Protein Stability, Folding and Misfolding in Human PGK1 Deficiency



Abstract
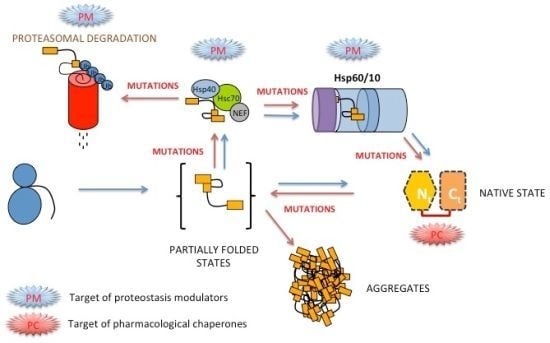
:
1. Introduction
2. Human PGK1 Deficiency
2.1. Clinical Presentation and Current Treatments
2.2. Genetics and Genotype/Phenotype Correlations in hPGK1 Deficiency
3. Functional and Stability Defects in Human PGK1 Deficiency
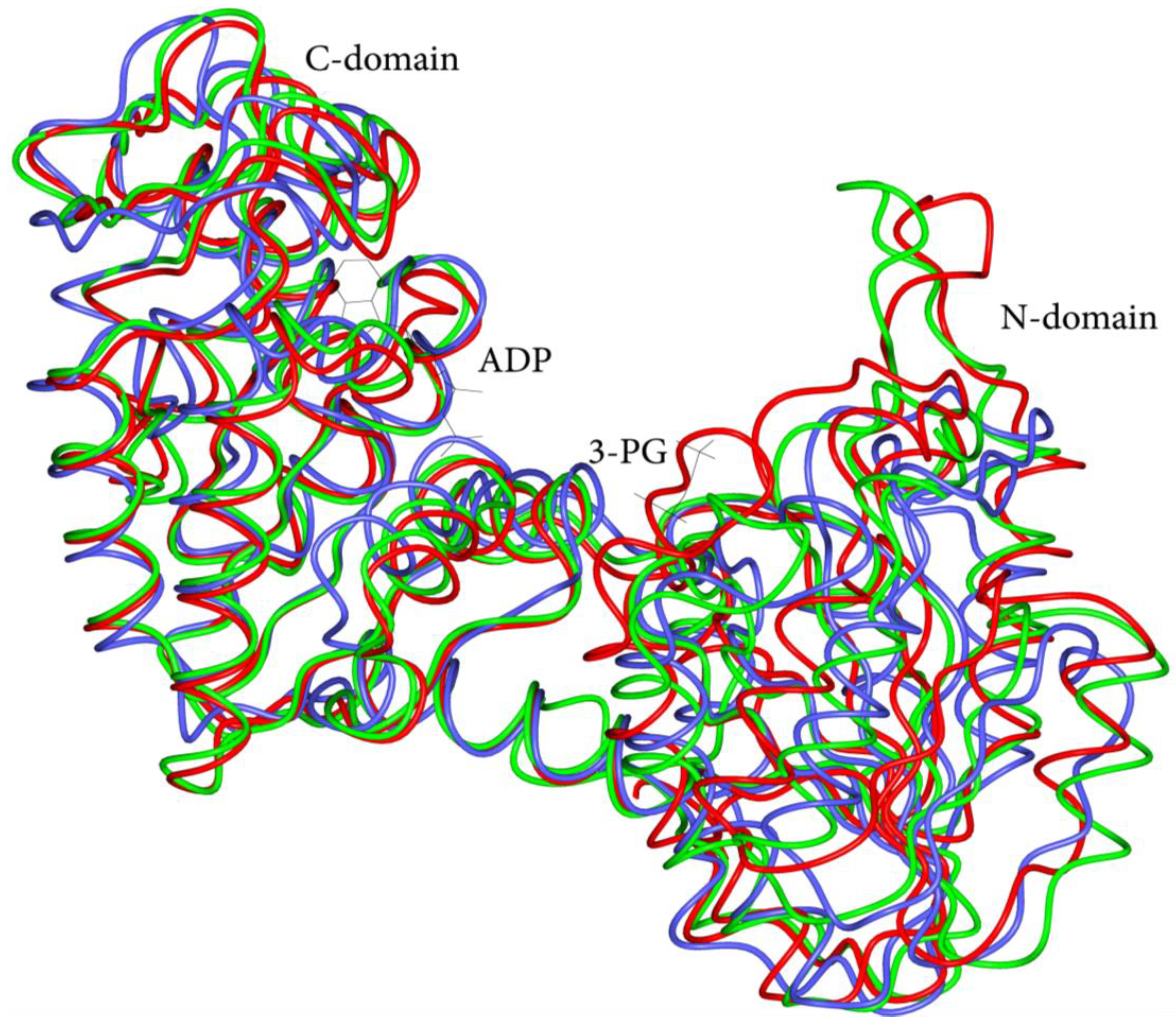
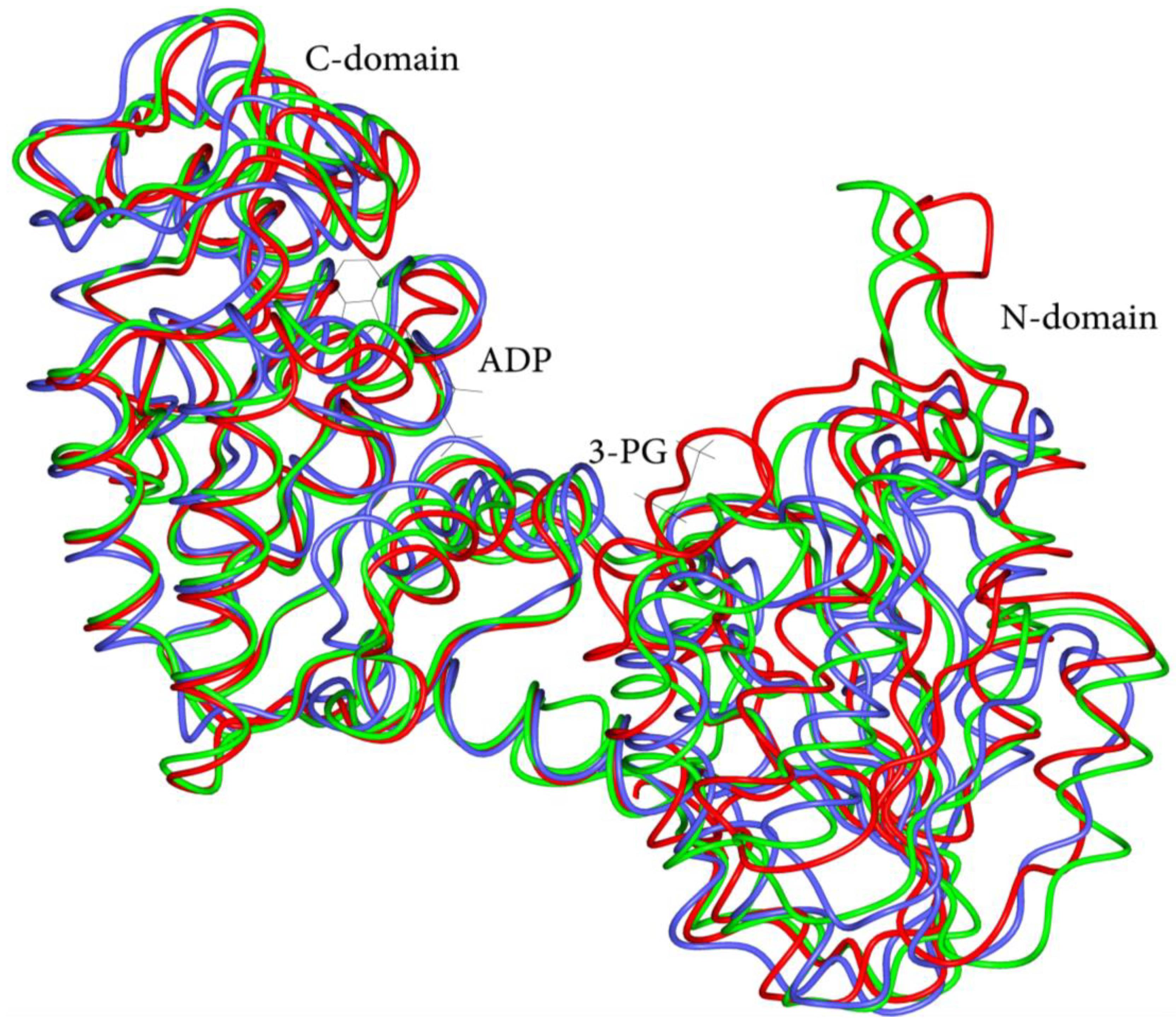
3.1. Overview of Human PGK1 Structure and Activity
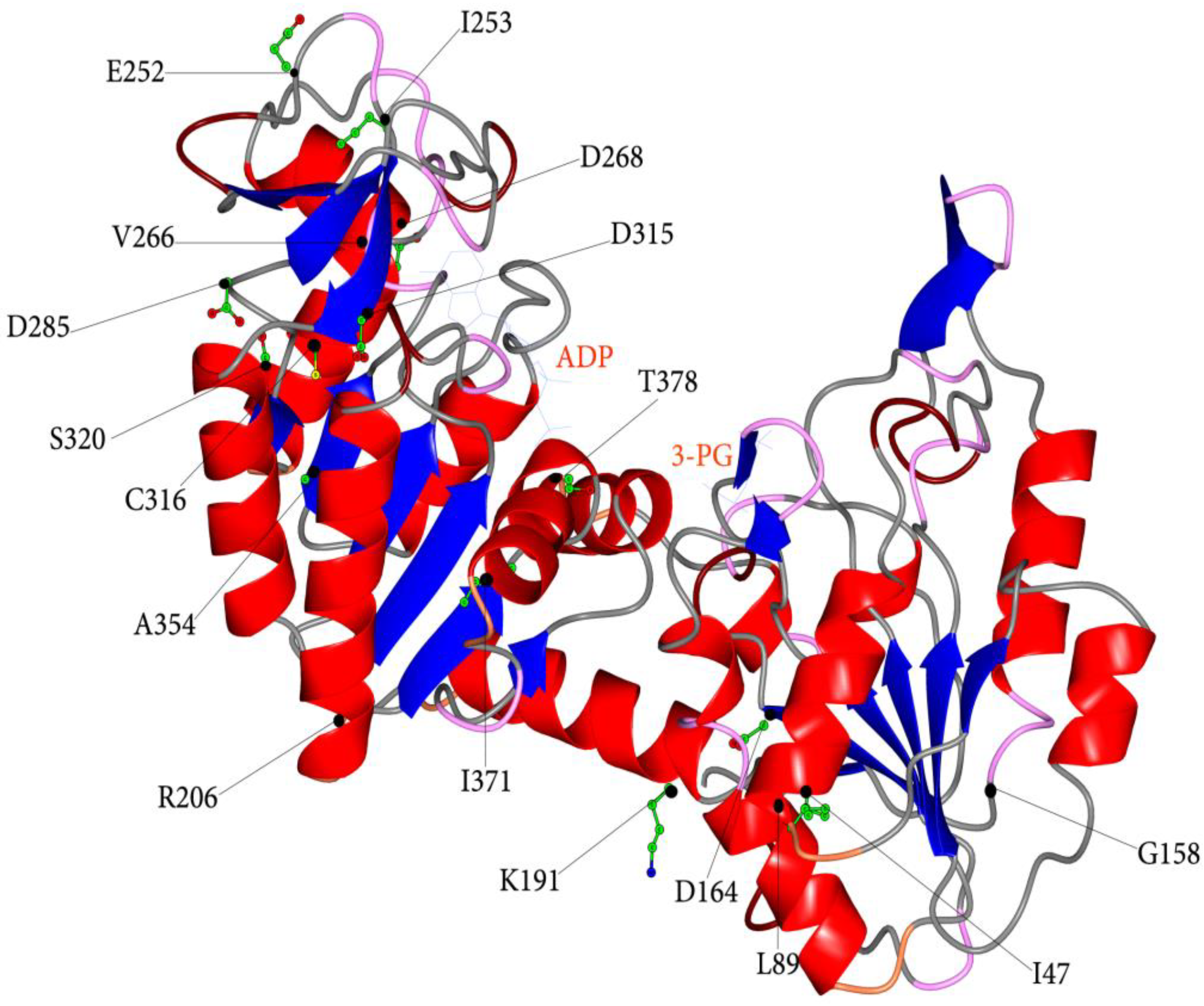
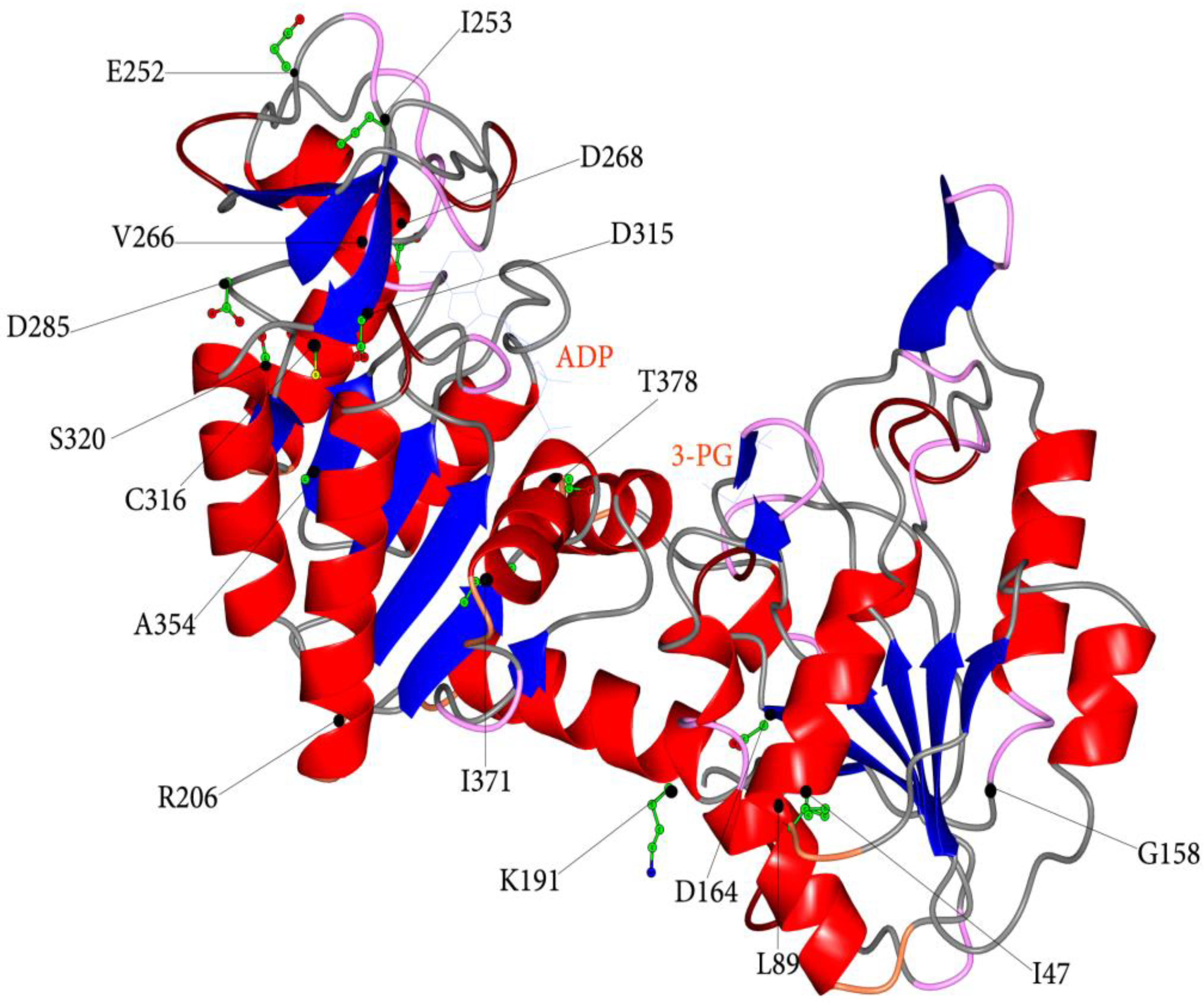
3.2. Structural and Functional Impact of Mutations in the Native hPGK1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Change | Amino Acid Change | a Localization | b Catalytic Properties | c Protein Stability | RBC Residual Activity (%) | Hb (g/dl) | Reticulocytes (%) | d Symptoms | References | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | M | N | |||||||||
| c.140 T > A | # p.I47N | α-helix 1b | ○ | ○○○ | 8 | 6.6–7.3 | N.A. | + | - | + | [37] |
| c.266 T > C | # p.L89P | α-helix 2 * | ○ | ○○○ | 5 | N.A. | N.A. | + | - | + | [40] |
| c.473 G > T | # p.G158V | loop α-helix 4, β-strand E | ○○ | ○○ | 1 | 12.8 | 2.5 | - | + | - | [65] |
| c.491 A > T | # p.D164V | β-strand E | ○○○ | ○○○ | 5 | 2.0–10.0 | 5.0-26-0 | + | +/- | + | [36,38,41,66] |
| c.571 > 573 del | # p.K191del | α-helix 7 * | ○○ | ○○ | 4 | 14.1 | 6.4 | - | - | - | [41] |
| c.617 G > C | # p.R206P | loop α-helix 7, β-strand G * | - | ○ | 10 | 5.6–13.7 | 2.0–20.0 | +/- | - | + | [67] |
| c.755 A > C | # p.E252A | loop α-helix 9, α-helix 10 * | - | - | 6 | 13.2 | N.A. | - | + | - | [68] |
| c.758 T > C | # p.I253T | loop α-helix 9, α-helix 10 * | - | - | 8 | N.A. | N.A. | - | + | + | [69] |
| c.796 G > A c.798 C > A | # p.V266M | α-helix 10a/b | - | - | 10 | 9.3 | 12.5 | + | - | + | [70] |
| c.802 G > A | # p.D268N | α-helix 10b * | - | - | 21 | N.A. | 0.4–1.3 | - | - | - | [71] |
| c.854 A > T | # p.D285V | β-strand o * | ○○ | ○○○ | 49 | 9.0–10.0 | 10–45 | - | - | - | [68] |
| c.943 G > A | # p.D315N | β-strand q | ○○ | ○○○ | 3 | 14.3 | N.A. | - | + | - | [72] |
| c.946 T > C | # p.C316R | β-strand q | ○ | ○○○ | 10 | 7.5-13.0 | 1.5–5.0 | +/- | - | + | [66] |
| c.959 G > A | # p.S320N | α-helix 11 | ○ | ○○○ | 36 | 7.6 | 9.0 | + | - | + | [73] |
| c.1060 G > C | # p.A354P | α-helix 12 * | ○ | ○○○ | 6 | 4.9–9.0 | 24 | + | + | + | [37] |
| c.1112 T > A | # p.I371K | β-strand K | ○○ | ○○ | 12 | 12.1–14.1 | 4.4–5.2 | +/- | + | + | [30] |
| c.1132 A > C | # p.T378P | α-helix 13 * | ○○ | - | 2 | 13.4–14.5 | N.A. | - | + | - | [28,29] |
| IVS4+1 G > T | splicing alteration | 3 | N.A. | 2.7 | - | + | + | [74] | |||
| c.637 > 640 delGGCG | frameshift | 6 | N.A. | N.A. | - | + | - | [75] | |||
| c.639 C > T | splicing alteration | 5 | N.A. | N.A. | - | + | - | [76,77] | |||
| IVS7+5 G > A | splicing alteration | 14 | N.A. | N.A. | - | + | + | [78] | |||
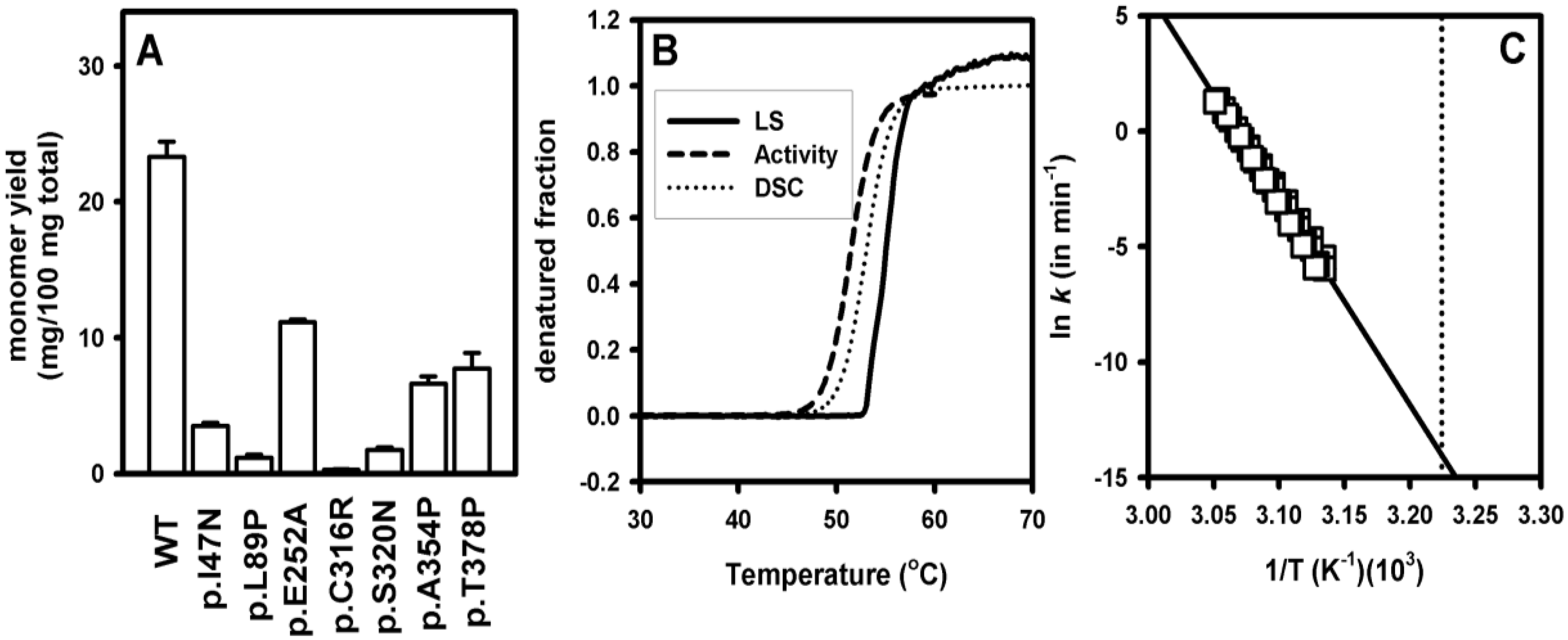
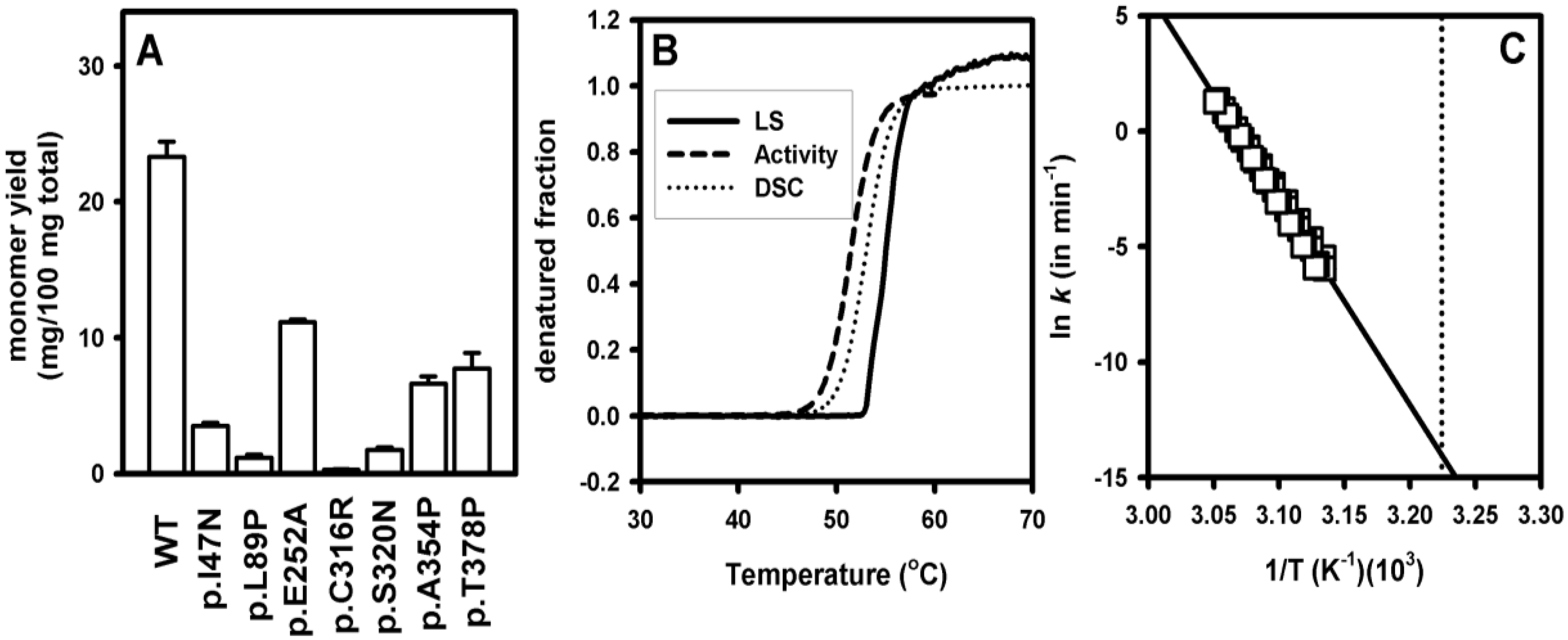
3.3. Increased Aggregation Propensity of Mutants Causing hPGK1 Deficiency: Biophysical and Expression Studies
3.4. Structural and Energetic Bases of Mutation Induced Kinetic Destabilization of hPGK1
| Chemical Denaturation | WT | p.T378P |
|---|---|---|
| Cm (M) | 2.43 ± 0.01 | 2.28 ± 0.06 |
| meq (kcal·mol−1·M−1) | 3.4 ± 0.2 | 1.5 ± 0.2 |
| ΔGU (kcal·mol−1) | 8.3 ± 0.5 | 3.5 ± 0.4 |
| Thermal denaturation | ||
| Tm (°C) | 52.8 ± 0.2 | 49.8 ± 0.4 |
| Ea (kcal·mol−1) | 191 ± 19 | 135 ± 12 |
| ΔH (kcal·mol−1) | 157 ± 18 | 127 ± 9 |
| Kinetic stability | ||
| Aggregation rate constant (kagg)(min−1) | 9 ± 5·10−7 | 1.6 ± 0.4·10−4 |
