
PFKFB3 Inhibitor 3PO Reduces Cardiac Remodeling after Myocardial Infarction by Regulating the TGF-β1/SMAD2/3 Pathway
 ,
,
Abstract
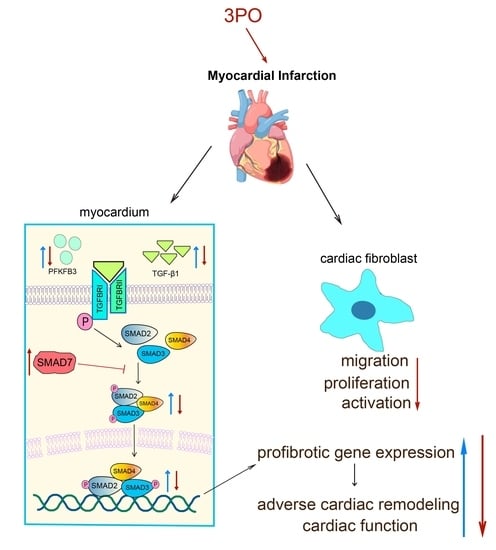
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Isolation and Culture of Neonatal Rat Cardiac Fibroblasts (NRCFs)
2.3. Induction of MI
2.4. Western Blotting
2.5. Immunofluorescence Analyses
2.6. Immunocytochemistry
2.7. Pathological Staining and TdT-Mediated dUTP Nick-End Labeling (TUNEL) Staining
2.8. Echocardiography
2.9. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.10. Migration and Proliferation Evaluation of NRCFs with Stimulation
2.11. Statistical Analysis
3. Results
3.1. PFKFB3 Expression Increases in Cardiac Tissue after MI
3.2. 3PO Ameliorates Cardiac Remodeling Induced by MI
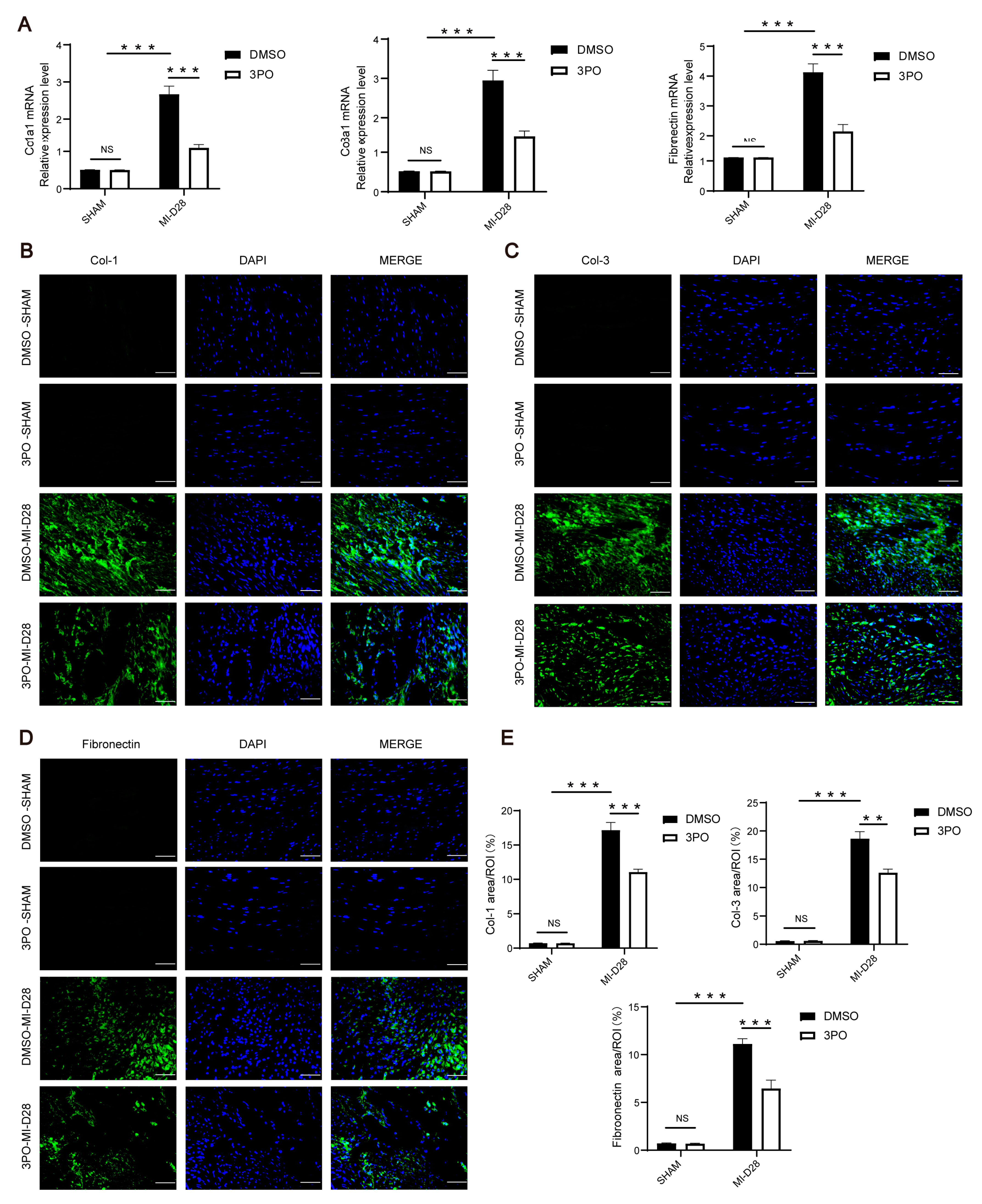
3.3. 3PO Attenuates Cardiac Fibrosis Induced by MI
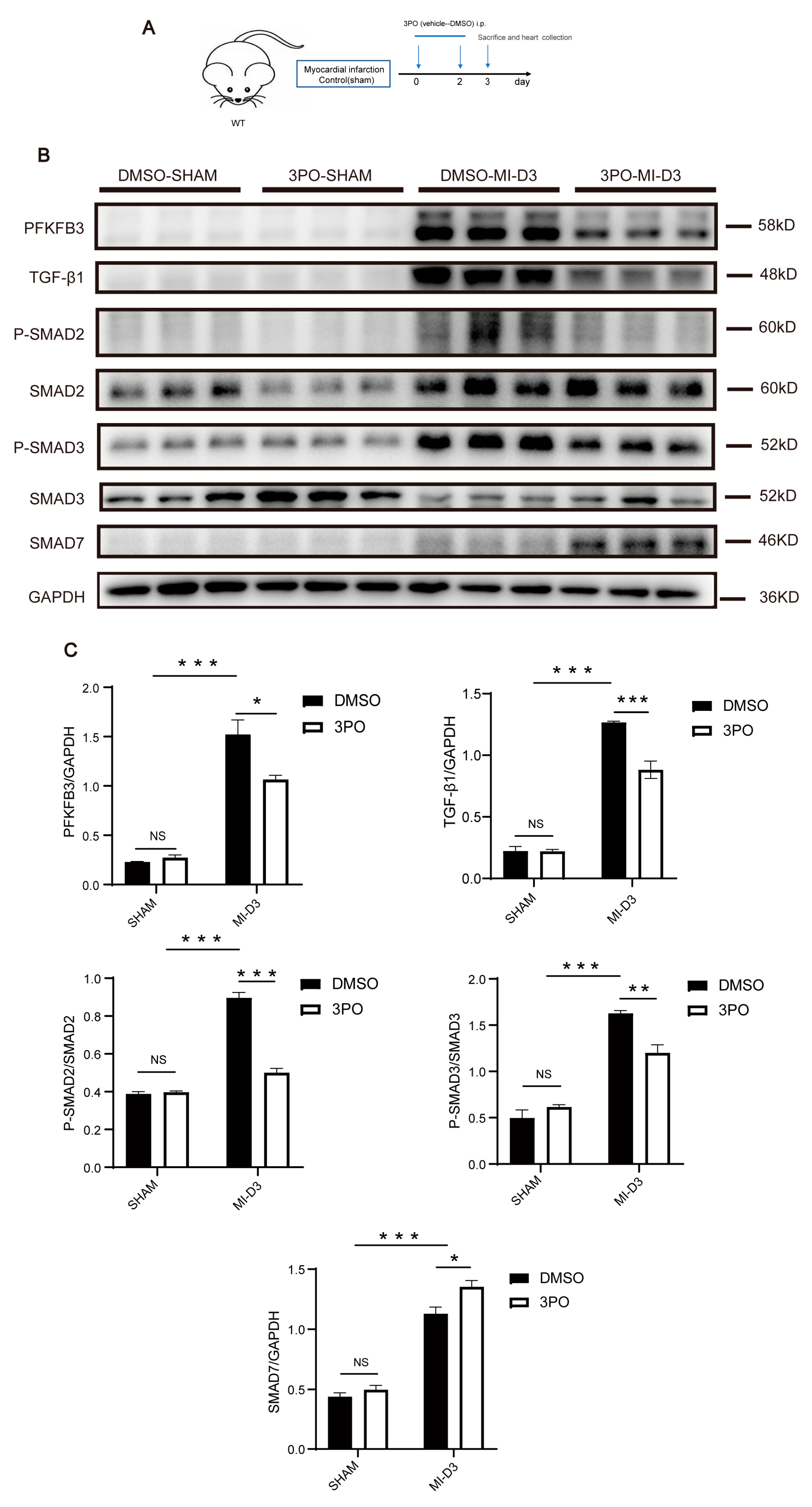
3.4. 3PO Decreases Expression of PFKFB3 and Inhibits TGF-β1/SMAD2/3 Pathway in Cardiac Tissue after MI
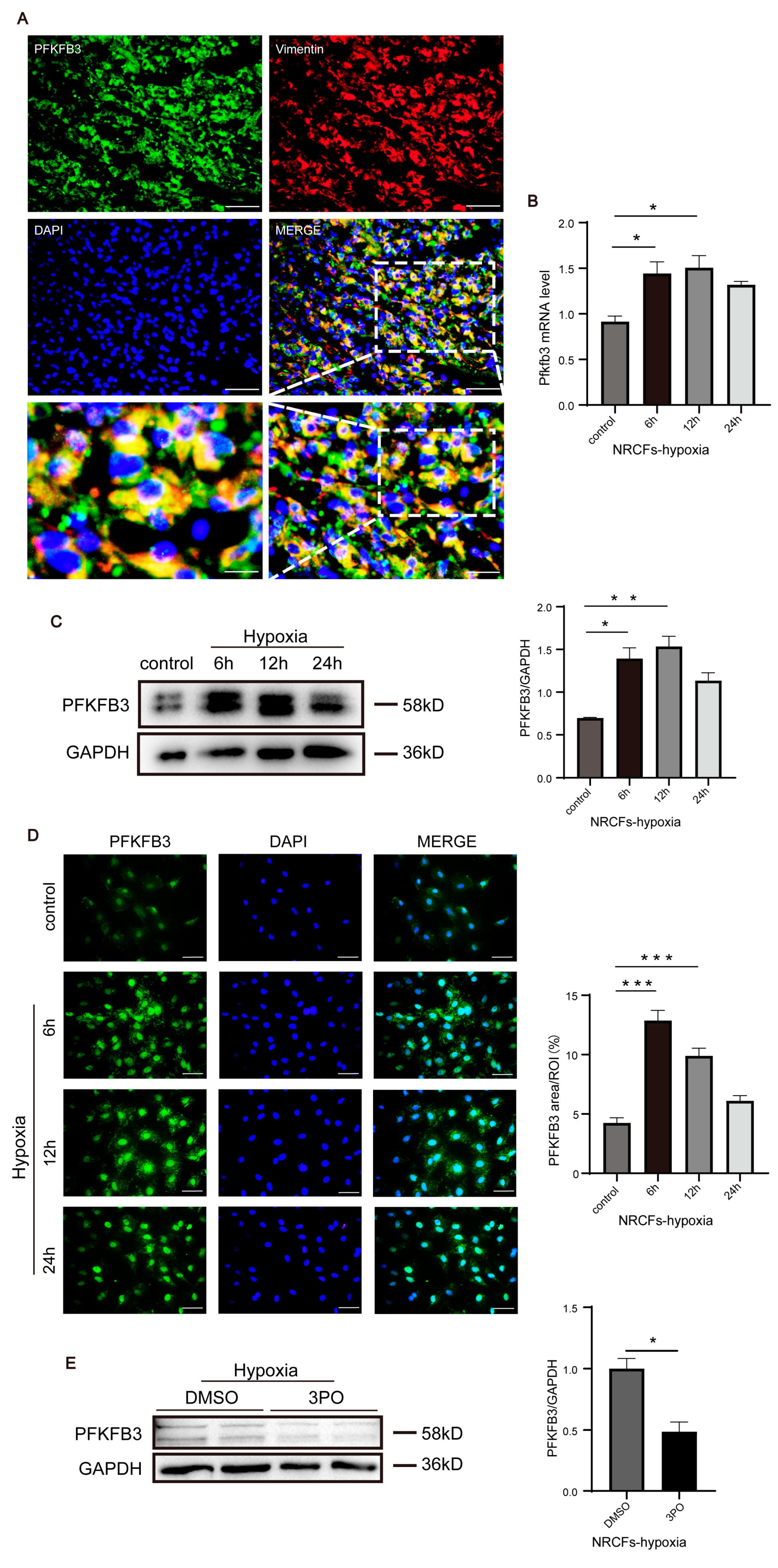
3.5. PFKFB3 Is Abundantly Expressed in CFs following MI and under Hypoxia
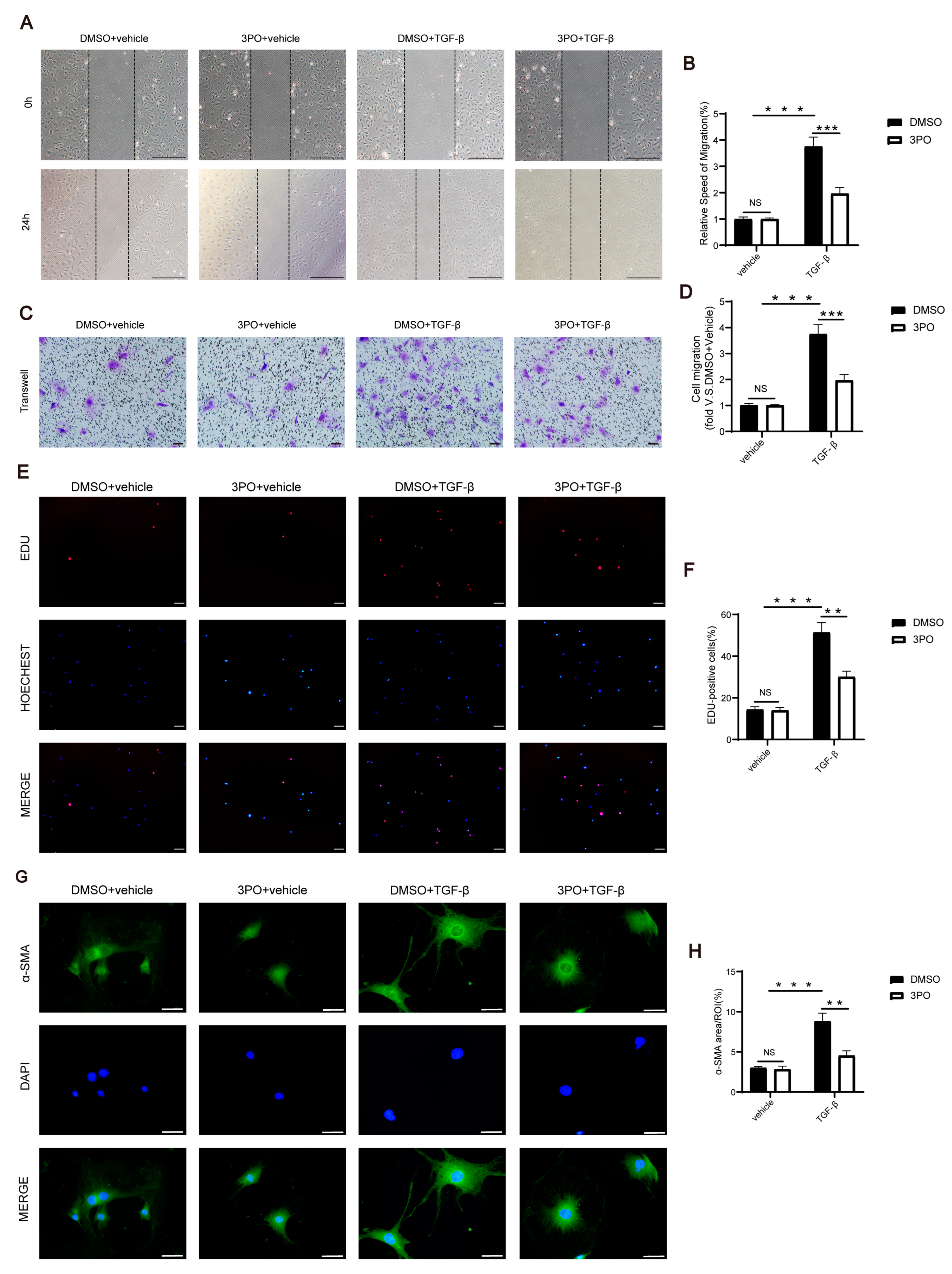
3.6. 3PO Mitigates the Migration, Proliferation, and Activation of CFs Induced by TGF-β1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Schumacher, D.; Kramann, R. Multiomic Spatial Mapping of Myocardial Infarction and Implications for Personalized Therapy. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Hundertmark, M.J.; Schulz-Menger, J.; Bengel, F.M.; Bauersachs, J. Left ventricular remodelling post-myocardial infarction: Pathophysiology, imaging, and novel therapies. Eur. Heart J. 2022, 43, 2549–2561. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Bacmeister, L.; Schwarzl, M.; Warnke, S.; Stoffers, B.; Blankenberg, S.; Westermann, D.; Lindner, D. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol. 2019, 114, 19. [Google Scholar] [CrossRef]
