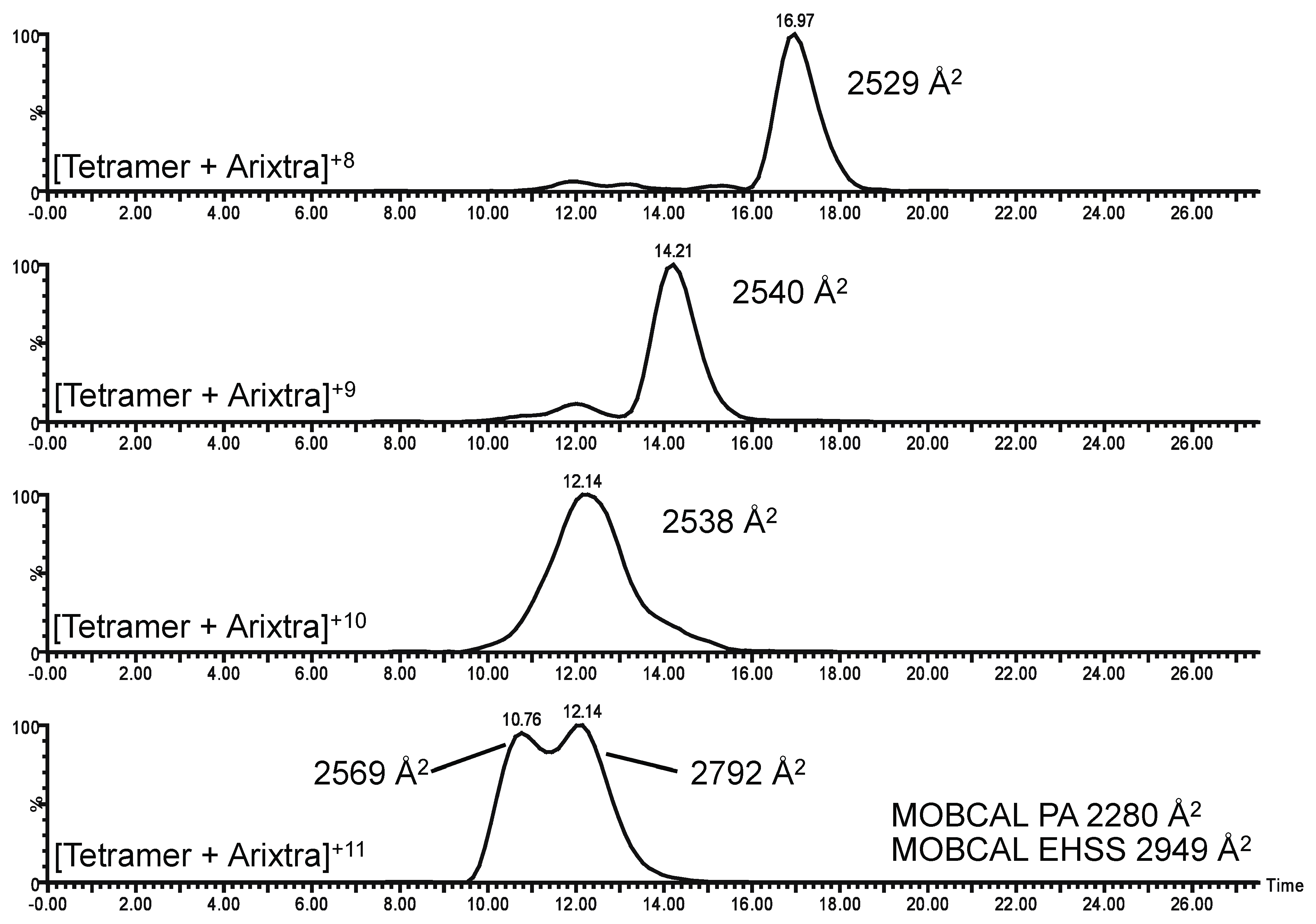
2.3. Ion Mobility Mass Spectrometry of CCL11 Tetrameric Species

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mass-to-Charge (Experimental) | Mass-to-Charge (Theoretical) | Intensity (Counts) | Charge | Species |
|---|---|---|---|---|
| 1,394.4 | 1,394.4 | 4.47E6 | 6 | Monomer |
| 1,645.8 | 1,645.6 | 8.25E4 | 6 | Monomer + Arixtra |
| 1,673.1 | 1,673.1 | 2.53E6 | 5 | Monomer |
| 1,974.8 | 1,974.5 | 3.73E4 | 5 | Monomer + Arixtra |
| 2,091.2 | 2,091.1 | 1.24E6 | 4 | Monomer |
| 8 | Dimer | |||
| 2,279.8 | 2,279.5 | 1.11E5 | 8 | Dimer + Arixtra |
| 2,389.8 | 2,389.7 | 7.51E3 | 7 | Dimer |
| 2,468.2 | 2,467.9 | 1.87E4 | 4 | Monomer + Arixtra |
| 2,605.3 | 2,605.0 | 1.28E4 | 7 | Dimer + Arixtra |
| 2,787.9 | 2,787.8 | 1.79E5 | 6 | Dimer |
| 9 | Trimer | |||
| 12 | Tetramer | |||
| 2,913.9 | 2,913.4 | 5.83E3 | 12 | Tetramer + Arixtra |
| 2,955.6 | 2,955.3 | 1.12E4 | 9 | Trimer + Arixtra |
| 3,039.4
3,041.5 3,136.4 | 3,039.0
3,041.2 3,136.2 | 2.97E4
8.50E3 1.18E4 | 6 | Dimer + Arixtra |
| 11 | Tetramer | |||
| 8 | Trimer | |||
| 3,178.5 | 3,178.2 | 1.42E4 | 11 | Tetramer + Arixtra |
| 3,324.9 | 3,324.6 | 7.29E3 | 8 | Trimer + Arixtra |
| 3,345.3 | 3,345.2 | 2.09E4 | 5 | Dimer |
| 10 | Tetramer | |||
| 3,495.9 | 3,495.9 | 1.82E3 | 10 | Tetramer + Arixtra |
| 3,584.3 | 3,584.1 | 1.47E4 | 7 | Trimer |
| 3,647.0 | 3,646.6 | 1.44E4 | 5 | Dimer + Arixtra |
| 3,717.0 | 3,716.8 | 2.27E3 | 9 | Tetramer |
| 3,800.0 | 3,799.4 | 8.58E3 | 7 | Trimer + Arixtra |
| 3,884.7 | 3,884.2 | 1.47E3 | 9 | Tetramer + Arixtra |
| 4,181.5 | 4,181.3 | 3.37E3 | 6 | Trimer |
| 8 | Tetramer | |||
| 4,370.1 | 4,369.6 | 1.10E3 | 8 | Tetramer + Arixtra |
| 4,433.0 | 4,432.4 | 2.06E3 | 6 | Trimer + Arixtra |
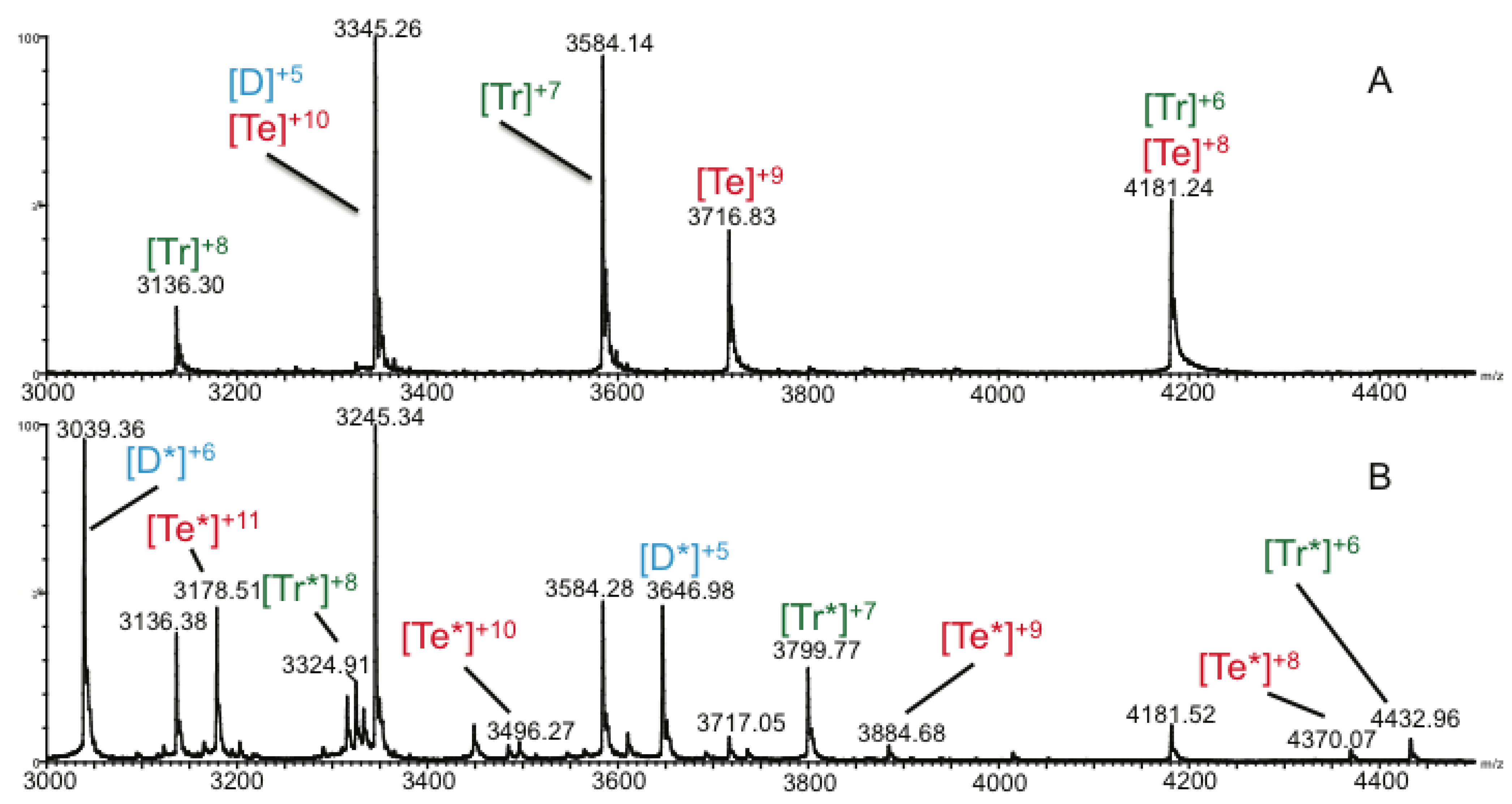

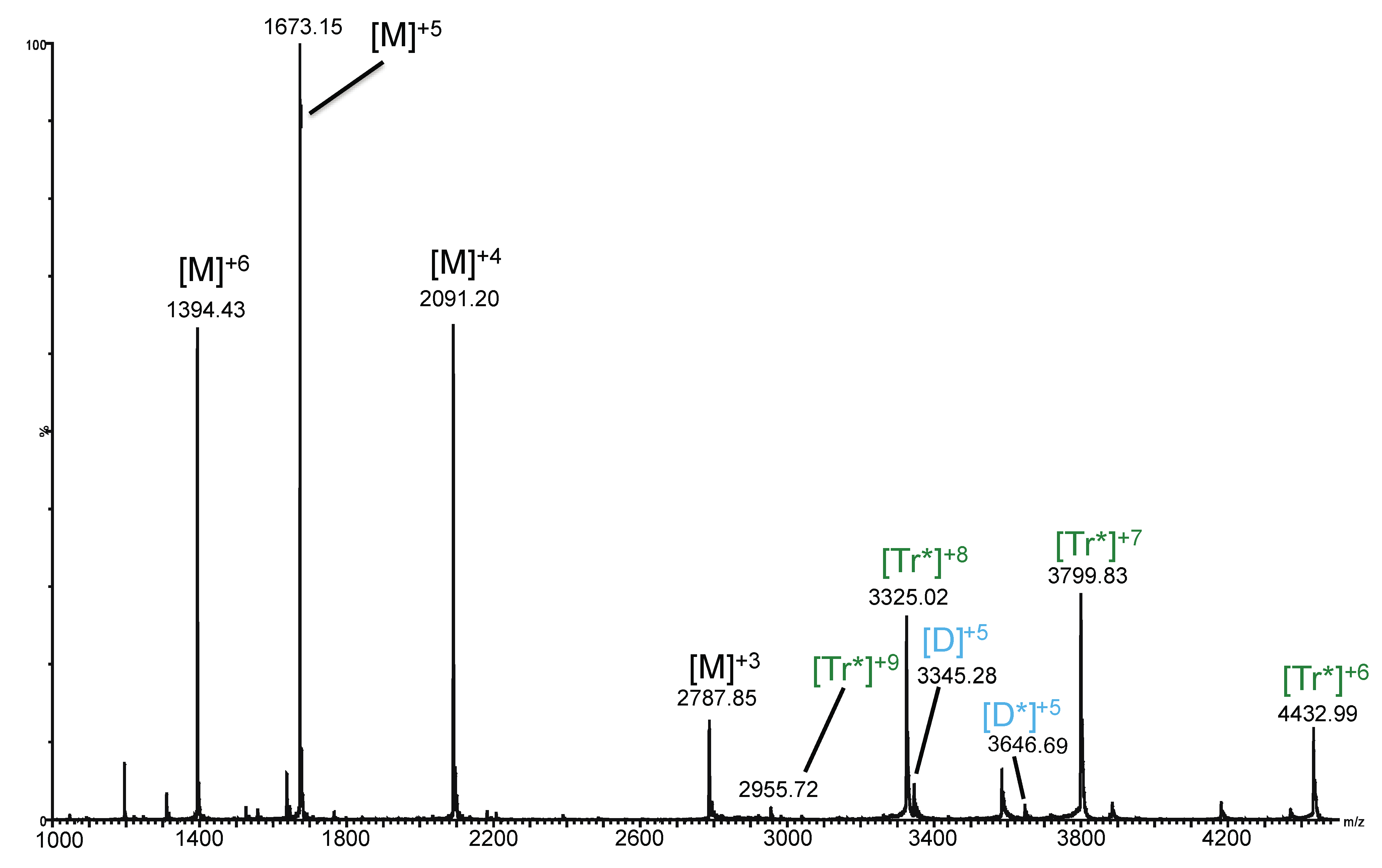
