Factor H: A Complement Regulator in Health and Disease, and a Mediator of Cellular Interactions


Abstract
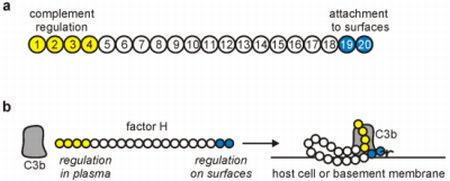
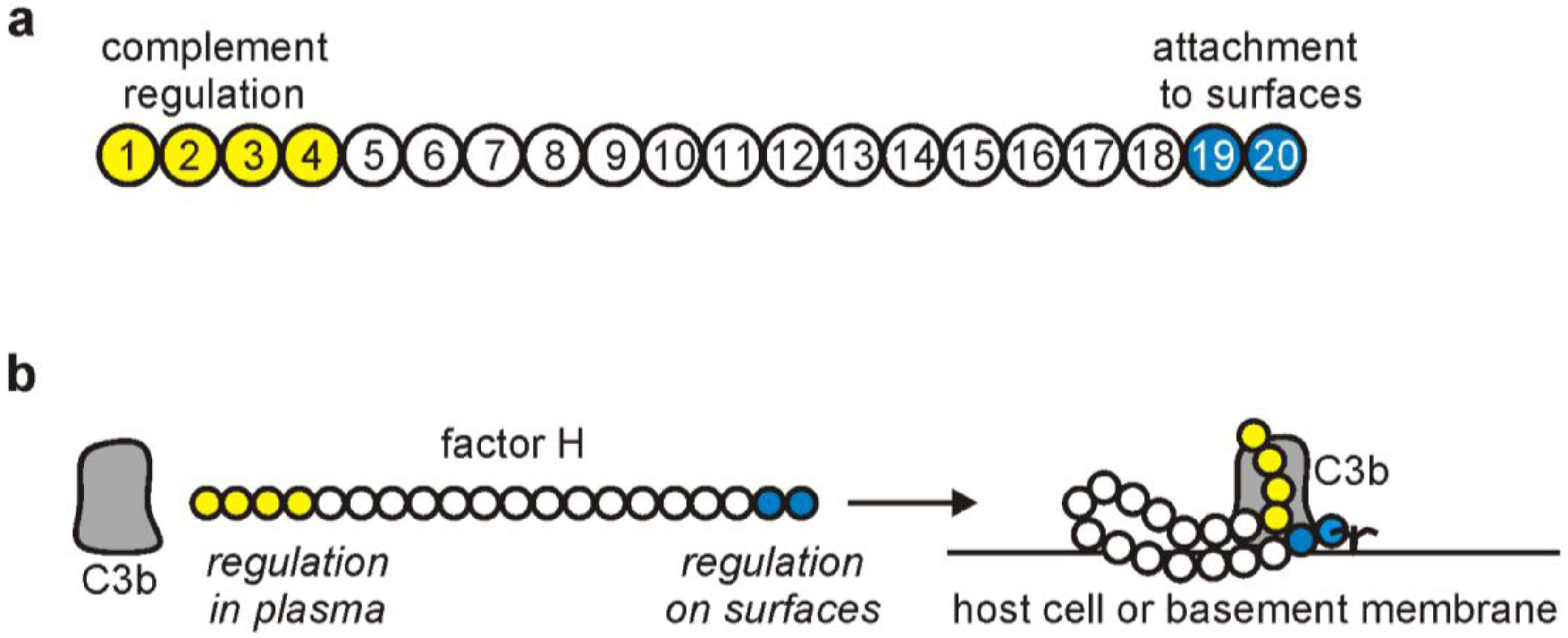
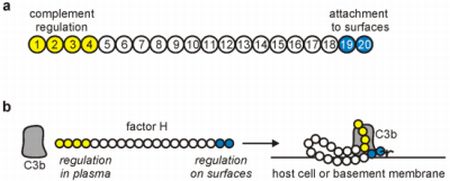
:
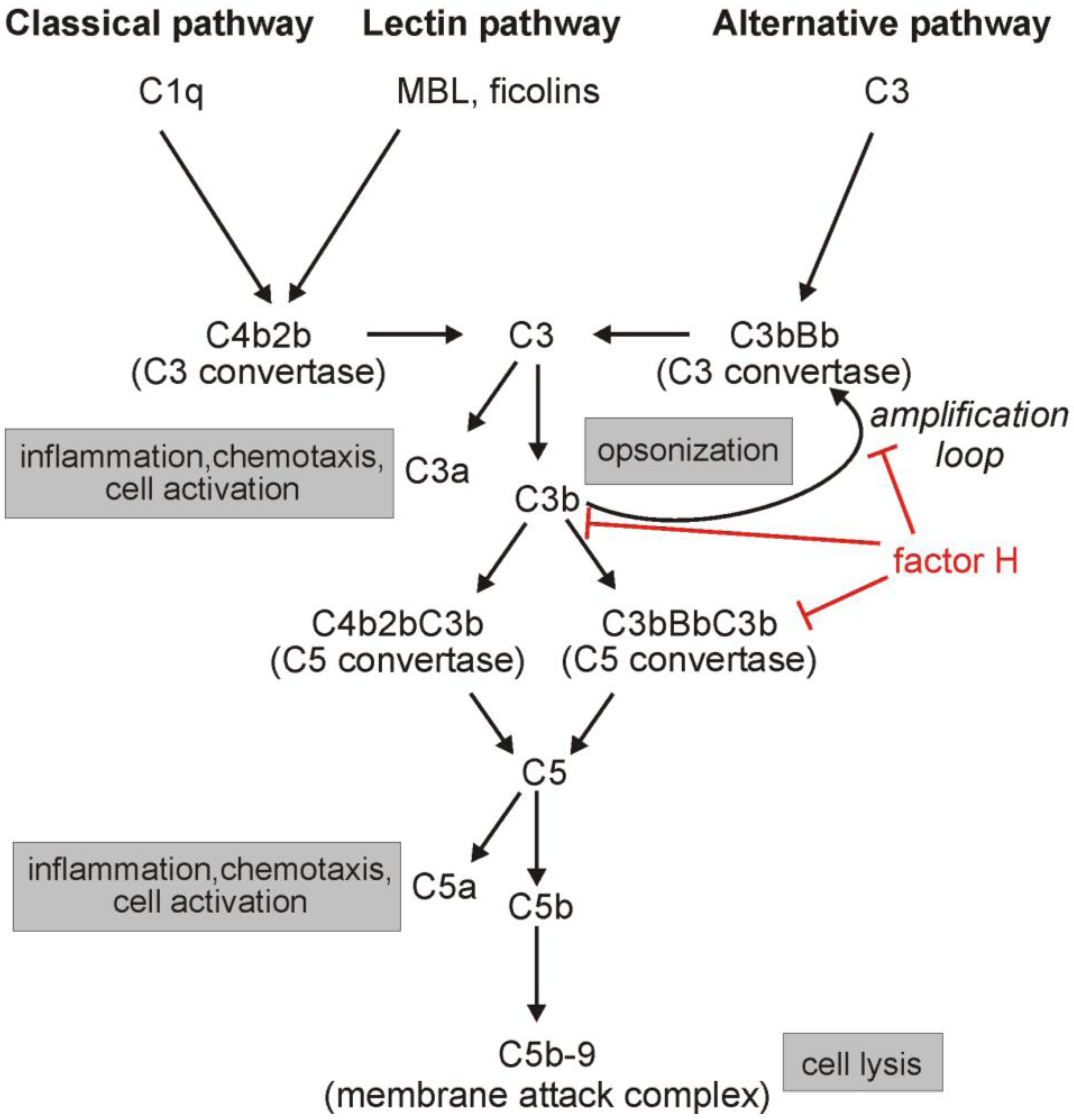
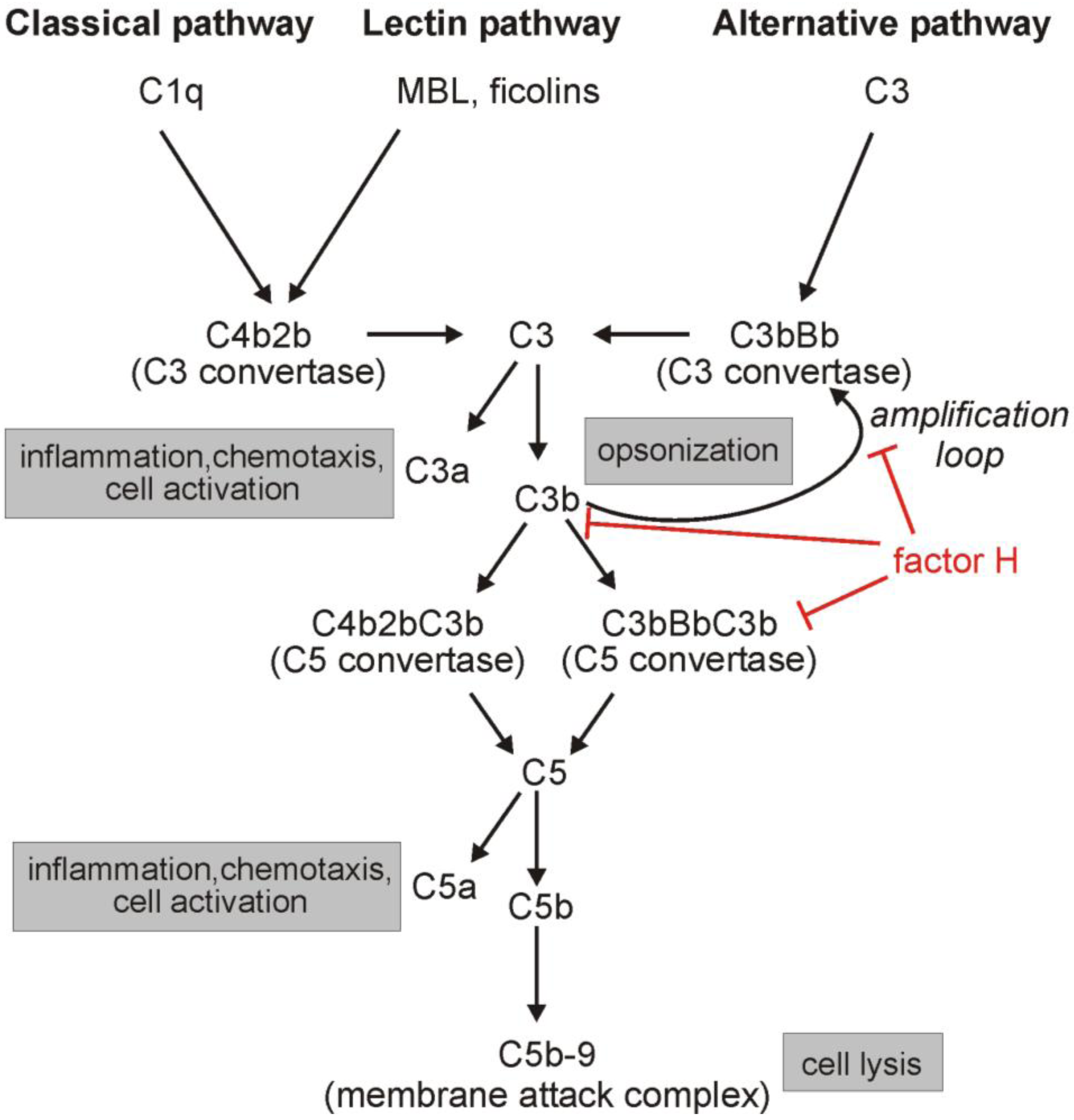
1. Introduction: The Complement System
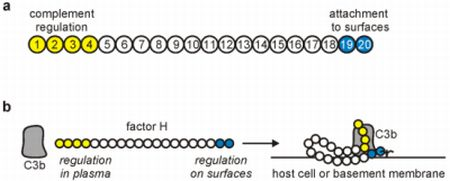
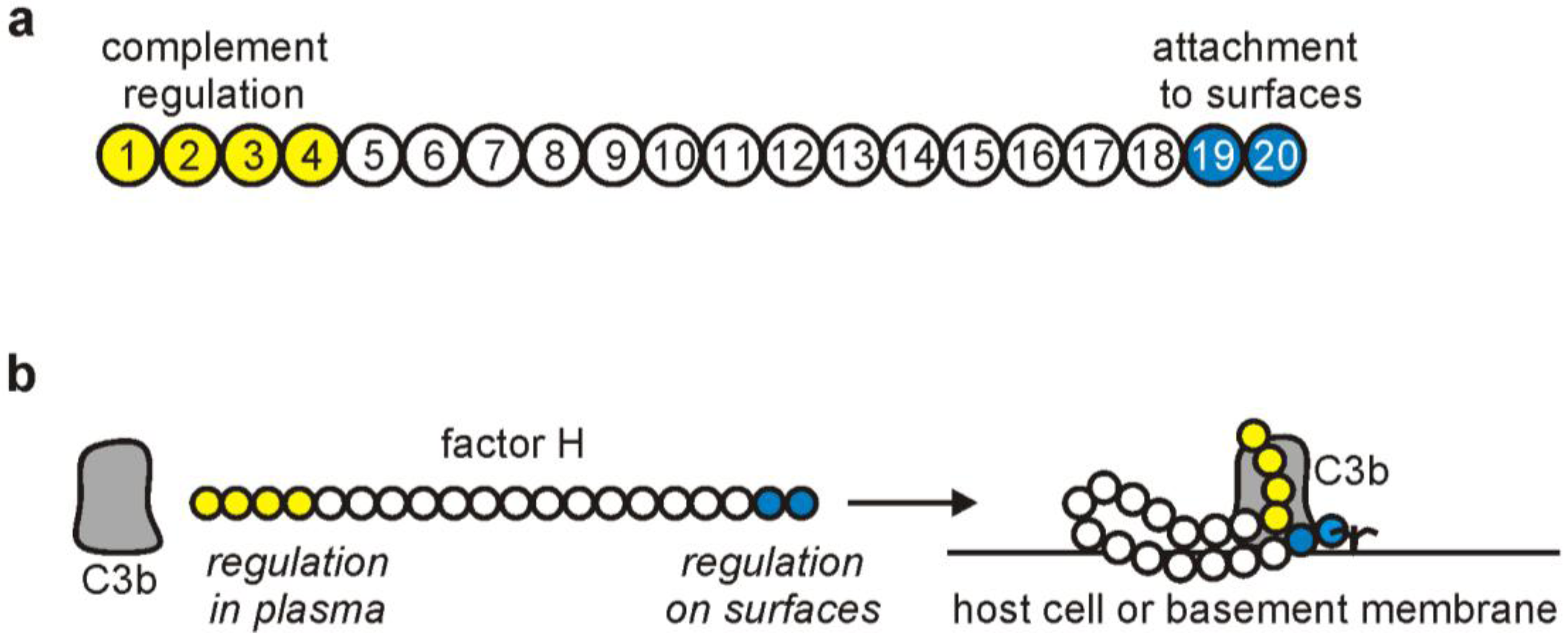
2. The Complement Regulator Factor H: Structure and Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Binding sites | Relevance | References | |
|---|---|---|---|---|
| Complement regulation | [36,37,38] | |||
| C3d | SCRs 19-20 | |||
| Heparin (Glycosaminoglycans, Sialic acid) | SCRs 7, 19-20 | Attachment to host cells | [29,38,39] | |
| Targeting the activity of factor H | [40,41,42] | |||
| Pentraxin 3 | SCRs 7, 19-20 | [43] | ||
| Promoting safe clearance, protection from autoimmunity | [41,44,45,46] | |||
| Histones | SCRs 1-4, 6-8, 8-15 | |||
| Regulation of inflammation (e.g., in rheumatoid arthritis) | [47,48,49] | |||
| Chondroadherin | ? | |||
| SCRs 7, 20 | Protection from oxidative stress | [50] | ||
| ? | Complement dysregulation may promote inflammation in the brain | [51] | ||
| SCRs 8-11, 15-20 | Modulation of adrenomedullin functions | [52,53] | ||
3. Factor H Interactions with Host Ligands
3.1. Factor H Interaction with C3b
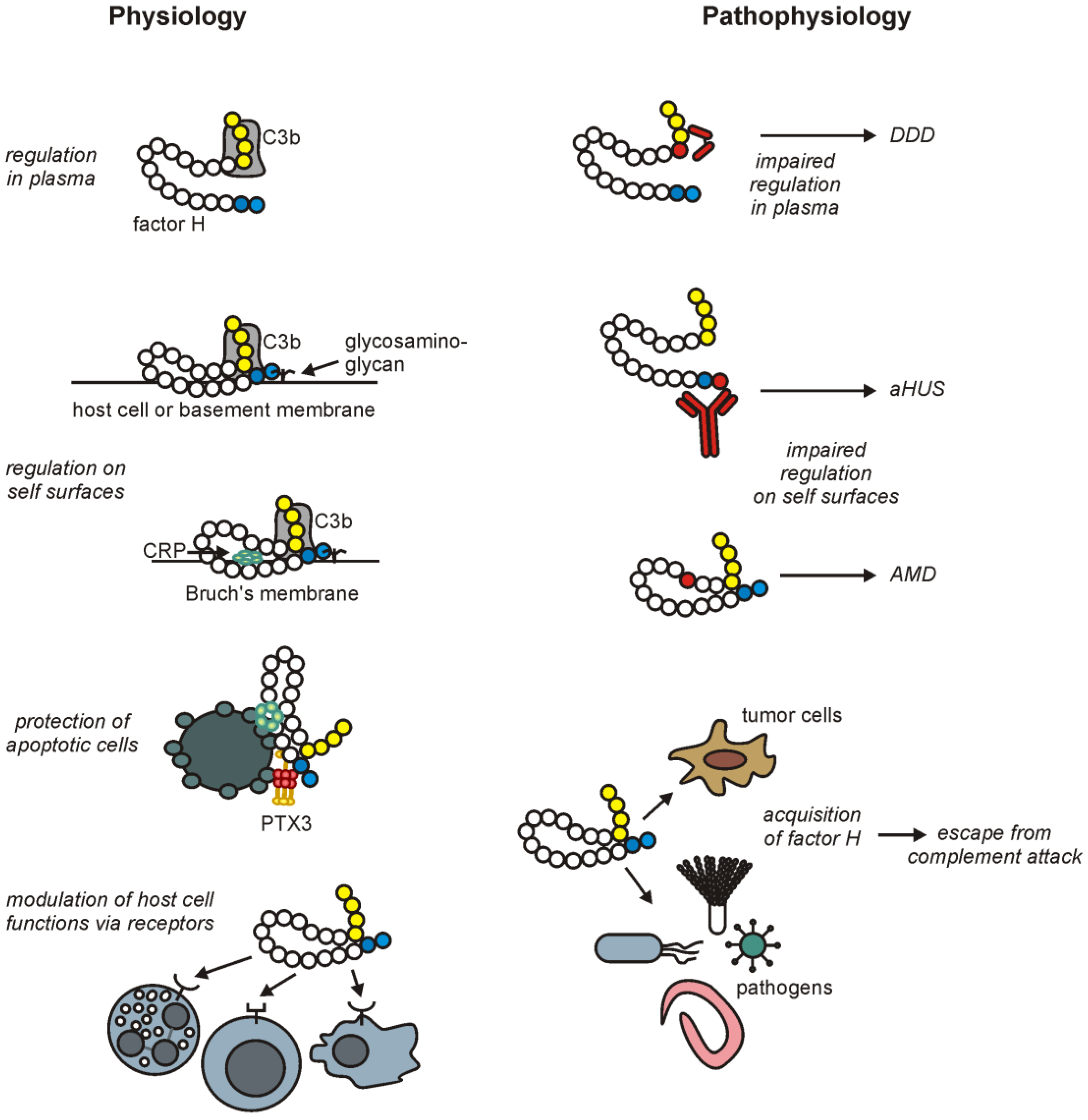
3.2. Factor H Attachment to Host Cells: An Important Role in Self-Nonself Discrimination by Complement
3.3. Factor H Binding to Apoptotic and Necrotic Cells
3.4. Factor H Interactions with Pentraxins
3.5. Factor H Binding to Extracellular Matrix (ECM)
4. Redundant and Non-Redundant Functions among Factor H Family Proteins
5. Factor H-Associated Diseases
5.1. Age-Related Macular Degeneration
5.2. Atypical Hemolytic Uremic Syndrome
5.3. Dense Deposit Disease
6. Misuse of Factor H by Pathogens and Tumor Cells
6.1. Factor H Binding to Pathogens
6.2. Factor H and Tumor Cells
7. Factor H in Cellular Interactions: Beyond Its Role as a Complement Regulator
8. Conclusions
Conflict of Interest
Acknowledgments
References
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. Second of two parts. N. Engl. J. Med. 2001, 344, 1140–1144. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Meri, S. Loss of self-control in the complement system and innate autoreactivity. Ann. NY Acad. Sci. 2007, 1109, 93–105. [Google Scholar]
- Rodríguez de Córdoba, S.; Goicoechea de Jorge, E. Translational mini-review series on complement factor H: Genetics and disease associations of human complement factor H. Clin. Exp. Immunol. 2008, 151, 1–13. [Google Scholar]
- Boon, C.J.; van de Kar, N.C.; Klevering, B.J.; Keunen, J.E.; Cremers, F.P.; Klaver, C.C.; Hoyng, C.B.; Daha, M.R.; den Hollander, A.I. The spectrum of phenotypes caused by variants in the CFH gene. Mol. Immunol. 2009, 46, 1573–1594. [Google Scholar] [CrossRef]
- Józsi, M.; Zipfel, P.F. Factor H family proteins and human diseases. Trends Immunol. 2008, 29, 380–387. [Google Scholar] [CrossRef]
- Nilsson, U.R.; Müller-Eberhard, H.J. Isolation of beta 1F-globulin from human serum and its characterization as the fifth component of complement. J. Exp. Med. 1965, 122, 277–298. [Google Scholar] [CrossRef]
- Rodríguez de Córdoba, S.; Esparza-Gordillo, J.; Goicoechea de Jorge, E.; Lopez-Trascasa, M.; Sánchez-Corral, P. The human complement factor H: Functional roles, genetic variations and disease associations. Mol. Immunol. 2004, 41, 355–367. [Google Scholar] [CrossRef]
- de Paula, P.F.; Barbosa, J.E.; Junior, P.R.; Ferriani, V.P.; Latorre, M.R.; Nudelman, V.; Isaac, L. Ontogeny of complement regulatory proteins - concentrations of factor H, factor I, C4b-binding protein, properdin and vitronectin in healthy children of different ages and in adults. Scand. J. Immunol. 2003, 58, 572–577. [Google Scholar] [CrossRef]
- Esparza-Gordillo, J.; Soria, J.M.; Buil, A.; Almasy, L.; Blangero, J.; Fontcuberta, J.; Rodríguez de Córdoba, S. Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics 2004, 56, 77–82. [Google Scholar] [CrossRef]
- Hakobyan, S.; Harris, C.L.; Tortajada, A.; Goicochea de Jorge, E.; García-Layana, A.; Fernández-Robredo, P.; Rodríguez de Córdoba, S.; Morgan, B.P. Measurement of factor H variants in plasma using variant-specific monoclonal antibodies: Application to assessing risk of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2008, 49, 1983–1990. [Google Scholar]
- Hakobyan, S.; Tortajada, A.; Harris, C.L.; Rodríguez de Córdoba, S.; Morgan, B.P. Variant-specific quantification of factor H in plasma identifies null alleles associated with atypical hemolytic uremic syndrome. Kidney Int. 2010, 78, 782–788. [Google Scholar] [CrossRef]
- Whaley, K. Biosynthesis of the complement components and the regulatory proteins of the alternative complement pathway by human peripheral blood monocytes. J. Exp. Med. 1980, 151, 501–516. [Google Scholar] [CrossRef]
- Katz, Y.; Strunk, R.C. Synthesis and regulation of complement protein factor H in human skin fibroblasts. J. Immunol. 1988, 141, 559–563. [Google Scholar]