- Grosche, J.; Meißner, J.; Eble, J.A. More than a syllable in fib-ROS-is: The role of ROS on the fibrotic extracellular matrix and on cellular contacts. Mol. Asp. Med. 2018, 63, 30–46. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.; Sassi, Y.; Troncone, L.; Benard, L.; Ishikawa, K.; Gordon, R.E.; Lamas, S.; Laborda, J.; Hajjar, R.J.; Lebeche, D. Deletion of delta-like 1 homologue accelerates fibroblast-myofibroblast differentiation and induces myocardial fibrosis. Eur. Heart J. 2019, 40, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Deten, A.; Hölzl, A.; Leicht, M.; Barth, W.; Zimmer, H.G. Changes in extracellular matrix and in transforming growth factor beta isoforms after coronary artery ligation in rats. J. Mol. Cell. Cardiol. 2001, 33, 1191–1207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Signal. Transduct. Target. 2022, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Christensen, G.; Herum, K.M.; Lunde, I.G. Sweet, yet underappreciated: Proteoglycans and extracellular matrix remodeling in heart disease. Matrix Biol. 2019, 75–76, 286–299. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Bao, Y.; Ding, J.; Li, H.; Liu, W.; Wang, X.; Guan, H.; Chen, Z. MicroRNA-130a attenuates cardiac fibrosis after myocardial infarction through TGF-β/Smad signaling by directly targeting TGF-β receptor 1. Bioengineered 2022, 13, 5779–5791. [Google Scholar] [CrossRef]
- Li, J.; Ge, F.; Wuken, S.; Jiao, S.; Chen, P.; Huang, M.; Gao, X.; Liu, J.; Tu, P.; Chai, X.; et al. Zerumbone, a humulane sesquiterpene from Syringa pinnatifolia, attenuates cardiac fibrosis by inhibiting of the TGF-β1/Smad signaling pathway after myocardial infarction in mice. Phytomedicine 2022, 100, 154078. [Google Scholar] [CrossRef] [PubMed]