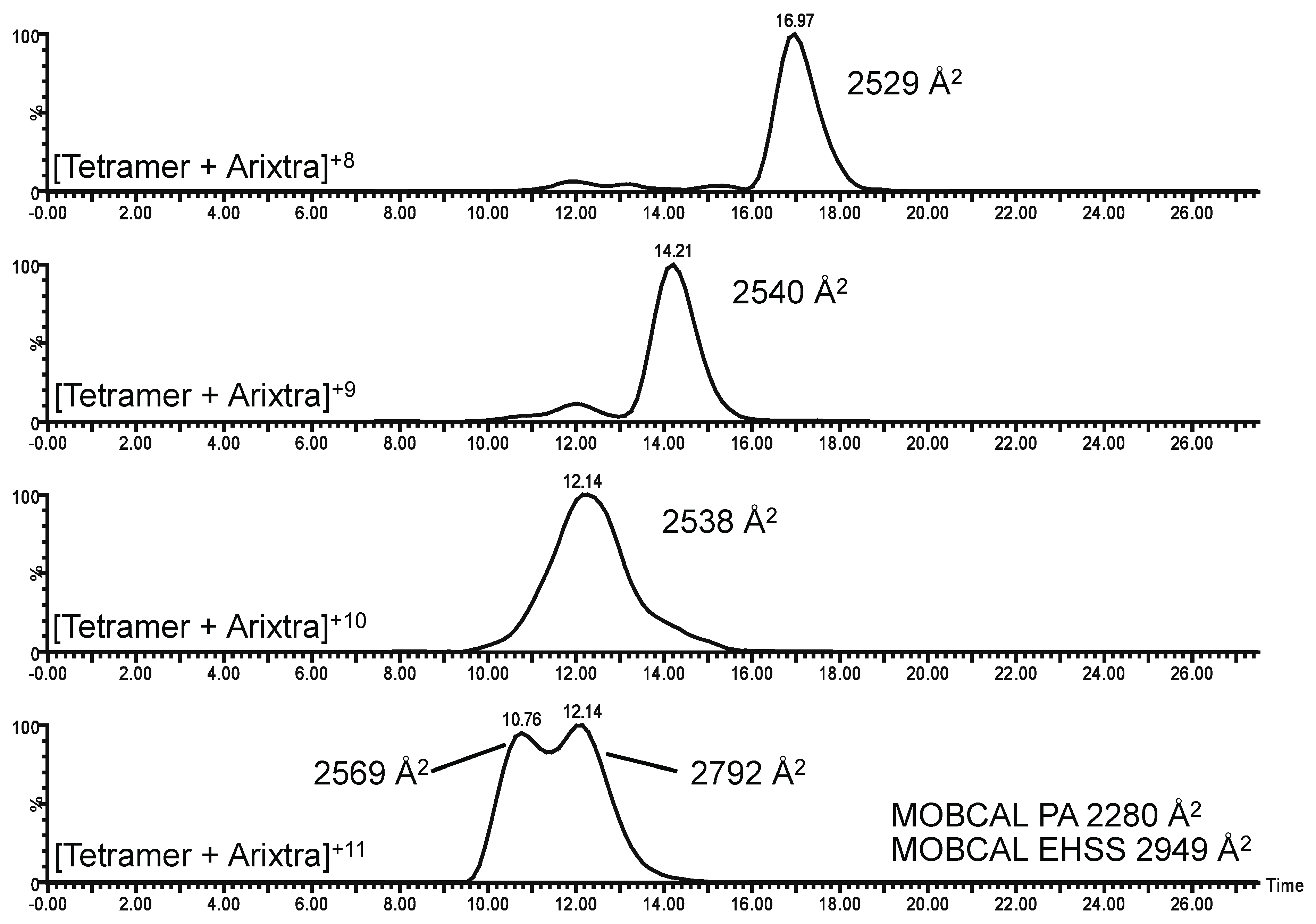
3. Experimental
3.1. Materials
3.2. Expression of Human CCL11 and Sample Preparation
3.3. Isothermal Titration Calorimetry
3.4. Ion Mobility Mass Spectrometry
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Conflicts of Interest
References
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Gomperts, B.N.; Strieter, R.M. Chemokine-directed metastasis. Contrib. Microbiol. 2006, 13, 170–190. [Google Scholar] [CrossRef]
- Matsushima, K.; Terashima, Y.; Toda, E.; Shand, F.; Ueha, S. Chemokines in inflammatory and immune diseases. Inflamm. Regen. 2011, 31, 11–22. [Google Scholar] [CrossRef]
- Balkwill, F.R. The chemokine system and cancer. J. Pathol. 2012, 226, 148–157. [Google Scholar] [CrossRef]
- Handel, T.M.; Johnson, Z.; Crown, S.E.; Lau, E.K.; Proudfoot, A.E. Regulation of protein function by glycosaminoglycans—As exemplified by chemokines. Annu. Rev. Biochem. 2005, 74, 385–410. [Google Scholar] [CrossRef]
- Rot, A. Endothelial cell binding of NAP-1/IL-8: Role in neutrophil emigration. Immunol. Today 1992, 13, 291–294. [Google Scholar] [CrossRef]
- Lau, E.K.; Paavola, C.D.; Johnson, Z.; Gaudry, J.P.; Geretti, E.; Borlat, F.; Kungl, A.J.; Proudfoot, A.E.; Handel, T.M. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: Implications for structure and function in vivo. J. Biol. Chem. 2004, 279, 22294–22305. [Google Scholar] [CrossRef]
- Hoogewerf, A.J.; Kuschert, G.S.; Proudfoot, A.E.; Borlat, F.; Clark-Lewis, I.; Power, C.A.; Wells, T.N. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 1997, 36, 13570–13578. [Google Scholar] [CrossRef]
- Yu, Y.; Sweeney, M.D.; Saad, O.M.; Crown, S.E.; Hsu, A.R.; Handel, T.M.; Leary, J.A. Chemokine-glycosaminoglycan binding: Specificity for CCR2 ligand binding to highly sulfated oligosaccharides using FTICR mass spectrometry. J. Biol. Chem. 2005, 280, 32200–32208. [Google Scholar]