- Brooimans, R.A.; Van der Ark, A.A.; Buurman, W.A.; Van Es, L.A.; Daha, M.R. Differential regulation of complement factor H and C3 production in human umbilical vein endothelial cells by IFN-gamma and IL-1. J. Immunol. 1990, 144, 3835–3840. [Google Scholar]
- Timár, K.K.; Pasch, M.C.; van den Bosch, N.H.A.; Jarva, H.; Junnikkala, S.; Meri, S.; Bos, J.D.; Asghar, S.S. Human keratinocytes produce the complement inhibitor factor H: Synthesis is regulated by interferon-γ. Mol. Immunol. 2006, 43, 317–325. [Google Scholar] [CrossRef]
- Devine, D.V.; Rosse, W.F. Regulation of the activity of platelet-bound C3 convertase of the alternative pathway of complement by platelet factor H. Proc. Natl. Acad. Sci. USA 1987, 84, 5873–5877. [Google Scholar] [CrossRef]
- Chen, M.; Forrester, J.V.; Xu, H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp. Eye Res. 2007, 84, 635–645. [Google Scholar] [CrossRef]
- Ripoche, J.; Day, A.J.; Harris, T.J.; Sim, R.B. The complete amino acid sequence of human complement factor H. Biochem. J. 1988, 249, 593–602. [Google Scholar]
- Weiler, J.M.; Daha, M.R.; Austen, K.F.; Fearon, D.T. Control of the amplification convertase of complement by the plasma protein beta1H. Proc. Natl. Acad. Sci. USA 1976, 73, 3268–3272. [Google Scholar]
- Whaley, K.; Ruddy, S. Modulation of the alternative complement pathway by beta 1H globulin. J. Exp. Med. 1976, 144, 1147–1163. [Google Scholar] [CrossRef]
- Pangburn, M.K.; Schreiber, R.D.; Müller-Eberhard, H.J. Human complement C3b inactivator: Isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J. Exp. Med. 1977, 146, 257–270. [Google Scholar] [CrossRef]
- Gordon, D.L.; Kaufman, R.M.; Blackmore, T.K.; Kwong, J.; Lublin, D.M. Identification of complement regulatory domains in human factor H. J. Immunol. 1995, 155, 348–356. [Google Scholar]
- Kühn, S.; Skerka, C.; Zipfel, P.F. Mapping of the complement regulatory domains in the human factor H-like protein 1 and in factor H1. J. Immunol. 1995, 155, 5663–5670. [Google Scholar]
- Pangburn, M.K. Cutting edge: Localization of the host recognition functions of complement factor H at the carboxyl-terminal: Implications for hemolytic uremic syndrome. J. Immunol. 2002, 169, 4702–4706. [Google Scholar]
- Oppermann, M.; Manuelian, T.; Józsi, M.; Brandt, E.; Jokiranta, T.S.; Heinen, S.; Meri, S.; Skerka, C.; Götze, O.; Zipfel, P.F. The C-terminus of complement regulator Factor H mediates target recognition: Evidence for a compact conformation of the native protein. Clin. Exp. Immunol. 2006, 144, 342–352. [Google Scholar] [CrossRef]
- Meri, S.; Pangburn, M.K. Discrimination between activators and nonactivators of the alternative pathway of complement: Regulation via a sialic acid/polyanion binding site on factor H. Proc. Natl. Acad. Sci. USA 1990, 87, 3982–3986. [Google Scholar]
- Pangburn, M.K. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology 2000, 49, 149–157. [Google Scholar] [CrossRef]
- Ferreira, V.P.; Herbert, A.P.; Hocking, H.G.; Barlow, P.N.; Pangburn, M.K. Critical role of the C-terminal domains of factor H in regulating complement activation at cell surfaces. J. Immunol. 2006, 177, 6308–6316. [Google Scholar]
- Józsi, M.; Oppermann, M.; Lambris, J.D.; Zipfel, P.F. The C-terminus of complement factor H is essential for host cell protection. Mol. Immunol. 2007, 44, 2697–2706. [Google Scholar] [CrossRef]
- Józsi, M.; Manuelian, T.; Heinen, S.; Oppermann, M.; Zipfel, P.F. Attachment of the soluble complement regulator factor H to cell and tissue surfaces: Relevance for pathology. Histol. Histopathol. 2004, 19, 251–258. [Google Scholar]
- Sjöberg, A.P.; Trouw, L.A.; Blom, A.M. Complement activation and inhibition: A delicate balance. Trends Immunol. 2008, 30, 83–90. [Google Scholar]
- Lambris, J.D.; Ricklin, D.; Geisbrecht, B.V. Complement evasion by human pathogens. Nat. Rev. Microbiol. 2008, 6, 132–142. [Google Scholar] [CrossRef]
- Sharma, A.K.; Pangburn, M.K. Identification of three physically and functionally distinct binding sites for C3b in human complement factor H by deletion mutagenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 10996–11001. [Google Scholar] [CrossRef]
- Jokiranta, T.S.; Hellwage, J.; Koistinen, V.; Zipfel, P.F.; Meri, S. Each of the three binding sites on complement factor H interacts with a distinct site on C3b. J. Biol. Chem. 2000, 275, 27657–27662. [Google Scholar]
- Schmidt, C.Q.; Herbert, A.P.; Kavanagh, D.; Gandy, C.; Fenton, C.J.; Blaum, B.S.; Lyon, M.; Uhrín, D.; Barlow, P.N. A new map of glycosaminoglycan and C3b binding sites on factor H. J. Immunol. 2008, 181, 2610–2619. [Google Scholar]
- Jokiranta, T.S.; Cheng, Z.Z.; Seeberger, H.; Józsi, M.; Heinen, S.; Noris, M.; Remuzzi, G.; Ormsby, R.; Gordon, D.L.; Meri, S.; et al. Binding of complement factor H to endothelial cells is mediated by the carboxy-terminal glycosaminoglycan binding site. Am. J. Pathol. 2005, 167, 1173–1181. [Google Scholar] [CrossRef]
- Jarva, H.; Jokiranta, T.S.; Hellwage, J.; Zipfel, P.F.; Meri, S. Regulation of complement activation by C-reactive protein: Targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8-11. J. Immunol. 1999, 163, 3957–3962. [Google Scholar]