- Mehdipoor, M.; Damirchi, A.; Tousi, S.; Babaei, P. Correction to: Concurrent vitamin D supplementation and exercise training improve cardiac fibrosis via TGF-β/Smad signaling in myocardial infarction model of rats. J. Physiol. Biochem. 2021, 77, 341. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, B.; Xu, Y.; Zhou, N.; Zhang, R.; Lu, L.; Feng, Z. MiR-208b/miR-21 Promotes the Progression of Cardiac Fibrosis Through the Activation of the TGF-β1/Smad-3 Signaling Pathway: An in vitro and in vivo Study. Front. Cardiovasc. Med. 2022, 9, 924629. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.C.; Pohlmann, P.R.; Clarke, R.; Sengupta, S. Treatment against glucose-dependent cancers through metabolic PFKFB3 targeting of glycolytic flux. Cancer Metastasis Rev. 2022, 41, 447–458. [Google Scholar] [CrossRef]
- Bousseau, S.; Vergori, L.; Soleti, R.; Lenaers, G.; Martinez, M.C.; Andriantsitohaina, R. Glycosylation as new pharmacological strategies for diseases associated with excessive angiogenesis. Pharmacol. Ther. 2018, 191, 92–122. [Google Scholar] [CrossRef]
- Wang, Y.; Qu, C.; Liu, T.; Wang, C. PFKFB3 inhibitors as potential anticancer agents: Mechanisms of action, current developments, and structure-activity relationships. Eur. J. Med. Chem. 2020, 203, 112612. [Google Scholar] [CrossRef]
- Yang, Z.; Goronzy, J.J.; Weyand, C.M. The glycolytic enzyme PFKFB3/phosphofructokinase regulates autophagy. Autophagy 2014, 10, 382–383. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Tang, J.; Lu, M.; Zhou, Z.; Li, Y.; Tian, H.; Liu, E.; Gao, B.; Liu, T.; Shao, P. Glycolysis Rate-Limiting Enzymes: Novel Potential Regulators of Rheumatoid Arthritis Pathogenesis. Front. Immunol. 