- Proudfoot, A.E.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef]
- Fernandez, E.J.; Lolis, E. Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 469–499. [Google Scholar] [CrossRef]
- Murphy, J.W.; Yuan, H.; Kong, Y.; Xiong, Y.; Lolis, E.J. Heterologous quaternary structure of CXCL12 and its relationship to the CC chemokine family. Proteins Struct. Funct. Bioinform. 2010, 78, 1331–1337. [Google Scholar] [CrossRef]
- Jabeen, T.; Leonard, P.; Jamaluddin, H.; Acharya, K.R. Structure of mouse IP-10, a chemokine. Acta Crystallogr. D 2008, 64, 611–619. [Google Scholar] [CrossRef]
- Jin, H.; Kagiampakis, I.; Li, P.; Liwang, P.J. Structural and functional studies of the potent anti-HIV chemokine variant P2-RANTES. Proteins Struct. Funct. Bioinform. 2010, 78, 295–308. [Google Scholar] [CrossRef]
- Lubkowski, J.; Bujacz, G.; Boque, L.; Domaille, P.J.; Handel, T.M.; Wlodawer, A. The structure of MCP-1 in two crystal forms provides a rare example of variable quaternary interactions. Nat. Struct. Biol. 1997, 4, 64–69. [Google Scholar] [CrossRef]
- Malkowski, M.G.; Wu, J.Y.; Lazar, J.B.; Johnson, P.H.; Edwards, B.F. The crystal structure of recombinant human neutrophil-activating peptide-2 (M6L) at 1.9-A resolution. J. Biol. Chem. 1995, 270, 7077–7087. [Google Scholar]
- Mayo, K.H.; Roongta, V.; Ilyina, E.; Milius, R.; Barker, S.; Quinlan, C.; La Rosa, G.; Daly, T.J. NMR solution structure of the 32-kDa platelet factor 4 ELR-motif N-terminal chimera: A symmetric tetramer. Biochemistry 1995, 34, 11399–11409. [Google Scholar] [CrossRef]
- Swaminathan, G.J.; Holloway, D.E.; Colvin, R.A.; Campanella, G.K.; Papageorgiou, A.C.; Luster, A.D.; Acharya, K.R. Crystal structures of oligomeric forms of the IP-10/CXCL10 chemokine. Structure 2003, 11, 521–532. [Google Scholar] [CrossRef]