- Mihlan, M.; Stippa, S.; Józsi, M.; Zipfel, P.F. Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H. Cell Death Differ. 2009, 16, 1630–1640. [Google Scholar] [CrossRef]
- Okemefuna, A.I.; Nan, R.; Miller, A.; Gor, J.; Perkins, S.J. Complement factor H binds at two independent sites to C-reactive protein in acute phase concentrations. J. Biol. Chem. 2010, 285, 1053–1065. [Google Scholar]
- Deban, L.; Jarva, H.; Lehtinen, M.J.; Bottazzi, B.; Bastone, A.; Doni, A.; Jokiranta, T.S.; Mantovani, A.; Meri, S. Binding of the long pentraxin PTX3 to factor H: Interacting domains and function in the regulation of complement activation. J. Immunol. 2008, 181, 8433–8440. [Google Scholar]
- Gershov, D.; Kim, S.; Brot, N.; Elkon, K.B. C-Reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: Implications for systemic autoimmunity. J. Exp. Med. 2000, 192, 1353–1364. [Google Scholar] [CrossRef]
- Trouw, L.A.; Bengtsson, A.A.; Gelderman, K.A.; Dahlbäck, B.; Sturfelt, G.; Blom, A.M. C4b-binding protein and factor H compensate for the loss of membrane-bound complement inhibitors to protect apoptotic cells against excessive complement attack. J. Biol. Chem. 2007, 282, 28540–28548. [Google Scholar]
- Leffler, J.; Herbert, A.P.; Norström, E.; Schmidt, C.Q.; Barlow, P.N.; Blom, A.M.; Martin, M. Annexin-II, DNA, and histones serve as factor H ligands on the surface of apoptotic cells. J. Biol. Chem. 2010, 285, 3766–3776. [Google Scholar]
- Sjöberg, A.; Onnerfjord, P.; Mörgelin, M.; Heinegård, D.; Blom, A.M. The extracellular matrix and inflammation: Fibromodulin activates the classical pathway of complement by directly binding C1q. J. Biol. Chem. 2005, 280, 32301–32308. [Google Scholar]
- Sjöberg, A.P.; Trouw, L.A.; Clark, S.J.; Sjölander, J.; Heinegård, D.; Sim, R.B.; Day, A.J.; Blom, A.M. The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. J. Biol. Chem. 2007, 282, 10894–10900. [Google Scholar]
- Sjöberg, A.; Manderson, G.A.; Mörgelin, M.; Day, A.J.; Heinegård, D.; Blom, A.M. Short leucine-rich glycoproteins of the extracellular matrix display diverse patterns of complement interaction and activation. Mol. Immunol. 2009, 46, 830–839. [Google Scholar] [CrossRef]
- Weismann, D.; Hartvigsen, K.; Lauer, N.; Bennett, K.L.; Scholl, H.P.; Charbel Issa, P.; Cano, M.; Brandstätter, H.; Tsimikas, S.; Skerka, C.; et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 2011, 478, 76–81. [Google Scholar]
- Sjöberg, A.P.; Nyström, S.; Hammarström, P.; Blom, A.M. Native, amyloid fibrils and beta-oligomers of the C-terminal domain of human prion protein display differential activation of complement and bind C1q, factor H and C4b-binding protein directly. Mol. Immunol. 2008, 45, 3213–3221. [Google Scholar] [CrossRef]
- Pio, R.; Martinez, A.; Unsworth, E.J.; Kowalak, J.A.; Bengoechea, J.A.; Zipfel, P.F.; Elsasser, T.H.; Cuttitta, F. Complement factor H is a serum-binding protein for adrenomedullin, and the resulting complex modulates the bioactivities of both partners. J. Biol. Chem. 2001, 276, 12292–12300. [Google Scholar]
- Martínez, A.; Pío, R.; Zipfel, P.F.; Cuttitta, F. Mapping of the adrenomedullin-binding domains in human complement factor H. Hypertens. Res. 2003, 26. Suppl:S55–Suppl:S59. [Google Scholar]
- Wu, J.; Wu, Y.Q.; Ricklin, D.; Janssen, B.J.; Lambris, J.D.; Gros, P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat. Immunol. 2009, 10, 728–733. [Google Scholar] [CrossRef]
- Kajander, T.; Lehtinen, M.J.; Hyvärinen, S.; Bhattacharjee, A.; Leung, E.; Isenman, D.E.; Meri, S.; Goldman, A.; Jokiranta, T.S. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc. Natl. Acad. Sci. USA 2011, 108, 2897–2902. [Google Scholar]
- Morgan, H.P.; Schmidt, C.Q.; Guariento, M.; Blaum, B.S.; Gillespie, D.; Herbert, A.P.; Kavanagh, D.; Mertens, H.D.; Svergun, D.I.; Johansson, C.M.; et al. Structural basis for engagement by complement factor H of C3b on a self surface. Nat. Struct. Mol. Biol. 2011, 18, 463–470. [Google Scholar] [CrossRef]
- Blackmore, T.K.; Sadlon, T.A.; Ward, H.M.; Lublin, D.M.; Gordon, D.L. Identification of a heparin binding domain in the seventh short consensus repeat of complement factor H. J. Immunol. 1996, 157, 5422–5427. [Google Scholar]
- Blackmore, T.K.; Hellwage, J.; Sadlon, T.A.; Higgs, N.; Zipfel, P.F.; Ward, H.M.; Gordon, D.L. Identification of the second heparin-binding domain in human complement factor H. J. Immunol. 1998, 160, 3342–3348. [Google Scholar]
- Pangburn, M.K.; Atkinson, M.A.; Meri, S. Localization of the heparin-binding site on complement factor H. J. Biol. Chem. 1991, 266, 16847–16853. [Google Scholar]
- Nauta, A.J.; Daha, M.R.; van Kooten, C.; Roos, A. Recognition and clearance of apoptotic cells: A role for complement and pentraxins. Trends Immunol. 2003, 24, 148–154. [Google Scholar] [CrossRef]
- Bottazzi, B.; Doni, A.; Garlanda, C.; Mantovani, A. An integrated view of humoral innate immunity: Pentraxins as a paradigm. Annu. Rev. Immunol. 2010, 28, 157–183. [Google Scholar] [CrossRef]
- Deban, L.; Jaillon, S.; Garlanda, C.; Bottazzi, B.; Mantovani, A. Pentraxins in innate immunity: Lessons from PTX3. Cell Tissue Res. 2011, 343, 237–249. [Google Scholar] [CrossRef]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Invest. 2003, 111, 1805–1812. [Google Scholar]