2021, 12, 779787. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in cancer. Signal Transduct. Target. 2017, 2, 17044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Zhang, X.; Wang, L.; Yang, Q.; Ma, Q.; Xu, J.; Wang, J.; Kovacs, L.; Ayon, R.J.; Liu, Z.; et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13394–13403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Li, A.; Fu, X.; Li, Z.; Cao, K.; Song, M.; Huang, S.; Li, Z.; Yan, J.; Wang, L.; et al. Gene-dosage effect of Pfkfb3 on monocyte/macrophage biology in atherosclerosis. Br. J. Pharm. 2022, 179, 4974–4991. [Google Scholar] [CrossRef] [PubMed]
- Kassa, B.; Kumar, R.; Mickael, C.; Sanders, L.; Vohwinkel, C.; Lee, M.H.; Gu, S.; Poth, J.M.; Stenmark, K.R.; Zhao, Y.Y.; et al. Endothelial cell PHD2-HIF1α-PFKFB3 contributes to right ventricle vascular adaptation in pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L675–L685. [Google Scholar] [CrossRef] [PubMed]
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer 2008, 7, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Kotowski, K.; Rosik, J.; Machaj, F.; Supplitt, S.; Wiczew, D.; Jabłońska, K.; Wiechec, E.; Ghavami, S.; Dzięgiel, P. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers 2021, 13, 909. [Google Scholar] [CrossRef]
- Hu, X.; Xu, Q.; Wan, H.; Hu, Y.; Xing, S.; Yang, H.; Gao, Y.; He, Z. PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab. Investig. 2020, 100, 801–811. [Google Scholar] [CrossRef]
- Mejias, M.; Gallego, J.; Naranjo-Suarez, S.; Ramirez, M.; Pell, N.; Manzano, A.; Suñer, C.; Bartrons, R.; Mendez, R.; Fernandez, M. CPEB4 Increases Expression of PFKFB3 to Induce Glycolysis and Activate Mouse and Human Hepatic Stellate Cells, Promoting Liver Fibrosis. Gastroenterology 2020, 159, 273–288. [Google Scholar] [CrossRef]