- Young, H.; Roongta, V.; Daly, T.J.; Mayo, K.H. NMR structure and dynamics of monomeric neutrophil-activating peptide 2. Biochem. J. 1999, 338, 591–598. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.; Bancroft, D.P.; Lai, C.K.; Maione, T.E. Crystal structure of recombinant human platelet factor 4. Biochemistry 1994, 33, 8361–8366. [Google Scholar] [CrossRef]
- Shaw, J.P.; Johnson, Z.; Borlat, F.; Zwahlen, C.; Kungl, A.; Roulin, K.; Harrenga, A.; Wells, T.N.; Proudfoot, A.E. The X-ray structure of RANTES: Heparin-derived disaccharides allows the rational design of chemokine inhibitors. Structure 2004, 12, 2081–2093. [Google Scholar] [CrossRef]
- Stringer, S.E.; Gallagher, J.T. Specific binding of the chemokine platelet factor 4 to heparan sulfate. J. Biol. Chem. 1997, 272, 20508–20514. [Google Scholar] [CrossRef]
- Stringer, S.E.; Forster, M.J.; Mulloy, B.; Bishop, C.R.; Graham, G.J.; Gallagher, J.T. Characterization of the binding site on heparan sulfate for macrophage inflammatory protein 1alpha. Blood 2002, 100, 1543–1550. [Google Scholar]
- Spillmann, D.; Witt, D.; Lindahl, U. Defining the interleukin-8-binding domain of heparan sulfate. J. Biol. Chem. 1998, 273, 15487–15493. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Yu, Y.; Leary, J.A. Effects of sulfate position on heparin octasaccharide binding to CCL2 examined by tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 1114–1119. [Google Scholar] [CrossRef]
- Salmivirta, M.; Lidholt, K.; Lindahl, U. Heparan sulfate: A piece of information. FASEB J. 1996, 10, 1270–1279. [Google Scholar]
- Zimmermann, N.; Hershey, G.K.; Foster, P.S.; Rothenberg, M.E. Chemokines in asthma: Cooperative interaction between chemokines and IL-13. J. Allergy Clin. Immunol. 2003, 111, 227–243. [Google Scholar] [CrossRef]