- Black, S.; Kushner, I.; Samols, D. C-reactive Protein. J. Biol. Chem. 2004, 279, 48487–48490. [Google Scholar]
- Volanakis, J.E.; Kaplan, M.H. Interaction of C-reactive protein complexes with the complement system. II. Consumption of guinea pig complement by CRP complexes: Requirement for human C1q. J. Immunol. 1974, 113, 9–17. [Google Scholar]
- Ng, P.M.; Le Saux, A.; Lee, C.M.; Tan, N.S.; Lu, J.; Thiel, S.; Ho, B.; Ding, J.L. C-reactive protein collaborates with plasma lectins to boost immune response against bacteria. EMBO J. 2007, 26, 3431–3440. [Google Scholar] [CrossRef]
- Lu, J.; Marnell, L.L.; Marjon, K.D.; Mold, C.; Du Clos, T.W.; Sun, P.D. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature 2008, 456, 989–992. [Google Scholar]
- Mihlan, M.; Hebecker, M.; Dahse, H.M.; Hälbich, S.; Huber-Lang, M.; Dahse, R.; Zipfel, P.F.; Józsi, M. Human complement factor H-related protein 4 binds and recruits native pentameric C-reactive protein to necrotic cells. Mol. Immunol. 2009, 46, 335–344. [Google Scholar] [CrossRef]
- Hammond, D.J., Jr.; Singh, S.K.; Thompson, J.A.; Beeler, B.W.; Rusiñol, A.E.; Pangburn, M.K.; Potempa, L.A.; Agrawal, A. Identification of acidic pH-dependent ligands of pentameric C-reactive protein. J. Biol. Chem. 2010, 285, 36235–36244. [Google Scholar]
- Ji, S.R.; Wu, Y.; Zhu, L.; Potempa, L.A.; Sheng, F.L.; Lu, W.; Zhao, J. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRP(m). FASEB J. 2007, 21, 284–294. [Google Scholar]
- Lauer, N.; Mihlan, M.; Hartmann, A.; Schlötzer-Schrehardt, U.; Keilhauer, C.; Scholl, H.P.; Charbel Issa, P.; Holz, F.; Weber, B.H.; Skerka, C.; Zipfel, P.F. Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. J. Immunol. 2011, 187, 4374–4383. [Google Scholar]
- Mold, C.; Gewurz, H.; Du Clos, T.W. Regulation of complement activation by C-reactive protein. Immunopharmacology 1999, 42, 23–30. [Google Scholar] [CrossRef]
- Bíró, A.; Rovó, Z.; Papp, D.; Cervenak, L.; Varga, L.; Füst, G.; Thielens, N.M.; Arlaud, G.J.; Prohászka, Z. Studies on the interactions between C-reactive protein and complement proteins. Immunology 2007, 121, 40–50. [Google Scholar] [CrossRef]
- Hakobyan, S.; Harris, C.L.; van den Berg, C.W.; Fernandez-Alonso, M.C.; Goicoechea de Jorge, E.; Rodríguez de Córdoba, S.; Rivas, G.; Mangione, P.; Pepys, M.B.; Morgan, B.P. Complement factor H binds to denatured rather than to native pentameric C-reactive protein. J. Biol. Chem. 2008, 283, 30451–30460. [Google Scholar]
- Nauta, A.J.; Bottazzi, B.; Mantovani, A.; Salvatori, G.; Kishore, U.; Schwaeble, W.J.; Gingras, A.R.; Tzima, S.; Vivanco, F.; Egido, J.; et al. Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur. J. Immunol. 2003, 33, 465–473. [Google Scholar] [CrossRef]
- Ma, Y.J.; Doni, A.; Skjoedt, M.O.; Honoré, C.; Arendrup, M.; Mantovani, A.; Garred, P. Heterocomplexes of mannose-binding lectin and the pentraxins PTX3 or serum amyloid P component trigger cross-activation of the complement system. J. Biol. Chem. 2011, 286, 3405–3417. [Google Scholar]
- Gout, E.; Moriscot, C.; Doni, A.; Dumestre-Pérard, C.; Lacroix, M.; Pérard, J.; Schoehn, G.; Mantovani, A.; Arlaud, G.J.; Thielens, N.M. M-ficolin interacts with the long pentraxin PTX3: A novel case of cross-talk between soluble pattern-recognition molecules. J. Immunol. 2011, 186, 5815–5822. [Google Scholar]
- Ma, Y.J.; Doni, A.; Hummelshøj, T.; Honoré, C.; Bastone, A.; Mantovani, A.; Thielens, N.M.; Garred, P. Synergy between ficolin-2 and pentraxin 3 boosts innate immune recognition and complement deposition. J. Biol. Chem. 2009, 284, 28263–28275. [Google Scholar]
- Braunschweig, A.; Józsi, M. Human pentraxin 3 binds to the complement regulator C4b-binding protein. PLoS One 2011, 6, e23991. [Google Scholar] [CrossRef]
- Braunschweig, A.; Józsi, M. Interaction of the long pentraxin PTX3 with soluble complement inhibitors. Mol. Immunol. 2010, 47, 2234–2235. [Google Scholar] [CrossRef]
- Braunschweig, A.; Strobel, S.; Józsi, M. Impaired binding of factor H to pentraxin 3 due to factor H mutations and autoantibodies associated with atypical hemolytic uremic syndrome. Mol. Immunol. 2011, 48, 1722. [Google Scholar]
- Clark, S.J.; Perveen, R.; Hakobyan, S.; Morgan, B.P.; Sim, R.B.; Bishop, P.N.; Day, A.J. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch's membrane in human retina. J. Biol. Chem. 2010, 285, 30192–30202. [Google Scholar]
- Didangelos, A.; Yin, X.; Mandal, K.; Baumert, M.; Jahangiri, M.; Mayr, M. Proteomics characterization of extracellular space components in the human aorta. Mol. Cell. Proteomics 2010, 9, 2048–2062. [Google Scholar] [CrossRef]
- Skerka, C.; Lauer, N.; Weinberger, A.A.; Keilhauer, C.N.; Sühnel, J.; Smith, R.; Schlötzer-Schrehardt, U.; Fritsche, L.; Heinen, S.; Hartmann, A.; et al. Defective complement control of factor H (Y402H) and FHL-1 in age-related macular degeneration. Mol. Immunol. 2007, 44, 3398–3406. [Google Scholar]
- Friese, M.A.; Hellwage, J.; Jokiranta, T.S.; Meri, S.; Peter, H.H.; Eibel, H.; Zipfel, P.F. FHL-1/reconectin and factor H: Two human complement regulators which are encoded by the same gene are differently expressed and regulated. Mol. Immunol. 1999, 36, 809–818. [Google Scholar] [CrossRef]