| Global unfolding rate constant (kunf(0M)) (min−1) | 0.09 ± 0.02 | 0.18 ± 0.04 |
| Proteolysis rate constant at high protease (k0) (min−1) | 0.11 ± 0.02 | 0.38 ± 0.01 |
| Proteolysis rate constant at low protease (k1) (min−1) | 0.27 ± 0.04 | 7.8 ± 0.5 |
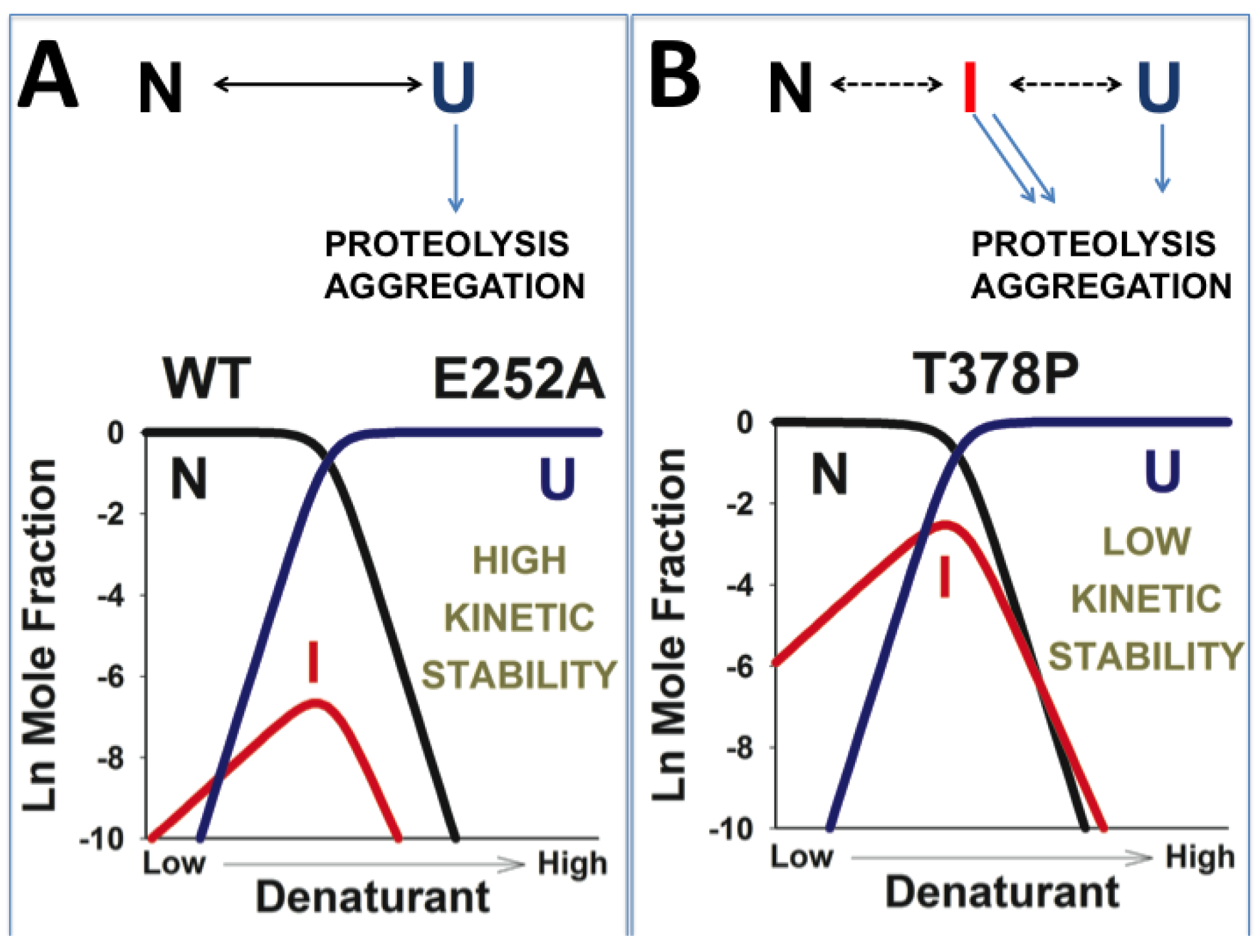
3.5. Dissection of the Thermodynamic and Kinetic Basis of hPGK1 Misfolding by Urea Denaturation and Proteolysis
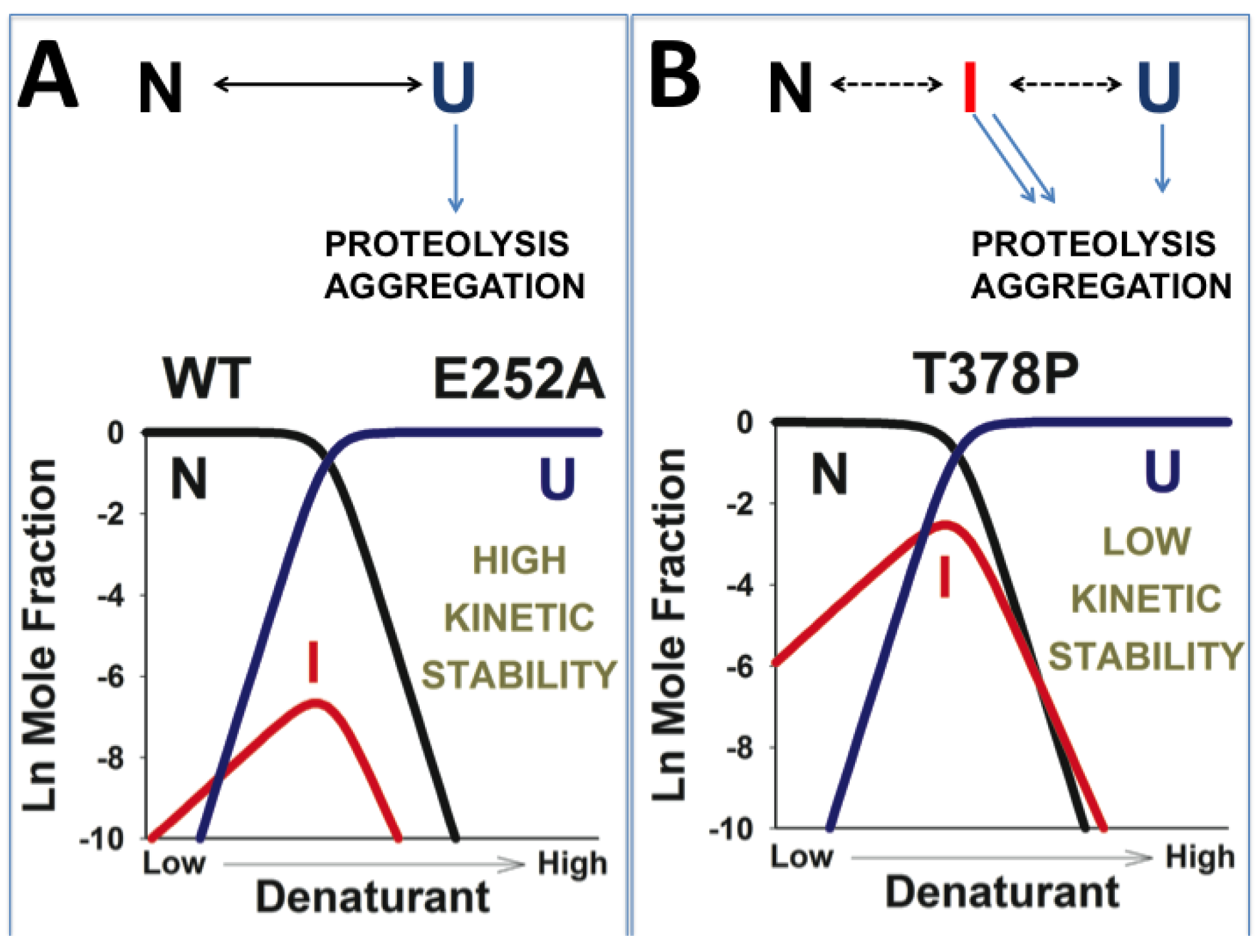
3.6. A Possible Role of Folding/Unfolding Cooperativity in the Loss-of-Function Mechanisms of hPGK1 Deficiency
3.7. hPGK1 Mutants may affect Spontaneous Refolding Kinetics
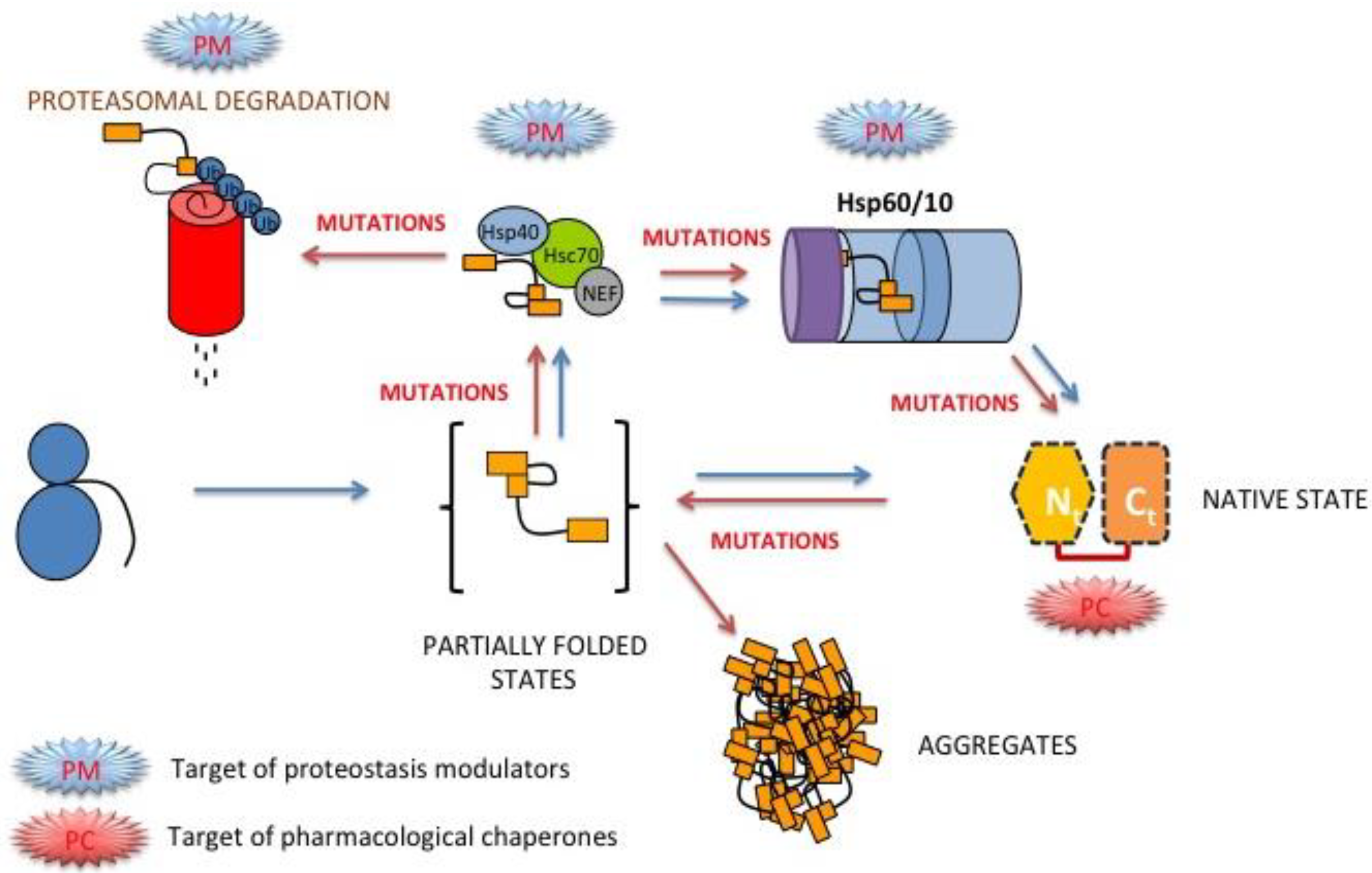
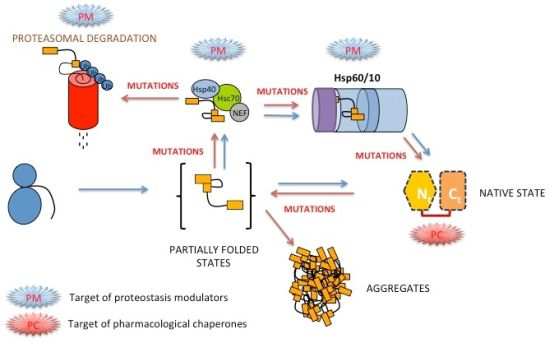
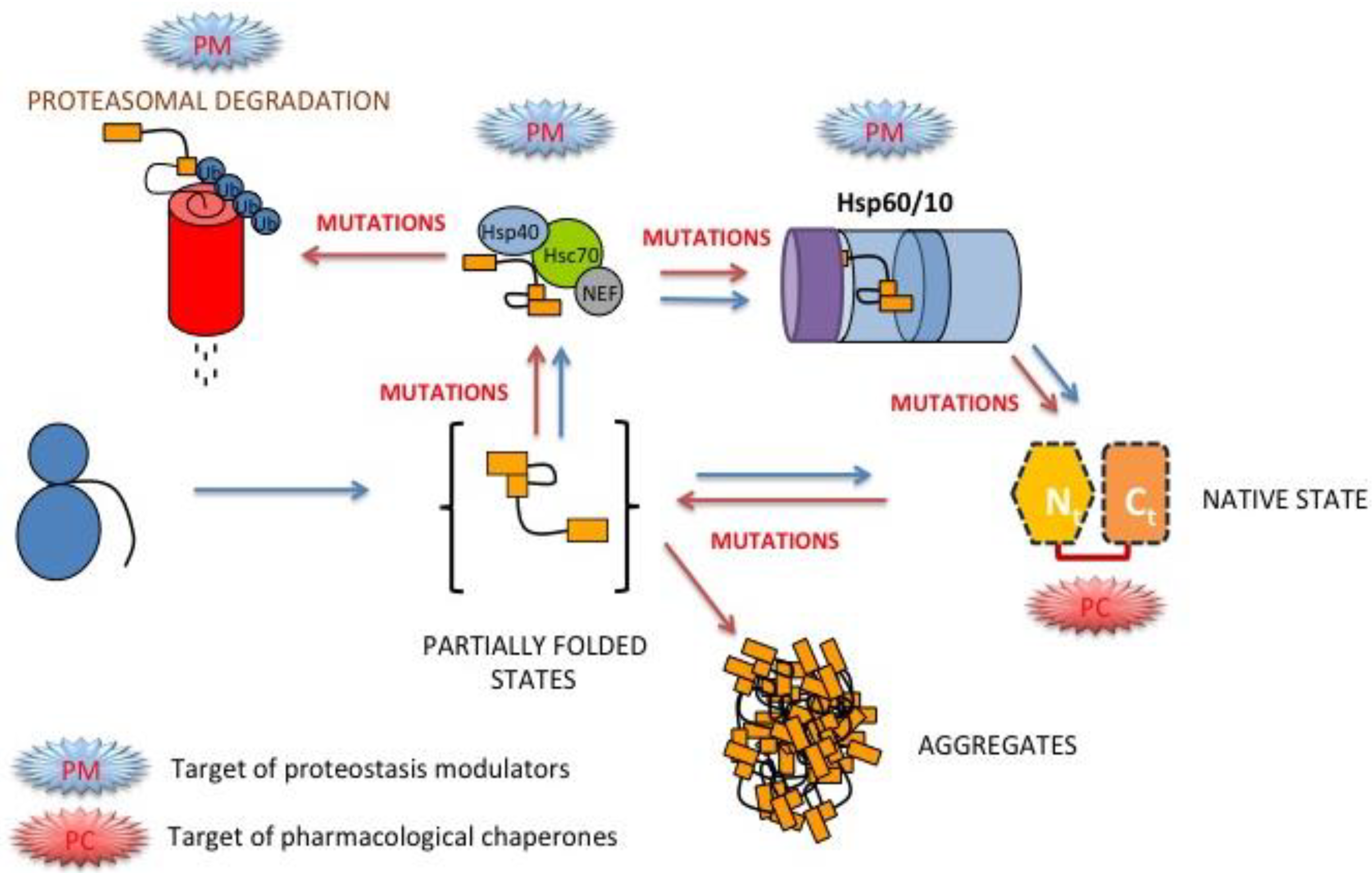
3.8. Folding, Stability and Function of PGK inside the Cell
4. Perspectives for Pharmacological Intervention in hPGK1 Deficiency
Acknowledgments
Conflicts of Interest
References
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol 2009, 16, 574–581. [Google Scholar] [CrossRef]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Gomes, C.M. Protein misfolding in disease and small molecule therapies. Curr. Top. Med. Chem. 2012, 12, 2460–2469. [Google Scholar] [CrossRef]
- Underhaug, J.; Aubi, O.; Martinez, A. Phenylalanine hydroxylase misfolding and pharmacological chaperones. Curr. Top. Med. Chem. 2012, 12, 2534–2545. [Google Scholar] [CrossRef]
- Rodrigues, J.V.; Henriques, B.J.; Lucas, T.G.; Gomes, C.M. Cofactors and metabolites as protein folding helpers in metabolic diseases. Curr. Top. Med. Chem. 2012, 12, 2546–2559. [Google Scholar]
- Ong, D.S.; Kelly, J.W. Chemical and/or biological therapeutic strategies to ameliorate protein misfolding diseases. Curr. Opin. Cell. Biol. 2011, 23, 231–238. [Google Scholar] [CrossRef]
- Beutler, E. PGK deficiency. Br. J. Haematol. 2007, 136, 3–11. [Google Scholar] [CrossRef]
- Willard, H.F.; Goss, S.J.; Holmes, M.T.; Munroe, D.L. Regional localization of the phosphoglycerate kinase gene and pseudogene on the human X chromosome and assignment of a related DNA sequence to chromosome 19. Hum. Genet. 1985, 71, 138–143. [Google Scholar]
- McCarrey, J.R.; Thomas, K. Human testis-specific PGK gene lacks introns and possesses characteristics of a processed gene. Nature 1987, 326, 501–505. [Google Scholar] [CrossRef]
- Cliff, M.J.; Bowler, M.W.; Varga, A.; Marston, J.P.; Szabo, J.; Hounslow, A.M.; Baxter, N.J.; Blackburn, G.M.; Vas, M.; Waltho, J.P. Transition state analogue structures of human phosphoglycerate kinase establish the importance of charge balance in catalysis. J. Am. Chem. Soc. 2010, 132, 6507–6516. [Google Scholar] [CrossRef]
- Pey, A.L.; Mesa-Torres, N.; Chiarelli, L.R.; Valentini, G. Structural and Energetic Basis of Protein Kinetic Destabilization in Human Phosphoglycerate Kinase 1 Deficiency. Biochemistry 2013, 52, 1160–1170. [Google Scholar] [CrossRef]
- Yon, J.M.; Desmadril, M.; Betton, J.M.; Minard, P.; Ballery, N.; Missiakas, D.; Gaillard-Miran, S.; Perahia, D.; Mouawad, L. Flexibility and folding of phosphoglycerate kinase. Biochimie 1990, 72, 417–429. [Google Scholar] [CrossRef]
- Young, T.A.; Skordalakes, E.; Marqusee, S. Comparison of proteolytic susceptibility in phosphoglycerate kinases from yeast and E. coli: Modulation of conformational ensembles without altering structure or stability. J. Mol. Biol. 2007, 368, 1438–1447. [Google Scholar] [CrossRef]
- Grattinger, M.; Dankesreiter, A.; Schurig, H.; Jaenicke, R. Recombinant phosphoglycerate kinase from the hyperthermophilic bacterium Thermotoga maritima: Catalytic, spectral and thermodynamic properties. J. Mol. Biol. 1998, 280, 525–533. [Google Scholar] [CrossRef]
