
From Hamamelitannin Synthesis to the Study of Enzymatic Acylations of D-Hamamelose

Institute of Chemistry, Slovak Academy of Sciences, Dúbravská Cesta 9, 845 38 Bratislava, Slovakia
*
Author to whom correspondence should be addressed.
Biomolecules 2023, 13(3), 519; https://doi.org/10.3390/biom13030519
Submission received: 23 February 2023
/
Revised: 8 March 2023
/
Accepted: 9 March 2023
/
Published: 12 March 2023
(This article belongs to the Special Issue Recent Advances in the Enzymatic Synthesis of Phenolic Compounds Derivatives)
Abstract
:The bioactive natural substance, hamamelitannin, was effectively synthesized in two ways. The chemical acylation of 2,3-O-isopropylidene-α,β-D-hamamelofuranose promoted by Bu2SnO using 3,4,5-tri-O-acetylgalloyl chloride, followed by the deprotection provided hamamelitannin in 79%. Pilot enzymatic benzoylation of D-hamamelose using vinyl benzoate (4 equiv.) and Lipozyme TL IM as a biocatalyst in t-butyl methyl ether (t-BuMeO) gave mainly benzoylated furanoses (89%), of which tribenzoates reached (52%). Enzymatic galloylation of 2,3-O-isopropylidene-α,β-D-hamamelofuranose with vinyl gallate under the catalysis of Lipozyme TL IM in t-butyl alcohol (t-BuOH) or t-BuMeO provided only the 5-O-galloylated product. The reaction in t-BuMeO proceeded in a shorter reaction time (61 h) and higher yield (82%). The more hydrophobic vinyl 3,4,5-tri-O-acetylgallate in the same reactions gave large amounts of acetylated products. Vinyl gallate and triacetylgallate in the enzymatic acylation of D-hamamelose with Lipozyme TL IM in t-BuMeO yielded 2′,5-diacylated hamamelofuranoses in a yield below 20%. The use of other vinyl gallates hydrophobized by methylation or benzylation provided 2′,5-diacylated hamamelofuranoses in good yields (65–84%). The reaction with silylated vinyl gallate did not proceed. The best results were obtained with vinyl 2,3,5-tri-O-benzyl gallate, and the only product, 2′,5-diacylated hamamelofuranoside precipitated from the reaction mixture (84% in 96 h). After debenzylation, hamamelitannin was obtained an 82% yield from hamamelose in two steps. This synthesis is preparatively undemanding and opens the way to multigram preparations of bioactive hamamelitannin and its analogues.
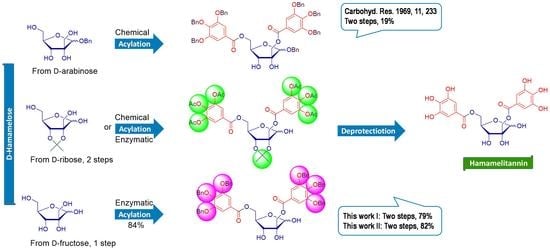
1. Introduction
Tannins are polyphenolic plant secondary metabolites that have enormous structural diversity [1]. Hydrolyzable tannins are often isolated from plants for their remarkable therapeutic effects [2]. Their structure generally consists of a central sugar core, typically a glucose unit, to which galloyl groups, often meta-depsidically bonded (gallotannins) or C-C bonded dehydrodigalloyl units (ellagitannins), are attached [3]. The total synthesis of such compounds tends to be complex [4,5,6].
Hamamelitannin (2′,5-di-O-galloyl-2-C-(hydroxymethyl)-D-ribofuranose or 2,5-di-O-galloyl-D-hamamelofuranose, 1, Figure 1) is the main component of the witch hazel bark extract (Hamamelis virginiana L.) [7,8,9]. Different galloylhamameloses, have been isolated to date from the bark of various Fagaceae spp. such as Castanea crenata [10,11], Castanopsis cuspidate [12], or Castanea sativa [13]. Gallotannin 1 is commercially available pure or as a component of various organic extracts. For commercial use, it is usually isolated from witch hazel (Hamamelis virginiana) or sweet chestnut (Castanea sativa).
Extracts and distillates of witch hazel bark, twigs, and leaves containing 1 are widely used as components of skin care products and in dermatological treatment of sunburn, irritated skin, acne, atopic eczema, and to promote wound healing through anti-inflammatory effects [14,15,16,17,18]. Pure gallotannin 1 inhibits the activity of α-TNF (tumor necrosis factor) [19], autoactivation of plasma hyaluronan-binding protein [20] and exhibits high scavenging and protective activity against cell damage by active oxygen and peroxide [21,22,23]. It also appears to be a promising chemotherapeutic agent, which might be used in the treatment of colon cancer without compromising the viability of healthy colon cells [24]. Very recently, 1 [25,26,27], cyclodextrin–hamamelitannin complexes [28], or different synthetic analogues of hamamelitannin analogues [29,30,31] in combination with antibiotics, were studied as perspective suppressors of staphylococcal infections by inhibiting virulence of bacterial biofilms through quorum sensing mechanisms.
The antiviral efficacy against influenza A virus and human papillomavirus of tannins from Hamamelis virginiana bark extract has also been demonstrated [32]. Gallotannin 1 has also become a relatively successful molecule in various in silico screening models aimed at studying the inhibition of proteins important in the process of carcinogenesis, atherosclerosis, or SARS-CoV-2 disease [33,34,35,36].
Despite numerous reports on the medical effects of hamamelitannin, only one total synthesis of 1 has been published so far, as early as 1969 [37]. The authors obtained the target di-O-acyl-glycoside in only 22% yield by conventional acylation of the prepared benzyl β-D-hamamelofuranoside with tri-O-benzylgalloyl chloride in a pyridine/tetrahydrofuran mixture. Acylation proceeded for 71 h at −40° to rt and afforded three products. The main product was hydrogenated over 10% palladium on charcoal to give compound 1 with a yield of 58%. The starting branched sugar D-hamamelose (2-C-hydroxymethyl-D-ribose) was prepared from D-arabinose in several steps via methyl 3,4-O-isopropylidene-β-D-erythro-pentopyranosidulose [38].
Regioselective acylation of polyhydroxylated molecules like sugars is often a challenge [39]. The solution is to use biocatalysts, especially lipases, and perform enzyme-catalyzed acylation [40,41]. Lipases tolerate a wide range of substrates and are able to work in an aqueous environment as well as in organic solvents [42]. Although they have primarily evolved to hydrolyze triacylglycerols with long fatty acids, some of them also tolerate phenolic substrates, making them similar in reactivity to feruloyl esterases [43]. In aprotic organic solvents, they can catalyze esterifications or transesterifications. The choice of a suitable solvent in reactions is important for their speed as well as selectivity [44,45]. Several reaction steps can be saved by the appropriate selection of the biocatalyst and reaction conditions in the acylation of carbohydrates. They work under mild conditions, so reactions of this type do not consume much energy. Moreover, they are commonly commercially available and can be used multiple times.