- Konhilas, J.P.; Leinwand, L.A. The effects of biological sex and diet on the development of heart failure. Circulation 2007, 116, 2747–2759. [Google Scholar] [CrossRef]
- Lin, J.; Steenbergen, C.; Murphy, E.; Sun, J. Estrogen receptor-beta activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation 2009, 120, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrotta, P.; Van der Veken, B.; Van Der Veken, P.; Pintelon, I.; Roosens, L.; Adriaenssens, E.; Timmerman, V.; Guns, P.J.; De Meyer, G.R.Y.; Martinet, W. Partial Inhibition of Glycolysis Reduces Atherogenesis Independent of Intraplaque Neovascularization in Mice. Arter. Thromb. Vasc. Biol. 2020, 40, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Jia, K.; Chen, C.; Li, Z.; Zhao, J.; Hu, J.; Zhang, H.; Fan, Q.; Huang, C.; Xie, H.; et al. DYRK1B-STAT3 Drives Cardiac Hypertrophy and Heart Failure by Impairing Mitochondrial Bioenergetics. Circulation 2022, 145, 829–846. [Google Scholar] [CrossRef]
- Gao, E.; Lei, Y.H.; Shang, X.; Huang, Z.M.; Zuo, L.; Boucher, M.; Fan, Q.; Chuprun, J.K.; Ma, X.L.; Koch, W.J. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 2010, 107, 1445–1453. [Google Scholar] [CrossRef]
- Fan, Q.; Tao, R.; Zhang, H.; Xie, H.; Lu, L.; Wang, T.; Su, M.; Hu, J.; Zhang, Q.; Chen, Q.; et al. Dectin-1 Contributes to Myocardial Ischemia/Reperfusion Injury by Regulating Macrophage Polarization and Neutrophil Infiltration. Circulation 2019, 139, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Z.S.; Tang, S.Q.; Xing, T.; Zhou, Y.; Lv, M.; Fu, H.X.; Wang, Y.; Xu, L.P.; Zhang, X.H.; Lee, H.Y.; et al. The glycolytic enzyme PFKFB3 determines bone marrow endothelial progenitor cell damage after chemotherapy and irradiation. Haematologica 2022, 107, 2365–2380. [Google Scholar] [CrossRef]