- Semisotnov, G.V.; Vas, M.; Chemeris, V.V.; Kashparova, N.J.; Kotova, N.V.; Razgulyaev, O.I.; Sinev, M.A. Refolding kinetics of pig muscle and yeast 3-phosphoglycerate kinases and of their proteolytic fragments. Eur J. Biochem. 1991, 202, 1083–1089. [Google Scholar] [CrossRef]
- Szilagyi, A.N.; Kotova, N.V.; Semisotnov, G.V.; Vas, M. Incomplete refolding of a fragment of the N-terminal domain of pig muscle 3-phosphoglycerate kinase that lacks a subdomain. Comparison with refolding of the complementary C-terminal fragment. Eur. J. Biochem. 2001, 268, 1851–1860. [Google Scholar] [CrossRef]
- Szilagyi, A.N.; Vas, M. Sequential domain refolding of pig muscle 3-phosphoglycerate kinase: Kinetic analysis of reactivation. Fold. Des. 1998, 3, 565–575. [Google Scholar] [CrossRef]
- Vas, M.; Sinev, M.A.; Kotova, N.V.; Semisotnov, G.V. Reactivation of 3-phosphoglycerate kinase from its unfolded proteolytic fragments. Eur. J. Biochem. 1990, 189, 575–579. [Google Scholar] [CrossRef]
- Galisteo, M.L.; Mateo, P.L.; Sanchez-Ruiz, J.M. Kinetic study on the irreversible thermal denaturation of yeast phosphoglycerate kinase. Biochemistry 1991, 30, 2061–2066. [Google Scholar] [CrossRef]
- Missiakas, D.; Betton, J.M.; Chaffotte, A.; Minard, P.; Yon, J.M. Kinetic studies of the refolding of yeast phosphoglycerate kinase: Comparison with the isolated engineered domains. Protein Sci. 1992, 1, 1485–1493. [Google Scholar] [CrossRef]
- Sabelko, J.; Ervin, J.; Gruebele, M. Observation of strange kinetics in protein folding. Proc. Natl. Acad. Sci. USA 1999, 96, 6031–6036. [Google Scholar] [CrossRef]
- Dhar, A.; Ebbinghaus, S.; Shen, Z.; Mishra, T.; Gruebele, M. The diffusion coefficient for PGK folding in eukaryotic cells. Biophys. J. 2010, 99, L69–L71. [Google Scholar] [CrossRef]
- Dhar, A.; Samiotakis, A.; Ebbinghaus, S.; Nienhaus, L.; Homouz, D.; Gruebele, M.; Cheung, M.S. Structure, function, and folding of phosphoglycerate kinase are strongly perturbed by macromolecular crowding. Proc. Natl. Acad. Sci. USA 2010, 107, 17586–17591. [Google Scholar] [CrossRef]
- Ebbinghaus, S.; Dhar, A.; McDonald, J.D.; Gruebele, M. Protein folding stability and dynamics imaged in a living cell. Nat. Methods 2010, 7, 319–323. [Google Scholar] [CrossRef]
- Fermo, E.; Bianchi, P.; Chiarelli, L.R.; Maggi, M.; Mandara, G.M.; Vercellati, C.; Marcello, A.P.; Barcellini, W.; Cortelezzi, A.; Valentini, G.; et al. A new variant of phosphoglycerate kinase deficiency (p.I371K) with multiple tissue involvement: Molecular and functional characterization. Mol. Genet. Metab 2012, 106, 455–461. [Google Scholar] [CrossRef]
- Sotiriou, E.; Greene, P.; Krishna, S.; Hirano, M.; DiMauro, S. Myopathy and parkinsonism in phosphoglycerate kinase deficiency. Muscle Nerve 2010, 41, 707–710. [Google Scholar]
- Spiegel, R.; Gomez, E.A.; Akman, H.O.; Krishna, S.; Horovitz, Y.; DiMauro, S. Myopathic form of phosphoglycerate kinase (PGK) deficiency: A new case and pathogenic considerations. Neuromuscul. Disord. 2009, 19, 207–211. [Google Scholar] [CrossRef]
- Morimoto, A.; Ueda, I.; Hirashima, Y.; Sawai, Y.; Usuku, T.; Kano, G.; Kuriyama, K.; Todo, S.; Sugimoto, T.; Kanno, H.; et al. A novel missense mutation (1060G --> C) in the phosphoglycerate kinase gene in a Japanese boy with chronic haemolytic anaemia, developmental delay and rhabdomyolysis. Br. J. Haematol. 2003, 122, 1009–1013. [Google Scholar] [CrossRef]
- Valentine, W.N.; Paglia, D.E. Erythrocyte enzymopathies, hemolytic anemia, and multisystem disease: An annotated review. Blood 1984, 64, 583–591. [Google Scholar]