In this study, we report two simpler and more efficient syntheses of hamamelitannin 1 from different starting compounds by conventional as well as lipase-promoted galloylation. The regioselectivity of various methods of galloylation of hamamelofuranose, including the enzymatic procedure, was studied.
2. Materials and Methods
2.1. General
The reactions were performed with commercial reagents purchased from Sigma-Aldrich (Saint-Louis, MI, USA), Acrōs Organics (part of Thermo Fisher Scientific, Waltham, MA, USA), Merck (Darmstadt, Germany), Fluorochem (Hadfield, UK). Molecular sieves with porosity 4Å were microwave-dried before use. Dichloromethane (P2O5), toluene (Na), acetonitrile (CaH2) were dried and distilled before use. D-Hamamelose was a gift from the Production Department of our Institute (Institute of Chemistry SAS, Bratislava, Slovakia). The Lipozyme TL IM, a product of Novozymes (Bagsværd, Denmark), was purchased from Biotech (Trnava, Slovakia). The solvents used in the enzymatic reactions t-butanol (t-BuOH), acetonitril (CH3CN) and t-butyl methyl ether (t-BuMeO) were of HPLC grade and predried over molecular sieves. Toluene (T), ethyl acetate (EtOAc), dichloromethane (CH2Cl2), tetrahydrofuran (THF) and methanol (MeOH) were dried (Na, P2O5, CaH2) and distilled before use. All reactions containing sensitive reagents were carried out under the argon atmosphere. TLC was performed on aluminum sheets pre-coated with silica gel 60 F254 (Merck, Darmstadt, Germany). The spots were visualized under UV lamp (λmax = 254 nm) and charred with 5% sulfuric acid in ethanol containing 1% orcinol and heating with a heat gun. Column chromatography was performed on Silica gel 60 (0.035–0.070 mm, pore diameter ca. 6 nm, Acrōs Organics). Melting points were recorded with a Kofler hot-block and were uncorrected. Optical rotations were measured on a Jasco P2000 polarimeter at 20 °C. The structures of products were determined by a combination of 1H and 13C NMR spectroscopy as well as by two-dimensional homonuclear and heteronuclear techniques (COSY, HSQC) recorded on a 400 MHz Bruker AVANCE III HD 400 MHz equipped with a Prodigy CryoProbe. Chemical shifts are reported in ppm (δ) and are referenced to internal CD3OD (δ 3.31, for 1H and δ 49.00, for 13C) or CHCl3 (δ 7.26, for 1H and δ 77.00, for 13C). Scalar couplings are reported in hertz (Hz). High-resolution mass determination was performed on the Orbitrap Velos Pro Thermo Scientific mass analyzer (ion source HESI, capillary temperature 350 °C, source heater temperature 300 °C).
2.2. Synthesis of Hamamelitannin from D-Ribose
2.2.1. 2,3-O-Isopropylidene-α,β-D-Hamamelofuranose (2)
Potassium carbonate (3.75 g) and an aqueous solution of formaldehyde (37% + 10% MeOH) (50 mL) were dissolved in methanol (75 mL), and 2,3-O-isopropylidene-α,β-D-ribofuranose [46] (9.51 g, 50 mmol) was added to the reaction solution. The reaction mixture was stirred at 80 °C under argon for 40 h, after which time it was neutralized with 1M H2SO4. Evaporation to dryness gave a residue that was extracted with hot ethyl acetate (3 × 100 mL), dried (Na2SO4), and the extracts were concentrated to a syrup. Purification of the crude product by column chromatography on silica gel (toluene:ethyl acetate, 2:1 → 0:1) gave 3 as a homogeneous syrup (8.88 g, 81%, α:β = 1:0.6); [α]D20 = +9.6° (c = 1.0, CH3OH), (lit. [47] [α]D23 = +9.3° (c = 3.0, H2O)). 1H NMR (400 MHz, CDCl3) δ: 5.42 (d, H-1α, transformed into a singlet on addition of D2O), 5.29 (d, H-1β, transformed into a singlet on addition of D2O), 4.58 (s, H-3α), 4.59 (d, J = 3.8 Hz, H-3β), 4.32 (dt, J = 4.3, 3.3, 1.1 Hz, H-4α), 4.24 (bdd, J = 3.3, 1.1 Hz, H-4β), 3.91 (d, J = 12.0 Hz, H-2′aα), 3.83 (d, J = 12.0 Hz, H-2′bα), 3.82 (s, H-2′aβ, H-2′bβ), 3.79–3.67 (m, H-5aβ, H-5bβ, H-5aα, H-5bα), 1.59 (s, CH3β), 1.50 (s, CH3α), 1.46 (s, CH3β),1.44 (s, CH3α). 13C NMR (101 MHz, CDCl3) δ: 114.9 (CMe2β), 113.5 (CMe2α), 103.6 (C-1α), 98.1 (C-1β), 94.5 (C-2α), 91.1 (C-2β), 87.5 (C-4α), 84.0 (C-3α), 83.2 (C-3β), 82.5 (C-4β), 63.3 (C-5β, C-5α), 62.8 (C-2′β), 62.7 (C-2′α), 28.1 (CH3α), 27.6 (CH3α), 27.1 (CH3β), 27.0 (CH3β). HRMS (ESI): m/z calcd for C9H16O6Na ([M + Na]+) 243.08446; found, 243.08389.
2.2.2. 3,4,5-Tri-O-Acetylgalloyl Chloride (3)
To vigorously stirred gallic acid (8.51 g, 50 mmol) in Ac2O (20 mL), 3 drops of concentrated H2SO4 were added at 5 °C. The mixture was then stirred for 60 min at room temperature and poured into an ice/water mixture (200 mL). After 2 h at room temperature, the precipitated white solid was washed with water until the filtrate was neutral, then filtered, dried, and crystallized (ethanol) to afford 12.56 g (85%) of 3,4,5-triacetoxybenzoic acid; Mp: 154–157 °C. To the acetylated gallic acid (5.92 g, 20 mmol) in toluene (80 mL), thionyl chloride (7.4 mL, 100 mmol) was added. The mixture was stirred for 3 h at 70 °C. After the evaporation of the liquids to one-third of the original volume, the mixture was cooled and the precipitated solid was washed with cyclohexane and dried. White solid (5.91 g, 94%) was obtained; Mp 104–105 °C.
2.2.3. Acylation Methods for Galloylation of 2 (Table 1)
Method A: Hamamelofuranose 2 (0.22 g, 1 mmol) and 3, 4,5-tri-O-acetylgalloyl chloride 3 (1.04 g, 3.3 mmol) were dissolved in dry CH2Cl2 (10 mL). Triethylamine (0.139 mL, 1 mmol) and 4-dimethylaminopyridine (0.031 g, 0.25 mmol) were added at 0 °C. The reaction mixture was then stirred for 3 h at laboratory temperature. The mixture was then diluted with CH2Cl2 (40 mL), washed with 1% HCl (10 mL), water (2 × 20 mL), dried over Na2SO4, and concentrated under reduced pressure. Products were isolated by column chromatography of the residue on silica gel (toluene/EtOAc, 2:1 → 1:2).