- Hellwage, J.; Kühn, S.; Zipfel, P.F. The human complement regulatory factor-H-like protein 1, which represents a truncated form of factor H, displays cell-attachment activity. Biochem. J. 1997, 326, 321–327. [Google Scholar]
- Heinen, S.; Hartmann, A.; Lauer, N.; Wiehl, U.; Dahse, H.M.; Schirmer, S.; Gropp, K.; Enghardt, T.; Wallich, R.; Hälbich, S.; et al. Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood 2009, 114, 2439–2447. [Google Scholar]
- Losse, J.; Zipfel, P.F.; Józsi, M. Factor H and factor H-related protein 1 bind to human neutrophils via complement receptor 3, mediate attachment to Candida albicans, and enhance neutrophil antimicrobial activity. J. Immunol. 2010, 184, 912–921. [Google Scholar] [CrossRef]
- Charbel Issa, P.; Chong, N.V.; Scholl, H.P. The significance of the complement system for the pathogenesis of age-related macular degeneration - current evidence and translation into clinical application. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 163–174. [Google Scholar] [CrossRef]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.; Spencer, K.L.; Kwan, S.Y.; Noureddine, M.; Gilbert, J.R.; et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005, 308, 419–421. [Google Scholar]
- Edwards, A.O.; Ritter, R., 3rd.; Abel, K.J.; Manning, A.; Panhuysen, C.; Farrer, L.A. Complement factor H polymorphism and age-related macular degeneration. Science 2005, 308, 421–424. [Google Scholar]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar]
- Hughes, A.E.; Orr, N.; Esfandiary, H.; Diaz-Torres, M.; Goodship, T.; Chakravarthy, U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat. Genet. 2006, 38, 1173–1177. [Google Scholar] [CrossRef]
- Laine, M.; Jarva, H.; Seitsonen, S.; Haapasalo, K.; Lehtinen, M.J.; Lindeman, N.; Anderson, D.H.; Johnson, P.T.; Järvelä, I.; Jokiranta, T.S.; et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J. Immunol. 2007, 178, 3831–3836. [Google Scholar]
- Yu, J.; Wiita, P.; Kawaguchi, R.; Honda, J.; Jorgensen, A.; Zhang, K.; Fischetti, V.A.; Sun, H. Biochemical analysis of a common human polymorphism associated with age-related macular degeneration. Biochemistry 2007, 46, 8451–8461. [Google Scholar]
- Herbert, A.P.; Deakin, J.A.; Schmidt, C.Q.; Blaum, B.S.; Egan, C.; Ferreira, V.P.; Pangburn, M.K.; Lyon, M.; Uhrín, D.; Barlow, P.N. Structure shows that a glycosaminoglycan and protein recognition site in factor H is perturbed by age-related macular degeneration-linked single nucleotide polymorphism. J. Biol. Chem. 2007, 282, 18960–18968. [Google Scholar]
- Ormsby, R.J.; Ranganathan, S.; Tong, J.C.; Griggs, K.M.; Dimasi, D.P.; Hewitt, A.W.; Burdon, K.P.; Craig, J.E.; Hoh, J.; Gordon, D.L. Functional and structural implications of the complement factor H Y402H polymorphism associated with age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2008, 49, 1763–1770. [Google Scholar]
- Clark, S.J.; Higman, V.A.; Mulloy, B.; Perkins, S.J.; Lea, S.M.; Sim, R.B.; Day, A.J. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J. Biol. Chem. 2006, 281, 24713–24720. [Google Scholar]
- Prosser, B.E.; Johnson, S.; Roversi, P.; Herbert, A.P.; Blaum, B.S.; Tyrrell, J.; Jowitt, T.A.; Clark, S.J.; Tarelli, E.; Uhrín, D.; et al. Structural basis for complement factor H linked age-related macular degeneration. J. Exp. Med. 2007, 204, 2277–2283. [Google Scholar] [CrossRef]
- Raychaudhuri, S.; Iartchouk, O.; Chin, K.; Tan, P.L.; Tai, A.K.; Ripke, S.; Gowrisankar, S.; Vemuri, S.; Montgomery, K.; Yu, Y.; et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011, 43, 1232–1236. [Google Scholar] [CrossRef]
- Manuelian, T.; Hellwage, J.; Meri, S.; Caprioli, J.; Noris, M.; Heinen, S.; Józsi, M.; Neumann, H.P.; Remuzzi, G.; Zipfel, P.F. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J. Clin. Invest. 2003, 111, 1181–1190. [Google Scholar]
- Heurich, M.; Martínez-Barricarte, R.; Francis, N.J.; Roberts, D.L.; Rodríguez de Córdoba, S.; Morgan, B.P.; Harris, C.L. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc. Natl. Acad. Sci. USA 2011, 108, 8761–8766. [Google Scholar]
- Haapasalo, K.; Jarva, H.; Siljander, T.; Tewodros, W.; Vuopio-Varkila, J.; Jokiranta, T.S. Complement factor H allotype 402H is associated with increased C3b opsonization and phagocytosis of Streptococcus pyogenes. Mol. Microbiol. 2008, 70, 583–594. [Google Scholar] [CrossRef]
- Haapasalo, K.; Vuopio, J.; Syrjänen, J.; Suvilehto, J.; Massinen, S.; Karppelin, M.; Järvelä, I.; Meri, S.; Kere, J.; Jokiranta, T.S. Acquisition of complement factor H is important for pathogenesis of streptococcus pyogenes infections: Evidence from bacterial in vitro survival and human genetic association. J. Immunol. 2012, 188, 426–435. [Google Scholar]
- Dhillon, B.; Wright, A.F.; Tufail, A.; Pappworth, I.; Hayward, C.; Moore, I.; Strain, L.; Kavanagh, D.; Barlow, P.N.; Herbert, A.P.; et al. Complement factor H autoantibodies and age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2010, 51, 5858–5863. [Google Scholar]
- Noris, M.; Remuzzi, G. Atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 1676–1687. [Google Scholar] [CrossRef]