- Berardo, A.; DiMauro, S.; Hirano, M. A diagnostic algorithm for metabolic myopathies. Curr. Neurol. Neurosci. Rep. 2010, 10, 118–126. [Google Scholar] [CrossRef]
- Climent, F.; Roset, F.; Repiso, A.; Perez de la Ossa, P. Red cell glycolytic enzyme disorders caused by mutations: An update. Cardiovasc. Hematol. Disord. Drug Targets 2009, 9, 95–106. [Google Scholar] [CrossRef]
- Parekh, R.; Care, D.A.; Tainter, C.R. Rhabdomyolysis: Advances in diagnosis and treatment. Emerg. Med. Pract. 2012, 14, 1–15, quiz 15. [Google Scholar]
- Volpi, L.; Ricci, G.; Orsucci, D.; Alessi, R.; Bertolucci, F.; Piazza, S.; Simoncini, C.; Mancuso, M.; Siciliano, G. Metabolic myopathies: Functional evaluation by different exercise testing approaches. Musculoskelet. Surg. 2011, 95, 59–67. [Google Scholar]
- Flanagan, J.M.; Rhodes, M.; Wilson, M.; Beutler, E. The identification of a recurrent phosphoglycerate kinase mutation associated with chronic haemolytic anaemia and neurological dysfunction in a family from USA. Br. J. Haematol 2006, 134, 233–237. [Google Scholar] [CrossRef]
- Noel, N.; Flanagan, J.M.; Ramirez Bajo, M.J.; Kalko, S.G.; Manu Mdel, M.; Garcia Fuster, J.L.; Perez de la Ossa, P.; Carreras, J.; Beutler, E.; Vives Corrons, J.L. Two new phosphoglycerate kinase mutations associated with chronic haemolytic anaemia and neurological dysfunction in two patients from Spain. Br. J. Haematol. 2006, 132, 523–529. [Google Scholar]
- Rhodes, M.; Ashford, L.; Manes, B.; Calder, C.; Domm, J.; Frangoul, H. Bone marrow transplantation in phosphoglycerate kinase (PGK) deficiency. Br. J. Haematol. 2011, 152, 500–502. [Google Scholar] [CrossRef]
- Fujii, H.; Miwa, S. Other erythrocyte enzyme deficiencies associated with non-haematological symptoms: Phosphoglycerate kinase and phosphofructokinase deficiency. Baillieres Best Pract. Res. Clin. Haematol. 2000, 13, 141–148. [Google Scholar] [CrossRef]
- Maeda, M.; Yoshida, A. Molecular defect of a phosphoglycerate kinase variant (PGK-Matsue) associated with hemolytic anemia: Leu----Pro substitution caused by T/A----C/G transition in exon 3. Blood 1991, 77, 1348–1352. [Google Scholar]
- Turner, G.; Fletcher, J.; Elber, J.; Yanagawa, Y.; Dave, V.; Yoshida, A. Molecular defect of a phosphoglycerate kinase variant associated with haemolytic anaemia and neurological disorders in a large kindred. Br. J. Haematol. 1995, 91, 60–65. [Google Scholar] [CrossRef]
- Michelson, A.M.; Blake, C.C.; Evans, S.T.; Orkin, S.H. Structure of the human phosphoglycerate kinase gene and the intron-mediated evolution and dispersal of the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 1985, 82, 6965–6969. [Google Scholar] [CrossRef]
- Kraus, A.P.; Langston, M.F., Jr.; Lynch, B.L. Red cell phosphoglycerate kinase deficiency. A new cause of non-spherocytic hemolytic anemia. Biochem. Biophys. Res. Commun. 1968, 30, 173–177. [Google Scholar] [CrossRef]
- Valentine, W.N. Hereditary hemolytic anemias associated with specific erythrocyte enzymopathies. Calif. Med. 1968, 108, 280–294. [Google Scholar]
- Chiarelli, L.R.; Morera, S.M.; Bianchi, P.; Fermo, E.; Zanella, A.; Galizzi, A.; Valentini, G. Molecular insights on pathogenic effects of mutations causing phosphoglycerate kinase deficiency. PLoS One 2012, 7, e32065. [Google Scholar]
- Pey, A.L. The interplay between protein stability and dynamics in conformational diseases: The case of hPGK1 deficiency. Biochim. Biophys. Acta 2013, 1834, 2502–2511. [Google Scholar] [CrossRef]