- Crump, M.P.; Rajarathnam, K.; Kim, K.S.; Clark-Lewis, I.; Sykes, B.D. Solution structure of eotaxin, a chemokine that selectively recruits eosinophils in allergic inflammation. J. Biol. Chem. 1998, 273, 22471–22479. [Google Scholar]
- Crown, S.E.; Yu, Y.; Sweeney, M.D.; Leary, J.A.; Handel, T.M. Heterodimerization of CCR2 chemokines and regulation by glycosaminoglycan binding. J. Biol. Chem. 2006, 281, 25438–25446. [Google Scholar] [CrossRef]
- Yu, Y.; Sweeney, M.D.; Saad, O.M.; Leary, J.A. Potential inhibitors of chemokine function: analysis of noncovalent complexes of CC chemokine and small polyanionic molecules by ESI FT-ICR mass spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 524–535. [Google Scholar] [CrossRef]
- Shvartsburg, A.A.; Jarrold, M.F. An exact hard-spheres scattering model for the mobilities of polyatomic ions. Chem. Phys. Lett. 1996, 261, 86–91. [Google Scholar] [CrossRef]
- Mesleh, M.F.; Hunter, J.M.; Shvartsburg, A.A.; Schatz, G.C.; Jarrold, M.F. Structural information from ion mobility measurements: Effects of the long-range potential. J. Phys. Chem. 1996, 100, 16082–16086. [Google Scholar]
- Ninonuevo, M.R.; Leary, J.A. Ion mobility mass spectrometry coupled with rapid protein threading predictor structure prediction and collision-induced dissociation for probing chemokine conformation and stability. Anal. Chem. 2012, 84, 3208–3214. [Google Scholar] [CrossRef]
- Ruotolo, B.T.; Robinson, C.V. Aspects of native proteins are retained in vacuum. Curr. Opin. Chem. Biol. 2006, 10, 402–408. [Google Scholar] [CrossRef]
- Pukala, T.L.; Ruotolo, B.T.; Zhou, M.; Politis, A.; Stefanescu, R.; Leary, J.A.; Robinson, C.V. Subunit architecture of multiprotein assemblies determined using restraints from gas-phase measurements. Structure 2009, 17, 1235–1243. [Google Scholar] [CrossRef]