- Liu, J.; Jin, J.; Liang, T.; Feng, X.H. To Ub or not to Ub: A regulatory question in TGF-β signaling. Trends Biochem. Sci. 2022, 47, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Xie, Z.; Liu, Q.; Zhuang, Y.; Xie, W.; Wang, X.; Gao, W.; Yang, F.; Li, Z.; et al. Inhibition of PFKFB Preserves Intestinal Barrier Function in Sepsis by Inhibiting NLRP3/GSDMD. Oxid. Med. Cell. Longev. 2022, 2022, 8704016. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Chen, S.; Li, S.; Wang, N.; Zhang, S.; Xu, L.; Zhu, S.; Li, H.; Gu, Q.; Xu, X.; et al. Enhancement of glycolysis-dependent DNA repair regulated by FOXO1 knockdown via PFKFB3 attenuates hyperglycemia-induced endothelial oxidative stress injury. Redox Biol. 2022, 59, 102589. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Wei, Q.; Livingston, M.J.; Dong, G.; Li, S.; Hu, X.; Li, Y.; Huo, Y.; Dong, Z. PFKFB3 mediates tubular cell death in cisplatin nephrotoxicity by activating CDK4. Transl. Res. 2023, 253, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Vohwinkel, C.U.; Burns, N.; Coit, E.; Yuan, X.; Vladar, E.K.; Sul, C.; Schmidt, E.P.; Carmeliet, P.; Stenmark, K.; Nozik, E.S.; et al. HIF1A-dependent induction of alveolar epithelial PFKFB3 dampens acute lung injury. JCI Insight 2022, 7, e157855. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Guo, H.S.; Li, C.Y.; Cao, W.; Wang, X.Y.; Mo, D.; Hao, X.W.; Feng, Y.D.; Sun, Y.; Lei, F.; et al. PFKFB3 promotes endotoxemia-induced myocardial dysfunction through inflammatory signaling and apoptotic induction. Toxicol. Appl. Pharmacol. 2019, 368, 26–36. [Google Scholar] [CrossRef]
- Calvieri, C.; Riva, A.; Sturla, F.; Dominici, L.; Conia, L.; Gaudio, C.; Miraldi, F.; Secchi, F.; Galea, N. Left Ventricular Adverse Remodeling in Ischemic Heart Disease: Emerging Cardiac Magnetic Resonance Imaging Biomarkers. J. Clin. Med. 2023, 12, 334. [Google Scholar] [CrossRef]