Method B: Hamamelofuranose 2 (0.22 g, 1 mmol) and dibutyltin oxide (0.548 g, 2.2 mmol) were dissolved in dry methanol (10 mL) and refluxed for 2 h. After evaporation of the solvent, the residue was dried under vacuum, dissolved in dichloromethane (10 mL), and cooled to 0 °C. The acylation reagent 2 (0.693 g, 2.2 mmol) in dichloromethane (6 mL) was added dropwise and then allowed to react at room temperature for 2 h. The resulting mixture was concentrated and directly purified by chromatography (toluene/EtOAc, 2:1 → 1:2).
Method C: Hamamelofuranose 2 (0.22 g, 1 mmol) and triacetylgalloyl chloride 3 (1.04 g, 3.3 mmol) were dissolved in dry CH3CN (5 mL). Zinc oxide (0.9 g, 11 mmol) was added in one portion. The heterogeneous mixture was stirred for 24 h at 40 °C, then diluted with ethyl acetate (10 mL) and filtered through Celite 545. The residue after concentration of the filtrate under reduced pressure was purified by column chromatography on silica gel (toluene/EtOAc, 2:1 → 1:2).
Method D: Isopropylidenated hamamelose 2 (0.220 g, 1 mmol) and vinyl 3,4,5-tri-O-acetylgallate (4a) [48] (0.588 g, 3.0 mmol, 3.0 equiv.) were dissolved in dry t-BuMeO (20 mL) at room temperature. Activated molecular sieves 4Å (0.5 g) and Lipozyme TL IM (0.4 g) were added and the reaction mixture was shaken at 450 rpm and 37 °C for 19 h. The reaction was stopped by filtration; the filter cake was washed with ethyl acetate, and combined organic phases were concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column eluted with toluene/EtOAc (2:1 → 1:2) to afford an unexpected product—2,3-O-isopropylidene-2′-O-acetyl-5-O-(3,4,5-tri-O-acetylgalloyl)-α,β-D-hamamelofuranose (5a-2′-Ac) (34%), 2,5-diacyl 6a (6%), and 5-monoacyl 5a (9%).
Method E: Isopropylidenated hamamelose 2 (0.220 g, 1 mmol) and vinyl gallate (4b) [48] (0.588 g, 3.0 mmol, 3.0 equiv.) were dissolved in dry t-BuOH (10 mL) or t-BuMeO (20 mL) at room temperature. Activated molecular sieves 4Å (0.5 g) and Lipozyme TL IM (0.8 g) were added and the reaction mixture was shaken at 450 rpm and 37 °C for 242 h (in t-BuOH) or 61 h (in t-BuMeO) and then finished by filtration. The filter cake was washed several times with ethyl acetate and combined organic phases were concentrated under reduced pressure. The residue was purified by chromatography on the column of silica-gel eluted with toluene/EtOAc (1:2 → 1:5) to afford 5-O-gallate 5b (0.245 g, 66% in t-BuOH or 0.304 g, 82% in t-BuMeO) as an amorphous white solid.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Acylation of 2 by acyl donors under various conditions.
| Entry | Method 1 | Acyl Donor/Equiv. | Catalyst (Equiv.) | Solvent | Temp. (°C) | Time (h) | 5a 2 (%) | 6a (%) | 7 (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | A | 3/2.2 | 2.0/0.5 | CH2Cl2 | 0-rt | 3 | 24 | 22 | 48 |
| 2 | A | 3/3.3 | 3.0/0.75 | CH2Cl2 | 0-rt | 3 | 4 | 9 | 81 |
| 3 | B | 3/2.2 | 1.0 | CH2Cl2 | 0-rt | 2.5 | 19 | 44 | 8 |
| 4 | B | 3/2.2 | 2.2 | CH3CN | 50 | 6 | 35 | 22 | n.d. 3 |
| 5 | B | 3/2.2 | 2.2 | CH2Cl2 | 0-rt | 2 | 5 | 84 | 4 |
| 6 | C | 3/2.2 | 4.9 | CH2Cl2 | rt | 12 | 49 | 21 | |
| 7 | C | 3/2.2 | 4.9 | CH3CN | 40 | 4 | 71 | 22 | 1 |
| 8 | C | 3/3.3 | 11 | CH3CN | 40 | 24 | 9 | 56 | 27 |
| 9 | D | 4a/3.0 | - | t-BuMeO | 37 | 19 | 9 + 34 4 | 6 | n.d. |
1 Method A—catalyst: Et3N/DMAP; method B—catalyst: Bu2SnO; method C—catalyst: ZnO; method D—biocatalyst: Lipozyme TL IM. 2 The yield of isolated monoacylated products mixture. 3 Not detected. 4 Position 2′-OH was acetylated.
2.2.4. Characterization Data of Acylated Products
2,3-O-Isopropylidene-5-O-(3,4,5-tri-O-acetylgalloyl)-α,β-D-hamamelofuranose (5a) from the enzymatic reaction (Method D). Colourless foam; α:β = 1:0.8; [α]D20 = +5.0 (c = 1.0, CH3OH). 1H NMR (400 MHz, CD3OD) δ: 7.85 (2xs, 4H, H-Arα, H-Arβ), 5.36 (s, 1H, H-1α), 5.19 (s, 1H, H-1β), 4.65 (bs, 2H, H-3α, H-3β), 4.51–4.33 (m, 6H, H-4α, H-4β, H-5aβ, H-5bβ, H-5aα, H-5bα), 3.84 (bd, J = 1.3 Hz, 2H, H-2′aα, H-2′bα), 3.73 (d, 1H, J = 11.8 Hz, H-2′aβ), 3.66 (d, 1H, J = 11.9 Hz, H-2′bβ), 2 × 2.32 (s), 4 × 2.32 (s) (6 × CH3CO), 1.58 (s, CH3β), 1.50 (s, CH3α), 1.46 (2 × s, CH3αβ). 13C NMR (101 MHz, CD3OD) δ: 169.4 (4 × COCH3), 168.2 (COOβ, COOα), 165.6 (2 × COCH3), 145.0 (2 × C-Ar), 140.4 (C-Ar), 129.2 (C-Arα), 129.1 (C-Arβ), 123.3 (2 × CH-Arαβ), 116.2 (CMe2β), 114.8 (CMe2α), 105.5 (C-1α), 99.1 (C-1β), 95.8 (C-2α), 92.6 (C-2β), 85.7 (C-3α), 84.7 (C-4α), 83.9 (C-3β), 80.8 (C-4β), 67.3 (C-5α), 65.7 (C-5β), 65.4 (C-2′β), 63.0 (C-2′α), 28.3 (2 × CH3α), 27.8 (CH3β), 27.4 (CH3β), 20.4 (4 × COCH3), 20.0 (2 × COCH3). HRMS (ESI): m/z calcd for C22H26O13Na ([M + Na]+) 521.12656; found 521.12687.