- Loirat, C.; Frémeaux-Bacchi, V. Atypical hemolytic uremic syndrome. Orphanet J. Rare Dis. 2011, 6, 60. [Google Scholar] [CrossRef]
- Saunders, R.E.; Abarrategui-Garrido, C.; Frémeaux-Bacchi, V.; Goicoechea de Jorge, E.; Goodship, T.H.; López Trascasa, M.; Noris, M.; Ponce Castro, I.M.; Remuzzi, G.; Rodríguez de Córdoba, S.; et al. The interactive Factor H-atypical hemolytic uremic syndrome mutation database and website: Update and integration of membrane cofactor protein and Factor I mutations with structural models. Hum. Mutat. 2007, 28, 222–234. [Google Scholar] [CrossRef]
- Sánchez-Corral, P.; Pérez-Caballero, D.; Huarte, O.; Simckes, A.M.; Goicoechea, E.; López-Trascasa, M.; Rodríguez de Córdoba, S. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am. J. Hum. Genet. 2002, 71, 1285–1295. [Google Scholar] [CrossRef]
- Józsi, M.; Heinen, S.; Hartmann, A.; Ostrowicz, C.W.; Hälbich, S.; Richter, H.; Kunert, A.; Licht, C.; Saunders, R.E.; Perkins, S.J.; et al. Factor H and atypical hemolytic uremic syndrome: Mutations in the C-terminus cause structural changes and defective recognition functions. J. Am. Soc. Nephrol. 2006, 17, 170–177. [Google Scholar]
- Heinen, S.; Sanchez-Corral, P.; Jackson, M.S.; Strain, L.; Goodship, J.A.; Kemp, E.J.; Skerka, C.; Jokiranta, T.S.; Meyers, K.; Wagner, E.; et al. De novo gene conversion in the RCA gene cluster (1q32) causes mutations in complement factor H associated with atypical hemolytic uremic syndrome. Hum. Mutat. 2006, 27, 292–293. [Google Scholar]
- Venables, J.P.; Strain, L.; Routledge, D.; Bourn, D.; Powell, H.M.; Warwicker, P.; Diaz-Torres, M.L.; Sampson, A.; Mead, P.; Webb, M.; et al. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med. 2006, 3, e431. [Google Scholar]
- Francis, N.J.; McNicholas, B.; Awan, A.; Waldron, M.; Reddan, D.; Sadlier, D.; Kavanagh, D.; Strain, L.; Marchbank, K.J.; Harris, C.L.; Goodship, T.H. A novel hybrid CFH/CFHR3 gene generated by a microhomology-mediated deletion in familial atypical hemolytic uremic syndrome. Blood 2012, 119, 591–601. [Google Scholar]
- Dragon-Durey, M.A.; Loirat, C.; Cloarec, S.; Macher, M.A.; Blouin, J.; Nivet, H.; Weiss, L.; Fridman, W.H.; Frémeaux-Bacchi, V. Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2005, 16, 555–563. [Google Scholar] [CrossRef]
- Józsi, M.; Licht, C.; Strobel, S.; Zipfel, S.L.; Richter, H.; Heinen, S.; Zipfel, P.F.; Skerka, C. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008, 111, 1512–1514. [Google Scholar]
- Dragon-Durey, M.A.; Blanc, C.; Marliot, F.; Loirat, C.; Blouin, J.; Sautes-Fridman, C.; Fridman, W.H.; Frémeaux-Bacchi, V. The high frequency of complement factor H related CFHR1 gene deletion is restricted to specific subgroups of patients with atypical haemolytic uraemic syndrome. J. Med. Genet. 2009, 46, 447–450. [Google Scholar]
- Abarrategui-Garrido, C.; Martínez-Barricarte, R.; López-Trascasa, M.; Rodríguez de Córdoba, S.; Sánchez-Corral, P. Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood 2009, 114, 4261–4271. [Google Scholar] [CrossRef]
- Moore, I.; Strain, L.; Pappworth, I.; Kavanagh, D.; Barlow, P.N.; Herbert, A.P.; Schmidt, C.Q.; Staniforth, S.J.; Holmes, L.V.; Ward, R.; et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood 2010, 115, 379–387. [Google Scholar]
- Józsi, M.; Strobel, S.; Dahse, H.M.; Liu, W.S.; Hoyer, P.F.; Oppermann, M.; Skerka, C.; Zipfel, P.F. Anti factor H autoantibodies block C-terminal recognition function of factor H in hemolytic uremic syndrome. Blood 2007, 110, 1516–1518. [Google Scholar]
- Strobel, S.; Abarrategui-Garrido, C.; Fariza-Requejo, E.; Seeberger, H.; Sánchez-Corral, P.; Józsi, M. Factor H-related protein 1 neutralizes anti-factor H autoantibodies in autoimmune hemolytic uremic syndrome. Kidney Int. 2011, 80, 397–404. [Google Scholar] [CrossRef]
- Strobel, S.; Hoyer, P.F.; Mache, C.J.; Sulyok, E.; Liu, W.S.; Richter, H.; Oppermann, M.; Zipfel, P.F.; Józsi, M. Functional analyses indicate a pathogenic role of factor H autoantibodies in atypical haemolytic uraemic syndrome. Nephrol. Dial. Transplant. 2010, 25, 136–144. [Google Scholar] [CrossRef]
- Tortajada, A.; Pinto, S.; Martínez-Ara, J.; López-Trascasa, M.; Sánchez-Corral, P.; Rodríguez de Córdoba, S. Complement factor H variants I890 and L1007 while commonly associated with atypical hemolytic uremic syndrome are polymorphisms with no functional significance. Kidney Int. 2012, 81, 56–63. [Google Scholar] [CrossRef]
- Smith, R.J.; Alexander, J.; Barlow, P.N.; Botto, M.; Cassavant, T.L.; Cook, H.T.; Rodríguez de Córdoba, S.; Hageman, G.S.; Jokiranta, T.S.; Kimberling, W.J.; et al. New approaches to the treatment of dense deposit disease. J. Am. Soc. Nephrol. 2007, 18, 2447–2456. [Google Scholar] [CrossRef]
- Licht, C.; Fremeaux-Bacchi, V. Hereditary and acquired complement dysregulation in membranoproliferative glomerulonephritis. Thromb. Haemost. 2009, 101, 271–278. [Google Scholar]
- Dragon-Durey, M.A.; Frémeaux-Bacchi, V.; Loirat, C.; Blouin, J.; Niaudet, P.; Deschenes, G.; Coppo, P.; Herman Fridman, W.; Weiss, L. Heterozygous and homozygous factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: Report and genetic analysis of 16 cases. J. Am. Soc. Nephrol. 2004, 15, 787–795. [Google Scholar] [CrossRef]