- Zieker, D.; Konigsrainer, I.; Weinreich, J.; Beckert, S.; Glatzle, J.; Nieselt, K.; Buhler, S.; Loffler, M.; Gaedcke, J.; Northoff, H.; et al. Phosphoglycerate kinase 1 promoting tumor progression and metastasis in gastric cancer - detected in a tumor mouse model using positron emission tomography/magnetic resonance imaging. Cell. Physiol. Biochem. 2010, 26, 147–154. [Google Scholar] [CrossRef]
- Lay, A.J.; Jiang, X.M.; Kisker, O.; Flynn, E.; Underwood, A.; Condron, R.; Hogg, P.J. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature 2000, 408, 869–873. [Google Scholar] [CrossRef]
- Ogino, T.; Iwama, M.; Kinouchi, J.; Shibagaki, Y.; Tsukamoto, T.; Mizumoto, K. Involvement of a cellular glycolytic enzyme, phosphoglycerate kinase, in Sendai virus transcription. J. Biol. Chem. 1999, 274, 35999–36008. [Google Scholar] [CrossRef]
- Popanda, O.; Fox, G.; Thielmann, H.W. Modulation of DNA polymerases alpha, delta and epsilon by lactate dehydrogenase and 3-phosphoglycerate kinase. Biochim. Biophys. Acta 1998, 1397, 102–117. [Google Scholar] [CrossRef]
- Myre, M.A.; O'Day, D.H. Calmodulin binds to and inhibits the activity of phosphoglycerate kinase. Biochim. Biophys. Acta 2004, 1693, 177–183. [Google Scholar] [CrossRef]
- Gallois-Montbrun, S.; Faraj, A.; Seclaman, E.; Sommadossi, J.P.; Deville-Bonne, D.; Veron, M. Broad specificity of human phosphoglycerate kinase for antiviral nucleoside analogs. Biochem. Pharmacol. 2004, 68, 1749–1756. [Google Scholar] [CrossRef]
- Krishnan, P.; Fu, Q.; Lam, W.; Liou, J.Y.; Dutschman, G.; Cheng, Y.C. Phosphorylation of pyrimidine deoxynucleoside analog diphosphates: Selective phosphorylation of L-nucleoside analog diphosphates by 3-phosphoglycerate kinase. J. Biol. Chem. 2002, 277, 5453–5459. [Google Scholar] [CrossRef]
- Mathe, C.; Gosselin, G. L-nucleoside enantiomers as antivirals drugs: A mini-review. Antivir. Res. 2006, 71, 276–281. [Google Scholar] [CrossRef]
- Gondeau, C.; Chaloin, L.; Lallemand, P.; Roy, B.; Perigaud, C.; Barman, T.; Varga, A.; Vas, M.; Lionne, C.; Arold, S.T. Molecular basis for the lack of enantioselectivity of human 3-phosphoglycerate kinase. Nucleic Acids Res. 2008, 36, 3620–3629. [Google Scholar] [CrossRef]
- Varga, A.; Chaloin, L.; Sagi, G.; Sendula, R.; Graczer, E.; Liliom, K.; Zavodszky, P.; Lionne, C.; Vas, M. Nucleotide promiscuity of 3-phosphoglycerate kinase is in focus: Implications for the design of better anti-HIV analogues. Mol. Biosyst. 2011, 7, 1863–1873. [Google Scholar] [CrossRef]
- Varga, A.; Szabo, J.; Flachner, B.; Roy, B.; Konarev, P.; Svergun, D.; Zavodszky, P.; Perigaud, C.; Barman, T.; Lionne, C.; et al. Interaction of human 3-phosphoglycerate kinase with L-ADP, the mirror image of D-ADP. Biochem. Biophys. Res. Commun. 2008, 366, 994–1000. [Google Scholar] [CrossRef]
- Szilagyi, A.N.; Ghosh, M.; Garman, E.; Vas, M. A 1.8 A resolution structure of pig muscle 3-phosphoglycerate kinase with bound MgADP and 3-phosphoglycerate in open conformation: New insight into the role of the nucleotide in domain closure. J. Mol. Biol. 2001, 306, 499–511. [Google Scholar] [CrossRef]
- Palmai, Z.; Chaloin, L.; Lionne, C.; Fidy, J.; Perahia, D.; Balog, E. Substrate binding modifies the hinge bending characteristics of human 3-phosphoglycerate kinase: A molecular dynamics study. Proteins 2009, 77, 319–329. [Google Scholar]
- Vas, M.; Varga, A.; Graczer, E. Insight into the mechanism of domain movements and their role in enzyme function: Example of 3-phosphoglycerate kinase. Curr. Protein Pept. Sci. 2010, 11, 118–147. [Google Scholar] [CrossRef]