2,3-O-Isopropylidene-2′-O-acetyl-5-O-(3,4,5-tri-O-acetylgalloyl)-α,β-D-hamamelofuranose (5a-2′-Ac). Colorless foam; α:β = 1:0.5; [α]D20 = +7.0 (c = 1.0, CH3OH). 1H NMR (400 MHz, CD3OD) δ: 7.83 (s, 2H, H-Arα), 7.79 (s, 2H, H-Arβ), 5.34 (s, 1H, H-1α), 5.15 (s, 1H, H-1β), 4.69 (d, J = 1.2 Hz, 1H, H-3α), 4.58 (d, 1H, J = 2.1 Hz, H-3β), 4.52–4.38 (m, 8H, H-4α, H-4β, H-5aβ, H-5bβ, H-5aα, H-5bα, H-2′aα, H-2′bα), 4.37 (d, 1H, J = 11.9 Hz, H-2′aβ), 4.19 (d, 1H, J = 11.9 Hz, H-2′bβ), 2 × 2.30 (s), 4 × 2.29 (s) (6 × CH3CO), 2.07 (s, 3H, COCH3α), 2.06 (s, 3H, COCH3β), 1.57 (s, CH3β), 1.48 (s, CH3α), 1.43 (s, CH3β),1.42 (s, CH3α). 13C NMR (101 MHz, CD3OD) δ: 172.5 (COCH3), 172.1 (COCH3), 169.4 (4 × COCH3), 168.2 (ArCOOβ, ArCOOα), 165.6 (2 × COCH3), 145.1 (2 × C-Arβ), 145.0 (2 × C-Arα), 140.4 (C-Arβ), 140.4 (C-Arα), 129.2 (C-Arα), 128.9 (C-Arβ), 123.3 (CH-Arα), 123.2 (CH-Arβ), 117.0 (CMe2β), 115.2 (CMe2α), 104.9 (C-1α), 99.0 (C-1β), 94.1 (C-2α), 91.2 (C-2β), 86.0 (C-3α), 84.7 (C-4α), 83.7 (C-3β), 80.9 (C-4β), 67.3 (C-5α), 65.8 (C-5β), 65.3 (C-2′β), 64.9 (C-2′α), 28.2 (2 × CH3α), 27.6 (CH3β), 27.2 (CH3β), 20.8 (COCH3), 20.7 (COCH3), 20.5 (COCH3), 20.4 (3 × COCH3), 20.0 (2 × COCH3). HRMS (ESI): m/z calcd for C24H28O14Na ([M + Na]+) 563.13768; found 563.13758.
2,3-O-Isopropylidene-5-O-galloyl-α,β-D-hamamelofuranose (5b) from enzyme reaction (Method E). Colorless foam; α:β = 1:0.7; [α]D20 = +8.7° (c = 1.0, CH3OH). 1H NMR (400 MHz, CD3OD) δ: 7.08 (s, 2H, H-Arα), 7.07 (s, 2H, H-Arβ), 5.34 (s, 1H, H-1α), 5.19 (s, 1H, H-1β), 4.62 (s, 1H, H-3α), 4.58 (d, 1H, J = 1.8 Hz, H-3β), 4.42–4.25 (m, 6H, H-4α, H-4β, H-5aβ, H-5bβ, H-5aα, H-5bα), 3.85 (d, 1H, J = 12.3 Hz, H-2′aα), 3.81 (d, 1H, J = 12.3 Hz, H-2′bα), 3.73 (d, 1H, J = 11.8 Hz, H-2′aβ), 3.68 (d, 1H, J = 11.8 Hz, H-2′bβ), 1.56 (s, CH3β), 1.48 (s, CH3α), 1.44 (s, CH3β),1.43 (s, CH3α). 13C NMR (101 MHz, CD3OD) δ: 168.0 (COOβ), 168.0 (COOα), 146.5 (2 × C-Arβ), 146.5 (2 × C-Arα), 140.0 (C-Arβ), 140.0 (C-Arα), 121.2 (C-Arα), 121.1 (C-Arβ), 116.2 (CMe2β), 114.8 (CMe2α), 110.2 (2 × CH-Ar), 105.4 (C-1α), 99.2 (C-1β), 95.8 (C-2α), 92.6 (C-2β), 86.0 (C-3α), 84.9 (C-4α), 84.4 (C-3β), 81.0 (C-4β), 66.3 (C-5α), 64.8 (C-5β), 63.6 (C-2′β), 63.2 (C-2′α), 28.3 (2 × CH3α), 27.7 (CH3β), 27.4 (CH3β). HRMS (ESI): m/z calcd for C16H20O10Na ([M + Na]+) 395.09487; found 395.09497.
2,3-O-Isopropylidene-2′,5-di-O-(3,4,5-tri-O-acetylgalloyl)-α,β-D-hamamelofuranose (6a). Colorless foam; α:β = 1:0.9, [α]D20 = −7.9° (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ: 7.83 (3 × s, H-Ar), 7.81 (s, H-Ar), 5.51 (d, J1,OH = 2.2 Hz, H-1α), 5.33 (dd, J1,OH = 9.3 Hz, H-1β), 4.71–4.37 (m, H-3α, H-3β, H-4α, H-4β, H-5aβ, H-5bβ, H-5aα, H-5bα, H-2′aα, H-2′bα, H-2′aβ, H-2′bβ), 3.82 (d, OHβ), 3.55 (d, OHα), 2 × 2.30, 3 × 2.29, 2.28, (6s, 6 × CH3CO), 1.61 (s, CH3β), 1.50 (s, CH3α), 1.47 (s, CH3β),1.42 (s, CH3α). 13C NMR (101 MHz, CDCl3) δ: 2 × 167.5, 166.3, 164.3, 164.1, 163.9, (COCH3), 143.5 (2 × C-Ar), 143.4 (2 × C-Ar), 138.9 (C-Ar), 128.0 (C-Ar), 127.7 (C-Ar), 127.5 (C-Ar), 127.3 (C-Ar), 122.4 (3 × CH-Ar), 122.2 (CH-Ar), 116.1 (CMe2), 114.6 (CMe2), 103.5 (C-1α), 98.1 (C-1β), 93.1, 89.3 (C-2α, C-2β), 84.6, 84.5, 83.5, 79.5, (C-3α, C-3β, C-4α, C-4β), 66.2, 65.1, 65.0, 64.4 (C-5α, C-5β, C-2′α, C-2′β), 28.2 (CH3), 27.7 (CH3), 27.3 (2 × CH3), 20.5 (COCH3), 20.1 (COCH3). HRMS (ESI): m/z calcd for C35H36O20Na ([M + Na]+) 799.16976; found 799.17012.