- Ault, B.H.; Schmidt, B.Z.; Fowler, N.L.; Kashtan, C.E.; Ahmed, A.E.; Vogt, B.A.; Colten, H.R. Human factor H deficiency. Mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J. Biol. Chem. 1997, 272, 25168–25175. [Google Scholar]
- Montes, T.; Goicoechea de Jorge, E.; Ramos, R.; Gomà, M.; Pujol, O.; Sánchez-Corral, P.; Rodríguez de Córdoba, S. Genetic deficiency of complement factor H in a patient with age-related macular degeneration and membranoproliferative glomerulonephritis. Mol. Immunol. 2008, 45, 2897–2904. [Google Scholar] [CrossRef]
- Licht, C.; Heinen, S.; Józsi, M.; Löschmann, I.; Saunders, R.E.; Perkins, S.J.; Waldherr, R.; Skerka, C.; Kirschfink, M.; Hoppe, B.; Zipfel, P.F. Deletion of Lys224 in regulatory domain 4 of Factor H reveals a novel pathomechanism for dense deposit disease (MPGN II). Kidney Int. 2006, 70, 42–50. [Google Scholar]
- Meri, S.; Koistinen, V.; Miettinen, A.; Törnroth, T.; Seppälä, I.J. Activation of the alternative pathway of complement by monoclonal lambda light chains in membranoproliferative glomerulonephritis. J. Exp. Med. 1992, 175, 939–950. [Google Scholar] [CrossRef]
- Jokiranta, T.S.; Solomon, A.; Pangburn, M.K.; Zipfel, P.F.; Meri, S. Nephritogenic lambda light chain dimer: A unique human miniautoantibody against complement factor H. J. Immunol. 1999, 163, 4590–4596. [Google Scholar]
- Schneider, M.C.; Prosser, B.E.; Caesar, J.J.; Kugelberg, E.; Li, S.; Zhang, Q.; Quoraishi, S.; Lovett, J.E.; Deane, J.E.; Sim, R.B.; et al. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature 2009, 458, 890–893. [Google Scholar]
- Jurianz, K.; Ziegler, S.; Garcia-Schüler, H.; Kraus, S.; Bohana-Kashtan, O.; Fishelson, Z.; Kirschfink, M. Complement resistance of tumor cells: Basal and induced mechanisms. Mol. Immunol. 1999, 36, 929–939. [Google Scholar] [CrossRef]
- Gasque, P.; Thomas, A.; Fontaine, M.; Morgan, B.P. Complement activation on human neuroblastoma cell lines in vitro: Route of activation and expression of functional complement regulatory proteins. J. Neuroimmunol. 1996, 66, 29–40. [Google Scholar] [CrossRef]
- Junnikkala, S.; Jokiranta, T.S.; Friese, M.A.; Jarva, H.; Zipfel, P.F.; Meri, S. Exceptional resistance of human H2 glioblastoma cells to complement-mediated killing by expression and utilization of factor H and factor H-like protein 1. J. Immunol. 2000, 164, 6075–6081. [Google Scholar]
- Junnikkala, S.; Hakulinen, J.; Jarva, H.; Manuelian, T.; Bjørge, L.; Bützow, R.; Zipfel, P.F.; Meri, S. Secretion of soluble complement inhibitors factor H and factor H-like protein (FHL-1) by ovarian tumour cells. Br. J. Cancer 2002, 87, 1119–1127. [Google Scholar] [CrossRef]
- Ajona, D.; Castaño, Z.; Garayoa, M.; Zudaire, E.; Pajares, M.J.; Martinez, A.; Cuttitta, F.; Montuenga, L.M.; Pio, R. Expression of complement factor H by lung cancer cells: Effects on the activation of the alternative pathway of complement. Cancer Res. 2004, 64, 6310–6318. [Google Scholar]
- Wilczek, E.; Rzepko, R.; Nowis, D.; Legat, M.; Golab, J.; Glab, M.; Gorlewicz, A.; Konopacki, F.; Mazurkiewicz, M.; Sladowski, D.; et al. The possible role of factor H in colon cancer resistance to complement attack. Int. J. Cancer 2008, 122, 2030–2037. [Google Scholar] [CrossRef]
- Cheng, Z.Z.; Corey, M.J.; Pärepalo, M.; Majno, S.; Hellwage, J.; Zipfel, P.F.; Kinders, R.J.; Raitanen, M.; Meri, S.; Jokiranta, T.S. Complement factor H as a marker for detection of bladder cancer. Clin. Chem. 2005, 51, 856–863. [Google Scholar]
- Cui, T.; Chen, Y.; Knösel, T.; Yang, L.; Zöller, K.; Galler, K.; Berndt, A.; Mihlan, M.; Zipfel, P.F.; Petersen, I. Human complement factor H is a novel diagnostic marker for lung adenocarcinoma. Int. J. Oncol. 2011, 39, 161–168. [Google Scholar]
- Fedarko, N.S.; Fohr, B.; Robey, P.G.; Young, M.F.; Fisher, L.W. Factor H binding to bone sialoprotein and osteopontin enables tumor cell evasion of complement-mediated attack. J. Biol. Chem. 2000, 275, 16666–16672. [Google Scholar]
- Amornsiripanitch, N.; Hong, S.; Campa, M.J.; Frank, M.M.; Gottlin, E.B.; Patz, E.F., Jr. Complement factor H autoantibodies are associated with early stage NSCLC. Clin. Cancer Res. 2010, 16, 3226–3231. [Google Scholar] [CrossRef]
- Lambris, J.D.; Dobson, N.J.; Ross, G.D. Release of endogenous C3b inactivator from lymphocytes in response to triggering membrane receptors for β1H globulin. J. Exp. Med. 1980, 152, 1625–1644. [Google Scholar] [CrossRef]
- Hammann, K.P.; Raile, A.; Schmitt, M.; Mussel, H.H.; Peters, H.; Scheiner, O.; Dierich, M.P. beta 1H stimulates mouse-spleen B lymphocytes as demonstrated by increased thymidine incorporation and formation of B cell blasts. Immunobiology 1981, 160, 289–301. [Google Scholar] [CrossRef]
- Tsokos, G.C.; Inghirami, G.; Tsoukas, C.D.; Balow, J.E.; Lambris, J.D. Regulation of immunoglobulin secretion by factor H of human complement. Immunobiology 1985, 55, 419–426. [Google Scholar]
- Lambris, J.D.; Ross, G.D. Characterization of the lymphocyte membrane receptor for factor H (β1H-globulin) with an antibody to anti-factor H idiotype. J. Exp. Med. 1982, 155, 1400–1411. [Google Scholar] [CrossRef]