- Ritco-Vonsovici, M.; Mouratou, B.; Minard, P.; Desmadril, M.; Yon, J.M.; Andrieux, M.; Leroy, E.; Guittet, E. Role of the C-terminal helix in the folding and stability of yeast phosphoglycerate kinase. Biochemistry 1995, 34, 833–841. [Google Scholar] [CrossRef]
- Varga, A.; Flachner, B.; Graczer, E.; Osvath, S.; Szilagyi, A.N.; Vas, M. Correlation between conformational stability of the ternary enzyme-substrate complex and domain closure of 3-phosphoglycerate kinase. FEBS J. 2005, 272, 1867–1885. [Google Scholar] [CrossRef]
- Kiefhaber, T.; Schmid, F.X. Kinetic coupling between protein folding and prolyl isomerization. II. Folding of ribonuclease A and ribonuclease T1. J. Mol. Biol. 1992, 224, 217–229. [Google Scholar] [CrossRef]
- Schmid, F.X.; Grafl, R.; Wrba, A.; Beintema, J.J. Role of proline peptide bond isomerization in unfolding and refolding of ribonuclease. Proc. Natl. Acad. Sci. USA 1986, 83, 872–876. [Google Scholar] [CrossRef]
- Fujii, H.; Kanno, H.; Hirono, A.; Shiomura, T.; Miwa, S. A single amino acid substitution (157 Gly----Val) in a phosphoglycerate kinase variant (PGK Shizuoka) associated with chronic hemolysis and myoglobinuria. Blood 1992, 79, 1582–1585. [Google Scholar]
- Cohen-Solal, M.; Valentin, C.; Plassa, F.; Guillemin, G.; Danze, F.; Jaisson, F.; Rosa, R. Identification of new mutations in two phosphoglycerate kinase (PGK) variants expressing different clinical syndromes: PGK Creteil and PGK Amiens. Blood 1994, 84, 898–903. [Google Scholar]
- Hjelm, M.; Wadam, B.; Yoshida, A. A phosphoglycerate kinase variant, PGK Uppsala, associated with hemolytic anemia. J. Lab. Clin. Med. 1980, 96, 1015–1021. [Google Scholar]
- Fujii, H.; Yoshida, A. Molecular abnormality of phosphoglycerate kinase-Uppsala associated with chronic nonspherocytic hemolytic anemia. Proc. Natl. Acad. Sci. USA 1980, 77, 5461–5465. [Google Scholar] [CrossRef]
- Ookawara, T.; Dave, V.; Willems, P.; Martin, J.J.; de Barsy, T.; Matthys, E.; Yoshida, A. Retarded and aberrant splicings caused by single exon mutation in a phosphoglycerate kinase variant. Arch. Biochem. Biophys. 1996, 327, 35–40. [Google Scholar] [CrossRef]
- Sugie, H.; Sugie, Y.; Ito, M.; Fukuda, T. A novel missense mutation (837T-->C) in the phosphoglycerate kinase gene of a patient with a myopathic form of phosphoglycerate kinase deficiency. J. Child. Neurol. 1998, 13, 95–97. [Google Scholar] [CrossRef]
- Fujii, H.; Chen, S.H.; Akatsuka, J.; Miwa, S.; Yoshida, A. Use of cultured lymphoblastoid cells for the study of abnormal enzymes: Molecular abnormality of a phosphoglycerate kinase variant associated with hemolytic anemia. Proc. Natl. Acad. Sci. USA 1981, 78, 2587–2590. [Google Scholar] [CrossRef]
- Valentin, C.; Birgens, H.; Craescu, C.T.; Brodum-Nielsen, K.; Cohen-Solal, M. A phosphoglycerate kinase mutant (PGK Herlev; D285V) in a Danish patient with isolated chronic hemolytic anemia: Mechanism of mutation and structure-function relationships. Hum. Mutat. 1998, 12, 280–287. [Google Scholar] [CrossRef]
- Maeda, M.; Bawle, E.V.; Kulkarni, R.; Beutler, E.; Yoshida, A. Molecular abnormalities of a phosphoglycerate kinase variant generated by spontaneous mutation. Blood 1992, 79, 2759–2762. [Google Scholar]
- Tsujino, S.; Tonin, P.; Shanske, S.; Nohria, V.; Boustany, R.M.; Lewis, D.; Chen, Y.T.; DiMauro, S. A splice junction mutation in a new myopathic variant of phosphoglycerate kinase deficiency (PGK North Carolina). Ann. Neurol. 1994, 35, 349–353. [Google Scholar] [CrossRef]
- Hamano, T.; Mutoh, T.; Sugie, H.; Koga, H.; Kuriyama, M. Phosphoglycerate kinase deficiency: An adult myopathic form with a novel mutation. Neurology 2000, 54, 1188–1190. [Google Scholar] [CrossRef]