From enzyme reaction (Method D) 6a, white solid, α:β = 1:0.5. 1H NMR (400 MHz, CD3OD, 40 °C) δ: 7.84 (s, H-Arβ), 7.83 (s, H-Arα), 7.82 (s, H-Arα), 7.80 (s, H-Arβ), 5.42 (s, H-1α), 5.26 (s, H-1β), 4.85 (d, J = 1.2 Hz, H-3α), 4.76 (d, J = 1.8 Hz, H-3β), 4.71 (d, J = 12.1 Hz, H-2′aα), 4.61 (d, J = 12.1 Hz, H-2′bα), 4.57–4.36 (m, H-4α, H-4β, H-5aβ, H-5bβ, H-5aα, H-5bα, H-2′aβ, H-2′bβ), 2 × 2.29 (s), 2 × 2.29 (s) 2.28 (s), 2 × 2.27 (s), 2.26 (s), 4 × 2.26 (s) (12 × CH3CO), 1.58 (s, CH3β), 1.49 (s, CH3α), 1.40 (s, CH3β),1.47 (s, CH3α). 13C NMR (101 MHz, CD3OD) δ: 8 × 169.4, 4 × 168.2, 3 × 165.7, 165.4 (12 × COCH3 and 4 × ArCOO), 4 × 145.1, 140.5 (C-Ar), 140.4 (3 × C-Ar), 129.3 (C-Arα), 129.2 (C-Arα), 129.0 (C-Arβ), 129.9 (C-Arβ), 123.3 (CH-Arβ), 123.3 (2 × CH-Arα), 123.2 (CH-Arβ), 117.1 (CMe2β), 115.4 (CMe2α), 105.0 (C-1α), 99.3 (C-1β), 94.2, 91.3 (C-2α, C-2β), 84.9 (C-3α), 84.5 (C-5α), 83.5 (C-3β), 79.5 (C-5β), 67.3 (C-5α), 66.0 (C-2′α,), 66.4, 65.9 (C-5β, C-2′β), 28.4 (CH3α), 28.1 (CH3α), 27.8 (CH3β), 27.2 (CH3β), 20.4 (COCH3), 20.0 (COCH3). HRMS (ESI): m/z calcd for C35H36O20Na ([M + Na]+) 799.16976; found 799.16946.
2,3-O-Isopropylidene-1,2′,5-tri-O-(3,4,5-tri-O-acetylgalloyl)-α-D-hamamelofuranose (7). White solid, mp 129–130 °C (EtOH); [α]D20 = −29.1° (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ: 7.80 (s, 2H, H-Ar), 7.79 (s, 2H, H-Ar), 7.73 (s, 2H, H-Ar), 6.65 (s, 1H, H-1), 4.78 (bs, 1H, H-3), 4.77 (d, J = 12.2 Hz, 1H, H-2′a), 4.68–4.60 (m, 2H, H-4, H-2′b), 4.48 (bd, J = 7.4 Hz, 2H, H-5a, H-5b), 2.29, 2.28, 2.28, 2.26 (4s, 27H, 9 × CH3CO), 1.55 (s, 3H, CH3), 1.44 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ: 167.5, 167.4, 166.2, 166.1, 163.8, 162.6 (9 × COCH3), 143.6 (C-Ar), 143.5 (2 × C-Ar), 139.3 (C-Ar), 139.0 (2 × C-Ar), 127.4 (2 × C-Ar), 127.0 (C-Ar), 122.4 (2 × CH-Ar), 122.2 (CH-Ar), 115.2 (CMe2), 102.9 (C-1), 92.9 (C-2), 85.3 (C-4), 84.2 (C-3), 64.7 (C-5), 64.4 (C-2′), 27.8 (CH3), 27.6 (CH3), 20.5 (COCH3), 20.1 (COCH3). HRMS (ESI): m/z calcd for C48H46O27Na ([M + Na]+) 1077.21242; found 1077.21210.
2.3. Enzymatic Acylation of D-Hamamelose
2.3.1. Preparation of New Derivatives of Vinyl Gallate
Vinyl 3,4,5-tri-O-(t-butyldimethylsilyl)gallate (4f). Vinyl gallate (0.690 g, 3.5 mmol) and t-butyldimethylsilyl chloride (1.662 g, 11.03 mmol) were dissolved in dry THF (5 mL), then Et3N (1.61 mL, 11.6 mmol) and DMAP (0.32 g, 2.63 mmol) were added at 0 °C. The reaction mixture was stirred for 1 h at rt., then toluene was added and the precipitated salts were filtered off and washed with toluene. After concentration of the filtrate, the product was purified by flash chromatography (toluene). Ester 4f (1.53 g, 81%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3,) δ 7.46 (dd, J = 14.0, 6.3 Hz, 1H, CH=), 7.27 (s, 2H, CH-gal), 4.99 (dd, J = 14.0, 1.6 Hz, 1H, =CH2a), 4.65 (dd, J = 6.3, 1.6 Hz, 1H, =CH2b), 0.99 (s, 9H, H-t-Bu), 0.96 (s, 18H, 2 × H-t-Bu), 0.25 (s, 12H, 4 × CH3), 0.15 (s, 6H, 2 × CH3). 13C NMR (101 MHz, CDCl3) δ 163.3 (COO), 148.6 (2 × C-Ar), 144.0 (C-Ar), 141.5 (CH=), 120.6 (C-Ar), 115.9 (CH-gal), 97.5 (=CH2), 26.2 (2 × C(CH3)3), 26.1 (C(CH3)3), 18.8 (2 × C(CH3)3), 18.5 (C(CH3)3), -3.7 (4 × CH3), -3.9 (2 × CH3). HRMS (ESI): m/z calcd for C27H50O5 Si3+H ([M + H]+) 539.30388; found 539.30400.
Vinyl 3,4,5-tri-O-benzylgallate (4g). 3,4,5-Tri-O-benzylgallic acid [49] (13.22 g, 30 mmol) was dissolved in dry THF (30 mL) and vinyl acetate (45 mL). Mercury (II) acetate (0.30 g) and BF3.OEt2 (10 drops) were added to the mixture. The reaction mixture was stirred for 4 h at 37 °C, then neutralized with anhydrous CH3COONa. After filtration, the mixture was concentrated and purified by chromatography (CHCl3). A white solid 9.38 g (67%) was obtained, mp 108 °C. 1H NMR (400 MHz, CDCl3,) δ 7.46 (dd, J = 14.1, 6.5 Hz, 1H, CH=), 7.45–7.42 (m, 4H, H-Ph), 7.43 (s, 2H, CH-gal), 7.40–7.31 (m, 8H, H-Ph), 7.28–7.22 (m, 3H, H-Ph), 5.14 (bs, 4H, 2 × CH2), 5.13 (s, 2H, CH2), 5.05 (dd, J = 14.0, 1.7 Hz, 1H, =CH2a), 4.69 (dd, J = 6.2, 1.7 Hz, 1H, =CH2b). 13C NMR (101 MHz, CDCl3) δ 163.2 (COO), 152.6 (2 × C-Ar), 143.1 (C-Ar), 141.5 (CH=), 137.3 (C-Ar), 136.5 (2 × C-Ar), 128.5 (4 × CH-Ph), 128.5 (2 × CH-Ph), 128.2 (2 × CH-Ph), 128.1 (2 × CH-Ph), 128.0 (CH-Ph), 127.6 (4 × CH-Ph), 123.8 (C-Ar), 109.5 (CH-gal), 98.2 (=CH2), 75.1 (2 × CH2), 71.3 (CH2). HRMS (ESI): m/z calcd for C30H26O5Na ([M + Na]+) 489.16725; found 489.16728.