- Erdei, A.; Sim, R.B. Complement Factor H-binding protein of Raji cells and tonsil B lymphocytes. Biochem. J. 1987, 246, 149–156. [Google Scholar]
- Avery, V.M.; Gordon, D.L. Characterization of factor H binding to human polymorphonuclear leukocytes. J. Immunol. 1993, 151, 5545–5553. [Google Scholar]
- DiScipio, R.G.; Daffern, P.J.; Schraufstätter, I.U.; Sriramarao, P. Human polymorphonuclear leukocytes adhere to complement factor H through an interaction that involves alphaMbeta2 (CD11b/CD18). J. Immunol. 1998, 160, 4057–4066. [Google Scholar]
- Agarwal, V.; Asmat, T.M.; Luo, S.; Jensch, I.; Zipfel, P.F.; Hammerschmidt, S. Complement regulator Factor H mediates a two-step uptake of Streptococcus pneumoniae by human cells. J. Biol. Chem. 2010, 285, 23486–23495. [Google Scholar]
- Agarwal, S.; Ram, S.; Ngampasutadol, J.; Gulati, S.; Zipfel, P.F.; Rice, P.A. Factor H facilitates adherence of Neisseria gonorrhoeae to complement receptor 3 on eukaryotic cells. J. Immunol. 2010, 185, 4344–4353. [Google Scholar] [CrossRef]
- Strohmeyer, R.; Ramirez, M.; Cole, G.J.; Mueller, K.; Rogers, J. Association of factor H of the alternative pathway of complement with agrin and complement receptor 3 in the Alzheimer’s disease brain. J. Neuroimmunol. 2002, 131, 135–146. [Google Scholar] [CrossRef]
- Schopf, R.E.; Hammann, K.P.; Scheiner, O.; Lemmel, E.M.; Dierich, M.P. Activation of human monocytes by both human beta 1H and C3b. Immunology 1982, 46, 307–312. [Google Scholar]
- Iferroudjene, D.; Schouft, M.T.; Lemercier, C.; Gilbert, D.; Fontaine, M. Evidence for an active hydrophobic form of factor H that is able to induce secretion of interleukin 1-beta or by human monocytes. Eur. J. Immunol. 1991, 21, 967–972. [Google Scholar] [CrossRef]
- Ohtsuka, H.; Imamura, T.; Matsushita, M.; Tanase, S.; Okada, H.; Ogawa, M.; Kambara, T. Thrombin generates monocyte chemotactic activity from complement factor H. Immunology 1993, 80, 140–145. [Google Scholar]
- Nabil, K.; Rihn, B.; Jaurand, M.C.; Vignaud, J.M.; Ripoche, J.; Martinet, Y.; Martinet, N. Identification of human complement factor H as a chemotactic protein for monocytes. Biochem. J. 1997, 326, 377–383. [Google Scholar]
- Hartung, H.P.; Hadding, U.; Bitter-Suermann, D.; Gemsa, D. Release of prostaglandin E and thromboxane from macrophages by stimulation with factor H. Clin. Exp. Immunol. 1984, 56, 453–458. [Google Scholar]
- Kang, Y.H.; Urban, B.C.; Sim, R.B.; Kishore, U. Human complement Factor H modulates C1q-mediated phagocytosis of apoptotic cells. Immunobiology 2011. [Google Scholar] [CrossRef]
- Svobodová, E.; Józsi, M. Characterization of factor H binding to monocytes. 2012; unpublished data. [Google Scholar]
- Malhotra, R.; Ward, M.; Sim, R.B.; Bird, M.I. Identification of human complement Factor H as a ligand for L-selectin. Biochem. J. 1999, 341, 61–69. [Google Scholar] [CrossRef]
- Vaziri-Sani, F.; Hellwage, J.; Zipfel, P.F.; Sjöholm, A.G.; Iancu, R.; Karpman, D. Factor H binds to washed human platelets. J. Thromb. Haemost. 2005, 3, 154–162. [Google Scholar] [CrossRef]
- Mnjoyan, Z.; Li, J.; Afshar-Kharghan, V. Factor H binds to platelet integrin alphaIIbbeta3. Platelets 2008, 19, 512–519. [Google Scholar] [CrossRef]
- Takeda, K.; Thurman, J.M.; Tomlinson, S.; Okamoto, M.; Shiraishi, Y.; Ferreira, V.P.; Cortes, C.; Pangburn, M.K.; Holers, V.M.; Gelfand, E.W. The Critical role of complement alternative pathway regulator factor H in allergen-induced airway hyperresponsiveness and inflammation. J. Immunol. 2012, 188, 661–667. [Google Scholar]
- Schmidt, C.Q.; Slingsby, F.C.; Richards, A.; Barlow, P.N. Production of biologically active complement factor H in therapeutically useful quantities. Protein Expr. Purif. 2011, 76, 254–263. [Google Scholar] [CrossRef]
- Büttner-Mainik, A.; Parsons, J.; Jérôme, H.; Hartmann, A.; Lamer, S.; Schaaf, A.; Schlosser, A.; Zipfel, P.F.; Reski, R.; Decker, E.L. Production of biologically active recombinant human factor H in Physcomitrella. Plant Biotechnol. J. 2011, 9, 373–383. [Google Scholar] [CrossRef]
- Tan, L.A.; Yang, A.C.; Kishore, U.; Sim, R.B. Interactions of complement proteins C1q and factor H with lipid A and Escherichia coli: Further evidence that factor H regulates the classical complement pathway. Protein Cell 2011, 2, 320–332. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kopp, A.; Hebecker, M.; Svobodová, E.; Józsi, M. Factor H: A Complement Regulator in Health and Disease, and a Mediator of Cellular Interactions. Biomolecules 2012, 2, 46-75. https://doi.org/10.3390/biom2010046
Kopp A, Hebecker M, Svobodová E, Józsi M. Factor H: A Complement Regulator in Health and Disease, and a Mediator of Cellular Interactions. Biomolecules. 2012; 2(1):46-75. https://doi.org/10.3390/biom2010046
Chicago/Turabian StyleKopp, Anne, Mario Hebecker, Eliška Svobodová, and Mihály Józsi. 2012. "Factor H: A Complement Regulator in Health and Disease, and a Mediator of Cellular Interactions" Biomolecules 2, no. 1: 46-75. https://doi.org/10.3390/biom2010046
APA StyleKopp, A., Hebecker, M., Svobodová, E., & Józsi, M. (2012). Factor H: A Complement Regulator in Health and Disease, and a Mediator of Cellular Interactions. Biomolecules, 2(1), 46-75. https://doi.org/10.3390/biom2010046