- Aasly, J.; van Diggelen, O.P.; Boer, A.M.; Bronstad, G. Phosphoglycerate kinase deficiency in two brothers with McArdle-like clinical symptoms. Eur. J. Neurol. 2000, 7, 111–113. [Google Scholar] [CrossRef]
- Svaasand, E.K.; Aasly, J.; Landsem, V.M.; Klungland, H. Altered expression of PGK1 in a family with phosphoglycerate kinase deficiency. Muscle Nerve 2007, 36, 679–684. [Google Scholar] [CrossRef]
- Shirakawa, K.; Takahashi, Y.; Miyajima, H. Intronic mutation in the PGK1 gene may cause recurrent myoglobinuria by aberrant splicing. Neurology 2006, 66, 925–927. [Google Scholar] [CrossRef]
- Mesa-Torres, N.; Fabelo-Rosa, I.; Riverol, D.; Yunta, C.; Albert, A.; Salido, E.; Pey, A.L. The role of protein denaturation energetics and molecular chaperones in the aggregation and mistargeting of mutants causing primary hyperoxaluria type I. PLoS One 2013, 8, e71963. [Google Scholar]
- Pey, A.L.; Desviat, L.R.; Gamez, A.; Ugarte, M.; Perez, B. Phenylketonuria: Genotype-phenotype correlations based on expression analysis of structural and functional mutations in PAH. Hum. Mutat. 2003, 21, 370–378. [Google Scholar] [CrossRef]
- Henriques, B.J.; Bross, P.; Gomes, C.M. Mutational hotspots in electron transfer flavoprotein underlie defective folding and function in multiple acyl-CoA dehydrogenase deficiency. Biochim. Biophys. Acta 2010, 1802, 1070–1077. [Google Scholar] [CrossRef]
- Majtan, T.; Liu, L.; Carpenter, J.F.; Kraus, J.P. Rescue of cystathionine beta-synthase (CBS) mutants with chemical chaperones: Purification and characterization of eight CBS mutant enzymes. J. Biol. Chem. 2010, 285, 15866–15873. [Google Scholar] [CrossRef]
- Pey, A.L.; Salido, E.; Sanchez-Ruiz, J.M. Role of low native state kinetic stability and interaction of partially unfolded states with molecular chaperones in the mitochondrial protein mistargeting associated with primary hyperoxaluria. Amino Acids 2011, 41, 1233–1245. [Google Scholar] [CrossRef]
- Sanchez-Ruiz, J.M.; Lopez-Lacomba, J.L.; Cortijo, M.; Mateo, P.L. Differential scanning calorimetry of the irreversible thermal denaturation of thermolysin. Biochemistry 1988, 27, 1648–1652. [Google Scholar] [CrossRef]
- Matouschek, A.; Fersht, A.R. Application of physical organic chemistry to engineered mutants of proteins: Hammond postulate behavior in the transition state of protein folding. Proc. Natl. Acad. Sci. USA 1993, 90, 7814–7818. [Google Scholar] [CrossRef]
- Costas, M.; Rodriguez-Larrea, D.; de Maria, L.; Borchert, T.V.; Gomez-Puyou, A.; Sanchez-Ruiz, J.M. Between-species variation in the kinetic stability of TIM proteins linked to solvation-barrier free energies. J. Mol. Biol. 2009, 385, 924–937. [Google Scholar] [CrossRef]
- Rodriguez-Larrea, D.; Minning, S.; Borchert, T.V.; Sanchez-Ruiz, J.M. Role of solvation barriers in protein kinetic stability. J. Mol. Biol. 2006, 360, 715–724. [Google Scholar] [CrossRef]
- Aguirre, Y.; Cabrera, N.; Aguirre, B.; Perez-Montfort, R.; Hernandez-Santoyo, A.; Reyes-Vivas, H.; Enriquez-Flores, S.; de Gomez-Puyou, M.T.; Gomez-Puyou, A.; Sanchez-Ruiz, J.M.; et al. Different contribution of conserved amino acids to the global properties of Triosephosphate isomerases. Proteins 2013. [Google Scholar] [CrossRef]
- Plaza del Pino, I.M.; Ibarra-Molero, B.; Sanchez-Ruiz, J.M. Lower kinetic limit to protein thermal stability: A proposal regarding protein stability in vivo and its relation with misfolding diseases. Proteins 2000, 40, 58–70. [Google Scholar] [CrossRef]
- Park, C.; Marqusee, S. Probing the high energy states in proteins by proteolysis. J. Mol. Biol. 2004, 343, 1467–1476. [Google Scholar] [CrossRef]