2.3.2. Enzymatic Benzoylation of 8
D-Hamamelose (0.36 g, 2 mmol) was suspended in t-BuMeO (40 mL). Molecular sieves 4Å (2g), vinyl benzoate (1.11 mL, 4 equiv.), and Lipozyme TL IM (0.4 g) were added. The tightly closed reaction mixture was shaken on a vibrating shaker at 450 rpm in an incubator at 37 °C. After 50 h, the reaction was filtered through Celite 545, the filter cake was washed several times with EtOAc, and the filtrate was concentrated. The reaction mixture was purified on a silica gel column eluted with toluene/EtOAc (3:1→1:2). Several products were obtained during the elution in the order: 13 (3%), 11c (35%), 12c (17%), 9c (37%), and 10c (1%).
2′,5-Di-O-benzoyl-α,β-D-hamamelofuranose (9c). White solid, mp 147–149 °C; α:β = 0.75:1; [α]D20 = +37.7° (c = 1.0, CH3OH). 1H NMR (400 MHz, CD3OD) δ: 8.08 (dt, J = 8.4, 1.2 Hz, H-Ph), 8.01 (td, J = 8.2, 1.4 Hz, H-Ph), 7.63–7.54 (m, H-Ph), 7.49–7.36 (m, H-Ph), 5.35 (s, H-1α), 5.26 (s, H-1β), 4.64 (dd, J = 12.2, 2.8 Hz, 5aα), 4.61 (dd, J = 11.9, 2.7 Hz, H-5aβ), 4.56 (d, J = 11.5 Hz, 2′aβ), 4.50 (d, J = 11.5 Hz, 2′bβ), 4.47–4.41 (m, H-5bα, H-5bβ), 4.39 (d, J = 11.5 Hz, 2′aα), 4.34 (d, J = 11.5 Hz, 2′bβ), 4.29 (ddd, J = 7.7, 4.8, 2.7 Hz, H-4α), 4.27–4.20 (m, H-3β, H-4β), 4.08 (d, J = 8.1 Hz, H-3α). 13C NMR (101 MHz, CD3OD) δ 168.3 (COOβ), 168.1 (COOβ), 167.9 (COOα), 167.7 (COOα), 134.4 (CH-Phα), 134.3 (CH-Phα), 134.2 (CH-Phβ), 134.2 (CH-Phβ), 131.5 (C-Phβ), 131.4 (C-Phβ), 131.2 (C-Phα), 131.1 (C-Phα), 130.7 (2 × CH-Phβ), 130.7 (2 × CH-Phβ), 130.64 (2 × CH-Phα), 130.5 (2 × CH-Phα), 129.6 (2 × CH-Phα), 129.6 (2 × CH-Phα), 129.5 (2 × CH-Phβ), 129.5 (2 × CH-Phβ), 103.0 (C-1β), 98.9 (C-1α), 81.4 (C-4β), 80.7 (C-2β), 80.4 (C-4α), 77.7 (C-2α), 74.1 (C-3β), 72.5 (C-3α), 67.7 (C-2′β), 66.9 (C-5β), 66.4 (C-2′α), 65.1(C-5α). HRMS (ESI): m/z calcd for C20H20O8Na ([M + Na]+) 411.10504; found 105.10543.
2′,3,5-Tri-O-benzoyl-α,β-D-hamamelofuranose (11c) and 1,2′,5-tri-O-benzoyl-α-D- hamamelofuranose (12cα). White amorphous solid; 12α:11α:11β = 0.4:0.6:1; [α]D20 = +25.8° (c = 1.0, CH3OH). 1H NMR (400 MHz, CD3OD) δ: 8.17–7.88 (m, H-Ph), 7.68–7.18 (m, H-Ph), 6.54 (s, H-1α-12), 5.82 (d, J = 7.1 Hz, H-3β-11), 5.52 (d, J = 6.8 Hz, H-3α-11), 5.40 (s, H-1α-11), 5.32 (s, H-1β-11), 4.71–4.45 (m, H-4α-11, H-4β-11, H-5aα-11, H-5aβ-11, H-5aα-12, H-5bα-11, H-5bβ-11, H-5bα-12, H-2′aβ-11, H-2′aα-11, H-2′aα-12, H-2′bβ-11, H-4α-12, H-2′bα-11), 4.42 (d, J = 11.6 Hz, H-2′bα-12), 4.18 (d, J = 6.6 Hz, H-3α-12). 13C NMR (101 MHz, CD3OD) δ 167.9 (COO), 167.7 (COO), 167.7 (2 × COO), 167.6 (COO), 167.6 (COO), 167.3 (COO), 167.3 (COO), 166.8 (COO), 134.6, 134.6, 134.5, 134.4, 134.4, 134.3, 134.3, 134.2, 134.1, 134.0 (CH-Ph), 3 × 131.0, 131.0, 131.0, 130.9, 130.9, 130.9, 130.8, 130.8, 130.7, 130.7, 3 × 130.7, 2 × 130.6, 130.6, 130.5 (CH-Ph and C-Ph), 129.6, 129.6, 2 × 129.6, 3 × 129.5, 129.4, 129.4, 129.3 (CH-Ph), 103.7 (C-1β-11), 99.4 (C-1α-12), 98.7 (C-1α-11), 83.9 (C-4α-12), 81.2 (C-2β-11), 79.1 (C-4β-11), 3 × 78.9 (C-4α-11, C-2α-11, C-2α-12), 77.1 (C-3β-11), 74.1 (C-3α-11), 71.5 (C-3α-12), 68.2 (C-2′β-11), 67.4 (C-2′α-11), 67.3 (C-2′α-12), 66.8 (C-5β-11), 65.1 (C-5α-11), 64.7 (C-5α-12). HRMS (ESI): m/z calcd for C27H24O9Na ([M + Na]+) 515.13125; found 515.13167.