- Wang, S.C.; Politis, A.; di Bartolo, N.; Bavro, V.N.; Tucker, S.J.; Booth, P.J.; Barrera, N.P.; Robinson, C.V. Ion mobility mass spectrometry of two tetrameric membrane protein complexes reveals compact structures and differences in stability and packing. J. Am. Chem. Soc. 2010, 132, 15468–15470. [Google Scholar] [CrossRef]
- Lane, L.A.; Fernandez-Tornero, C.; Zhou, M.; Morgner, N.; Ptchelkine, D.; Steuerwald, U.; Politis, A.; Lindner, D.; Gvozdenovic, J.; Gavin, A.C.; et al. Mass spectrometry reveals stable modules in holo and apo RNA polymerases I and III. Structure 2011, 19, 90–100. [Google Scholar] [CrossRef]
- Wang, X.; Watson, C.; Sharp, J.S.; Handel, T.M.; Prestegard, J.H. Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure 2011, 19, 1138–1148. [Google Scholar] [CrossRef]
- Clore, G.M.; Appella, E.; Yamada, M.; Matsushima, K.; Gronenborn, A.M. Three-dimensional structure of interleukin 8 in solution. Biochemistry 1990, 29, 1689–1696. [Google Scholar] [CrossRef]
- Burrows, S.D.; Doyle, M.L.; Murphy, K.P.; Franklin, S.G.; White, J.R.; Brooks, I.; Mcnulty, D.E.; Scott, M.O.; Knutson, J.R.; Porter, D.; et al. Determination of the monomer-dimer equilibrium of interleukin-8 reveals it is a monomer at physiological concentrations. Biochemistry 1994, 33, 12741–12745. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schaffer, A.A.; Yu, Y.K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Schenauer, M.R.; Leary, J.A. An ion mobility-mass spectrometry investigation of monocyte chemoattractant protein-1. Int. J. Mass Spectrom. 2009, 287, 70–76. [Google Scholar] [CrossRef]
- Bleiholder, C.; Dupuis, N.F.; Wyttenbach, T.; Bowers, M.T. Ion mobility-mass spectrometry reveals a conformational conversion from random assembly to beta-sheet in amyloid fibril formation. Nat. Chem. 2011, 3, 172–177. [Google Scholar] [CrossRef]