- Tur-Arlandis, G.; Rodriguez-Larrea, D.; Ibarra-Molero, B.; Sanchez-Ruiz, J.M. Proteolytic scanning calorimetry: A novel methodology that probes the fundamental features of protein kinetic stability. Biophys. J. 2010, 98, L12–L14. [Google Scholar] [CrossRef]
- Myers, J.K.; Pace, C.N.; Scholtz, J.M. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995, 4, 2138–2148. [Google Scholar] [CrossRef]
- Whitten, S.T.; Wooll, J.O.; Razeghifard, R.; Garcia-Moreno, E.B.; Hilser, V.J. The origin of pH-dependent changes in m-values for the denaturant-induced unfolding of proteins. J. Mol. Biol. 2001, 309, 1165–1175. [Google Scholar] [CrossRef]
- Pey, A.L.; Majtan, T.; Sanchez-Ruiz, J.M.; Kraus, J.P. Human cystathionine beta-synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): Complex regulation of CBS activity and stability by SAM. Biochem. J. 2013, 449, 109–121. [Google Scholar] [CrossRef]
- Ruzafa, D.; Conejero-Lara, F.; Morel, B. Modulation of the stability of amyloidogenic precursors by anion binding strongly influences the rate of amyloid nucleation. Phys. Chem. Chem. Phys. 2013, 15, 15508–15517. [Google Scholar] [CrossRef]
- Sanchez-Romero, I.; Ariza, A.; Wilson, K.S.; Skjot, M.; Vind, J.; de Maria, L.; Skov, L.K.; Sanchez-Ruiz, J.M. Mechanism of protein kinetic stabilization by engineered disulfide crosslinks. PLoS One 2013, 8, e70013. [Google Scholar] [CrossRef]
- Kerner, M.J.; Naylor, D.J.; Ishihama, Y.; Maier, T.; Chang, H.C.; Stines, A.P.; Georgopoulos, C.; Frishman, D.; Hayer-Hartl, M.; Mann, M.; et al. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell 2005, 122, 209–220. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Dhar, A.; Girdhar, K.; Singh, D.; Gelman, H.; Ebbinghaus, S.; Gruebele, M. Protein stability and folding kinetics in the nucleus and endoplasmic reticulum of eucaryotic cells. Biophys. J. 2011, 101, 421–430. [Google Scholar] [CrossRef]
- Ellis, R.J.; Minton, A.P. Protein aggregation in crowded environments. Biol. Chem. 2006, 387, 485–497. [Google Scholar]
- Banks, R.D.; Blake, C.C.; Evans, P.R.; Haser, R.; Rice, D.W.; Hardy, G.W.; Merrett, M.; Phillips, A.W. Sequence, structure and activity of phosphoglycerate kinase: A possible hinge-bending enzyme. Nature 1979, 279, 773–777. [Google Scholar] [CrossRef]
- Casanueva, M.O.; Burga, A.; Lehner, B. Fitness trade-offs and environmentally induced mutation buffering in isogenic C. elegans. Science 2012, 335, 82–85. [Google Scholar] [CrossRef]
- Sancho, J.; Meiering, E.M.; Fersht, A.R. Mapping transition states of protein unfolding by protein engineering of ligand-binding sites. J. Mol. Biol. 1991, 221, 1007–1014. [Google Scholar] [CrossRef]
- Calamini, B.; Silva, M.C.; Madoux, F.; Hutt, D.M.; Khanna, S.; Chalfant, M.A.; Saldanha, S.A.; Hodder, P.; Tait, B.D.; Garza, D.; et al. Small-molecule proteostasis regulators for protein conformational diseases. Nat. Chem. Biol. 2011, 8, 185–196. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Valentini, G.; Maggi, M.; Pey, A.L. Protein Stability, Folding and Misfolding in Human PGK1 Deficiency. Biomolecules 2013, 3, 1030-1052. https://doi.org/10.3390/biom3041030
Valentini G, Maggi M, Pey AL. Protein Stability, Folding and Misfolding in Human PGK1 Deficiency. Biomolecules. 2013; 3(4):1030-1052. https://doi.org/10.3390/biom3041030
Chicago/Turabian StyleValentini, Giovanna, Maristella Maggi, and Angel L. Pey. 2013. "Protein Stability, Folding and Misfolding in Human PGK1 Deficiency" Biomolecules 3, no. 4: 1030-1052. https://doi.org/10.3390/biom3041030