1,2′,5-Tri-O-benzoyl-β-D-hamamelofuranose (12cβ). White solid, mp 126–128 °C; [α]D20 = −17.9° (c = 1.0, CH3OH). 1H NMR (400 MHz, CD3OD) δ 7.94–7.79 (m, 6H, H-Ph), 7.57–7.17 (m, 9H, H-Ph), 6.38 (s, 1H, H-1), 4.73 (dd, J = 12.4, 2.7 Hz, 1H, H-5a), 4.69 (d, J = 11.8 Hz, 1H, H-2′a), 4.56 (d, J = 8.4 Hz, 1H, H-3), 4.55 (d, J = 11.8 Hz, 1H, H-2′b), 4.47 (dd, J = 12.4, 3.7 Hz, 1H, H-5b), 4.41 (ddd, J = 8.4, 3.7, 2.7 Hz, 1H, H-4). 13C NMR (101 MHz, CD3OD) δ 167.7 (COO), 167.6 (COO), 166.3 (COO), 134.5 (CH-Ph), 134.3 (CH-Ph), 134.2 (CH-Ph), 130.9 (C-Ph), 130.9 (C-Ph), 130.6 (4 × CH-Ph), 130.6 (C-Ph), 130.5 (2 × CH-Ph), 129.6 (2 × CH-Ph), 129.5 (4 × CH-Ph), 101.6 (C-1), 82.8 (C-4), 80.7 (C-2), 72.3 (C-3), 66.9 (C-2′), 64.1 (C-5). HRMS (ESI): m/z calcd for C27H24O9Na ([M + Na]+) 515.13125; found 515.13155.
1,2′,4-Tri-O-benzoyl-D-hamamelopyranose (13). Amorphous white solid. 1H NMR (400 MHz, CD3OD) δ 8.18–7.95 (m, 6H, H-Ph), 7.64–7.39 (m, 9H, H-Ph), 6.50 (s, 1H, H-1), 5.50–5.43 (m, 1H, H-4), 4.81 (d, J = 11.8 Hz, 1H, H-2′a), 4.48 (d, J = 4.0 Hz, 2H, H-3), 4.47 (d, J = 11.8 Hz, 1H, H-2′b), 4.25 (dd, J = 13.1, 2.3 Hz, 1H, H-5a), 4.07 (dd, J = 13.0, 3.2 Hz, 1H, H-5b). 13C NMR (101 MHz, CD3OD) δ 167.8 (COO), 167.6 (COO), 166.9 (COO), 134.9 (CH-Ph), 134.4 (CH-Ph), 134.3 (CH-Ph), 131.3 (C-Ph), 131.0 (2 × CH-Ph), 131.0 (C-Ph), 130.8 (2 × CH-Ph), 130.7 (2 × CH-Ph), 130.4 (C-Ph), 129.7 (2 × CH-Ph), 129.5 (2 × CH-Ph), 129.4 (2 × CH-Ph), 95.5 (C-1), 73.9 (C-2), 71.8 (C-4), 67.4, 67.4 (C-3, C-2′), 63.9 (C-5).
2.3.3. Enzymatic Acylation of 8 by Vinyl Gallates 4d–g
D-Hamamelose 8 (0.18 g, 1 mmol) was suspended in t-BuMeO (20 mL) or in t-BuOH (20 mL). Molecular sieves 4Å (1g), derivatized vinyl gallate (3 equiv.) and Lipozyme TL IM (0.2 g) were added. The reaction mixture was shaken on a vibrating shaker at 450 rpm in an incubator at 37 °C. After the time indicated in Table 2, the reaction was filtered through Celite 545, the filter cake was washed several times with EtOAc, and the filtrate was concentrated. The reaction mixture was purified on a silica gel column eluted with toluene/EtOAc (3:1→1:2). The data of the products are presented in Table 2.
2.3.4. Characterization Data of Acylated Hamameloses
2′,5-Di-O-syringoyl-α,β-D-hamamelofuranose (9d). White foam; α:β = 0.4:1; [α]D20 = +27.3° (c = 1.0, CH3OH). 1H NMR (400 MHz, CD3OD) δ: 7.43 (s, H-Arβ), 7.39 (s, H-Arβ), 7.26 (s, H-Arα), 7.24 (s, H-Arα), 5.31 (s, H-1α), 5.27 (s, H-1β), 4.56 (dd, J = 12.1, 2.9 Hz, 5aα), 4.53–4.46 (m, H-5aβ, H-2′aβ, H-2′bβ, H-5bα), 4.42 (dd, J = 11.9, 4.6 Hz, H-5bβ), 4.37 (d, J = 11.4 Hz, H-2′aα), 4.33 (d, J = 7.8 Hz, H-3β), 4.31–4.27 (m, H-4α), 4.28 (d, J = 11.4 Hz, 2′bα), 4.23 (ddd, J = 7.6, 4.5, 2.8 Hz, H-4β), 4.04 (d, J = 7.6 Hz, H-3α), 3.88 (s, OCH3β), 3.87 (s, OCH3β), 3.84 (s, OCH3α), 3.81 (s, OCH3α). 13C NMR (101 MHz, CD3OD) δ 168.2 (COOβ), 168.1 (COOβ), 167.8 (COOα), 167.6 (COOα), 148.9 (2 × C-OCH3β), 148.8 (2 × C-OCH3α), 142.0 (C-OHβ), 141.9 (C-OHα), 121.4 (C-Arβ), 121.3 (C-Arβ), 120.9 (C-Arα), 120.9 (C-Arα), 108.4 (CH-Arβ), 108.3 (CH-Arβ), 108.2 (CH-Arα), 108.0 (CH-Arα), 102.9 (C-1β), 99.2 (C-1α), 81.6 (C-4β), 81.1 (C-4α), 80.8 (C-2β), 77.9 (C-2α), 73.6 (C-3β), 72.5 (C-3α), 67.7 (C-2′β), 66.9 (C-2′α), 65.8 (C-5β), 64.9 (C-5α), 56.8 (2 × OCH3β), 56.7 (OCH3α), 56.6 (OCH3α). HRMS (ESI): m/z calcd for C24H28O14Na ([M + Na]+) 563.13713; found 563.13708.
Mono-O-syringoyl-α,β-D-hamamelofuranoses (10d). Colourless waxy solid; [α]D20 = −22.3° (c = 1.0, CH3OH). Selected NMR signals are presented in Table 3. HRMS (ESI): m/z calcd for C15H20O10H ([M + H]+) 361.11292; found 361.11278; calcd for C15H20O10Na ([M + Na]+) 383.09487; found 383.09494.