- Sutton, M.G.; Sharpe, N. Left ventricular remodeling after myocardial infarction: Pathophysiology and therapy. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef]
- Greuter, T.; Yaqoob, U.; Gan, C.; Jalan-Sakrikar, N.; Kostallari, E.; Lu, J.; Gao, J.; Sun, L.; Liu, M.; Sehrawat, T.S.; et al. Mechanotransduction-induced glycolysis epigenetically regulates a CXCL1-dominant angiocrine signaling program in liver sinusoidal endothelial cells in vitro and in vivo. J. Hepatol. 2022, 77, 723–734. [Google Scholar] [CrossRef]
- Henderson, J.; Duffy, L.; Stratton, R.; Ford, D.; O’Reilly, S. Metabolic reprogramming of glycolysis and glutamine metabolism are key events in myofibroblast transition in systemic sclerosis pathogenesis. J. Cell. Mol. Med. 2020, 24, 14026–14038. [Google Scholar] [CrossRef]
- Xu, J.; Li, J.; Yu, Z.; Rao, H.; Wang, S.; Lan, H. HMGB1 promotes HLF-1 proliferation and ECM production through activating HIF1-α-regulated aerobic glycolysis. Pulm. Pharmacol. Ther. 2017, 45, 136–141. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, H.; Fan, Q.; Hu, J.; Tao, R.; Chen, Q.; Iwakura, Y.; Shen, W.; Lu, L.; Zhang, Q.; et al. Dectin-2 Deficiency Modulates Th1 Differentiation and Improves Wound Healing After Myocardial Infarction. Circ. Res. 2017, 120, 1116–1129. [Google Scholar] [CrossRef]
- Hofmann, U.; Knorr, S.; Vogel, B.; Weirather, J.; Frey, A.; Ertl, G.; Frantz, S. Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction. Circ. Heart Fail. 2014, 7, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The role of transforming growth factor (TGF)-β in the infarcted myocardium. J. Thorac. Dis. 2017, 9 (Suppl. S1), S52–S63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquière, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boscaro, C.; Baggio, C.; Carotti, M.; Sandonà, D.; Trevisi, L.; Cignarella, A.; Bolego, C. Targeting of PFKFB3 with miR-206 but not mir-26b inhibits ovarian cancer cell proliferation and migration involving FAK downregulation. FASEB J. 2022, 36, e22140. [Google Scholar] [CrossRef]
- Dou, Q.; Grant, A.K.; Callahan, C.; Coutinho de Souza, P.; Mwin, D.; Booth, A.L.; Nasser, I.; Moussa, M.; Ahmed, M.; Tsai, L.L. PFKFB3-mediated Pro-glycolytic Shift in Hepatocellular Carcinoma Proliferation. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 61–75. [Google Scholar] [CrossRef]
- Thirusangu, P.; Ray, U.; Sarkar Bhattacharya, S.; Oien, D.B.; Jin, L.; Staub, J.; Kannan, N.; Molina, J.R.; Shridhar, V. Correction to: PFKFB3 regulates cancer stemness through the hippo pathway in small cell lung carcinoma. Oncogene 2023, 42, 79–82. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Zong, X.; Zhuang, L.; Pan, R.; Tudi, X.; Fan, Q.; Tao, R. PFKFB3 Inhibitor 3PO Reduces Cardiac Remodeling after Myocardial Infarction by Regulating the TGF-β1/SMAD2/3 Pathway. Biomolecules 2023, 13, 1072. https://doi.org/10.3390/biom13071072
Yang Q, Zong X, Zhuang L, Pan R, Tudi X, Fan Q, Tao R. PFKFB3 Inhibitor 3PO Reduces Cardiac Remodeling after Myocardial Infarction by Regulating the TGF-β1/SMAD2/3 Pathway. Biomolecules. 2023; 13(7):1072. https://doi.org/10.3390/biom13071072
Chicago/Turabian StyleYang, Qian, Xiao Zong, Lingfang Zhuang, Roubai Pan, Xierenayi Tudi, Qin Fan, and Rong Tao. 2023. "PFKFB3 Inhibitor 3PO Reduces Cardiac Remodeling after Myocardial Infarction by Regulating the TGF-β1/SMAD2/3 Pathway" Biomolecules 13, no. 7: 1072. https://doi.org/10.3390/biom13071072