- Crown, S.E. Characterization of Chemokine Heterodimerization and Glycosaminoglycan Interactions. Ph.D. Dissertation, University of California, Berkeley, CA, USA, 2006. [Google Scholar]
- Blaszczyk, J.; Coillie, E.V.; Proost, P.; Damme, J.V.; Opdenakker, G.; Bujacz, G.D.; Wang, J.M.; Ji, X. Complete crystal structure of monocyte chemotactic protein-2, a CC chemokine that interacts with multiple receptors. Biochemistry 2000, 39, 14075–14081. [Google Scholar] [CrossRef]
- Handel, T.M.; Domaille, P.J. Heteronuclear (1H, 13C, 15N) NMR assignments and solution structure of the monocyte chemoattractant protein-1 (MCP-1) dimer. Biochemistry 1996, 35, 6569–6584. [Google Scholar] [CrossRef]
- Nesmelova, I.V.; Sham, Y.; Dudek, A.Z.; van Eijk, L.I.; Wu, G.; Slungaard, A.; Mortari, F.; Griffioen, A.W.; Mayo, K.H. Platelet factor 4 and interleukin-8 CXC chemokine heterodimer formation modulates function at the quaternary structural level. J. Biol. Chem. 2005, 280, 4948–4958. [Google Scholar]
- Das, P.; Ziada, K.; Steinhubl, S.R.; Moliterno, D.J.; Hamdalla, H.; Jozic, J.; Mukherjee, D. Heparin-induced thrombocytopenia and cardiovascular diseases. Am. Heart J. 2006, 152, 19–26. [Google Scholar] [CrossRef]
- Savi, P.; Chong, B.H.; Greinacher, A.; Gruel, Y.; Kelton, J.G.; Warkentin, T.E.; Eichler, P.; Meuleman, D.; Petitou, M.; Herault, J.P.; et al. Effect of fondaparinux on platelet activation in the presence of heparin-dependent antibodies: A blinded comparative multicenter study with unfractionated heparin. Blood 2005, 105, 139–144. [Google Scholar] [CrossRef]
- Potekhina, A.V.; Arefieva, T.I.; Krasnikova, T.L.; Provatorov, S.I.; Masenko, V.P.; Osyaeva, M.K.; Noeva, E.A. Changes in the concentration of monocytic chemotaxic protein-1 in patients with unstable angina treated with arixtra. Bull. Exp. Biol. Med. 2011, 150, 656–658. [Google Scholar] [CrossRef]
- Shriver, Z.; Raman, R.; Venkataraman, G.; Drummond, K.; Turnbull, J.; Toida, T.; Linhardt, R.; Biemann, K.; Sasisekharan, R. Sequencing of 3-O sulfate containing heparin decasaccharides with a partial antithrombin III binding site. Proc. Natl. Acad. Sci. USA 2000, 97, 10359–10364. [Google Scholar] [CrossRef]
- Pringle, S.D.; Giles, K.; Wildgoose, J.L.; Williams, J.P.; Slade, S.E.; Thalassinos, K.; Bateman, R.H.; Bowers, M.T.; Scrivens, J.H. An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/oa-ToF instrument. Int. J. Mass Spectrom. 2007, 261, 1–12. [Google Scholar]
- Thalassinos, K.; Grabenauer, M.; Slade, S.E.; Hilton, G.R.; Bowers, M.T.; Scrivens, J.H. Characterization of phosphorylated peptides using traveling wave-based and drift cell ion mobility mass spectrometry. Anal. Chem. 2009, 81, 248–254. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dykstra, A.B.; Sweeney, M.D.; Leary, J.A. Structural Evidence for the Tetrameric Assembly of Chemokine CCL11 and the Glycosaminoglycan Arixtra™. Biomolecules 2013, 3, 905-922. https://doi.org/10.3390/biom3040905
Dykstra AB, Sweeney MD, Leary JA. Structural Evidence for the Tetrameric Assembly of Chemokine CCL11 and the Glycosaminoglycan Arixtra™. Biomolecules. 2013; 3(4):905-922. https://doi.org/10.3390/biom3040905
Chicago/Turabian StyleDykstra, Andrew B., Matt D. Sweeney, and Julie A. Leary. 2013. "Structural Evidence for the Tetrameric Assembly of Chemokine CCL11 and the Glycosaminoglycan Arixtra™" Biomolecules 3, no. 4: 905-922. https://doi.org/10.3390/biom3040905
APA StyleDykstra, A. B., Sweeney, M. D., & Leary, J. A. (2013). Structural Evidence for the Tetrameric Assembly of Chemokine CCL11 and the Glycosaminoglycan Arixtra™. Biomolecules, 3(4), 905-922. https://doi.org/10.3390/biom3040905