2′,5-Di-O-(3,4,5-tri-O-methylgalloyl)-α,β-D-hamamelofuranose (9e). Colourless waxy solid; α:β = 0.4:1; [α]D20 = +20.2° (c = 1.0, CH3OH). 7.42 (s, H-Arβ), 7.39 (s, H-Arβ), 7.26 (s, H-Arα), 7.25 (s, H-Arα), 5.32 (s, H-1α), 5.27 (s, H-1β), 4.60 (dd, J = 12.2, 2.7 Hz, 5aα), (d, 4.56–4.48 (m, H-5aβ, H-5bα), 4.54, J = 11.5, H-2′aβ), 4.50, J = 11.6, H-2′bβ), 4.43 (dd, J = 12.0, 4.4 Hz, H-5bβ), 4.38 (d, J = 11.5 Hz, H-2′aα), 4.35 (d, J = 7.9 Hz, H-3β), 4.32–4.27 (m, H-4α), 4.32 (d, J = 11.5 Hz, 2′bα), 4.24 (ddd, J = 7.5, 4.3, 2.9 Hz, H-4β), 4.04 (d, J = 7.7 Hz, H-3α), 3.86 (s, 2 × OCH3), 3.85 (s, 2 × OCH3), 3.82 (s, 2 × OCH3), 3.81 (s, 4 × OCH3), 3.80 (s, OCH3α), 3.79 (s, OCH3β). 13C NMR (101 MHz, CD3OD) δ 167.7 (COOβ), 167.6 (COOβ), 167.3 (COOα), 167.1 (COOα), 154.3 (4 × C-OCH3β), 154.3 (4 × C-OCH3α), 143.7 (C-OCH3α), 143.6 (C-OCH3α), 143.6 (C-OCH3β), 143.5 (C-OCH3β), 126.6 (C-Arβ), 126.4 (C-Arβ), 126.1 (C-Arα), 126.0 (C-Arα), 108.3 (CH-Arβ), 108.2 (CH-Arβ), 108.1 (CH-Arα), 107.9 (CH-Arα), 102.9 (C-1β), 99.1 (C-1α), 81.5 (C-4β), 80.8 (C-4α), 80.7 (C-2β), 77.8 (C-2α), 73.5 (C-3β), 72.5 (C-3α), 67.9 (C-2′β), 67.0 (C-2′α), 65.9 (C-5β), 65.3 (C-5α), 61.1 (2 × OCH3β, 2 × OCH3α), 56.7 (4 × OCH3β), 56.6 (4 × OCH3α). HRMS (ESI): m/z calcd for C26H32O14Na ([M + Na]+) 591.16843; found 591.16848.
Mono-O-(3,4,5-tri-O-methylgalloyl)-α,β-D-hamamelofuranose (10e). Colourless waxy solid; [α]D20 = −8.8° (c = 1.0, CH3OH). Selected NMR signals are presented in Table 3. HRMS (ESI): m/z calcd for C16H22O10Na ([M + Na]+) 397.11052; found 397.11090.
2′,3,5-Tri-O-(3,4,5-tri-O-methylgalloyl)-α,β-D-hamamelofuranose (11e) and 1,2′,5-tri-O-(3,4,5-tri-O-methylgalloyl)-α,β-D-hamamelofuranose (12e). Colorless waxy solid; 12eα:11eα:12eβ:11eβ = 0.1:0.4:0.8:1; Selected signals 1H NMR (400 MHz, CD3OD) δ 6.54 (s, H-1α-12e), 6.31 (s, 1H, H-1β-12e), 5.40 (s, H-1α-11e), 5.32 (s, H-1β-11e), 13C NMR (101 MHz, CD3OD) δ 103.8 (C-1β-11), 98.9 (C-1α-11), 101.8 (C-1β-12). HRMS (ESI): m/z calcd for C36H42O18Na ([M + Na]+) 785.22634; found 785.22703.
2′,5-Di-O-(3,4,5-tri-O-benzylgalloyl)-α,β-D-hamamelofuranose (9g). White solid; α:β = 1:0.6; [α]D20 = +41.9° (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3+CD3OD, 40 °C) δ: 7.49, 7.41, 7.38, 7.34 (4 × s, 2 × H-Arβ, 2 × H-Arα), 7.45–7.30 (m, 48 × CH-Ph), 7.29–7.21 (12 × m, CH-Ph), 5.34 (s, H-1α), 5.28 (s, H-1β), 5.14–5.03 (m, 12 × CH2), 4.66 (dd, J = 12.2, 2.8 Hz, 5aα), 4.61–4.47 (m, H-5aβ, 2′aβ, 2′bβ, H-5bβ), 4.43 (dd, J = 12.3, 5.7 Hz, H-5bα),4.34 (s, H-2′aα,H-2′bβ), 4.29 (d, J = 7.9 Hz, H-3β), 4.29–4.23 (m, H-4α, H-4β), 3.89 (d, J = 8.1 Hz, H-3α). 13C NMR (101 MHz, CDCl3+CD3OD, 40 °C) δ 166.7 (COOβ), 166.5 (COOβ), 166.2 (COOα), 166.0 (COOα), 152.5, 152.4 (4 × C-OCH2α, 4 × C-OCH2β), 142.6 (C-OCH2α), 142.5 (C-OCH2α), 142.4 (C-OCH2β), 142.3 (C-OCH2β), 137.2 (8 × C-Ph), 136.5 (8 × C-Ph), 128.5, 128.5, 128.4, 128.4 (24 × CH-Ph) 128.1, 128.1 (8 × CH-Ph), 128.0, 127.9, 127.9, 127.9 (12 × CH-Ph), 127.5, 127.5 (16 × CH-Ph), 124.9 (C-Arβ), 124.8 (C-Arβ), 124.7 (C-Arα), 124.3 (C-Arα), 109.2 (CH-Arβ), 109.1 (CH-Arβ, CH-Arα), 109.0 (CH-Arα), 101.5 (C-1β), 97.4 (C-1α), 80.7 (C-4β), 79.4 (C-2β), 79.1 (C-4α), 77.3 (C-2α), 75.1 (4 × CH2-Ph), 72.7 (C-3β), 71.7 (C-3α), 3 × 71.1, 71.0 (8 × CH2-Ph), 66.7 (C-2′β), 66.1 (C-2′α), 65.5 (C-5β), 64.3 (C-5α). HRMS (ESI): m/z calcd for C62H56O14H ([M + H]+) 1025.37428; found 1025.37038; calcd for C62H56O14Na ([M + Na]+) 1047.35678; found 1047.35623.
Mixture of mono-O-(3,4,5-Tri-O-benzylgalloyl)-α,β-D-hamamelofuranoses (10g). White solid; [α]D20 = −10.9° (c = 1.0, CH3OH). Selected NMR signals are presented in Table 3. HRMS (ESI): m/z calcd for C34H34O10Na ([M + Na]+) 625.20442; found 625.20470.
2.4. Removal of Protecting Groups
2.4.1. Simultaneous Deacetylation and Deisopropylidenation of 6a and 7
Diacylated compound 6a (0.1 mmol) or triacylated compound 7 (0.1 mmol) were dissolved in CH3CN (2 mL), and 3M HCl (2 mL) was added. The reaction mixture was stirred at laboratory temperature for 72 h. After the reaction, the organic solvent was removed under reduced pressure and the aqueous residue was extracted with ethyl acetate (3 × 50 mL). The organic layer was washed with brine (3 × 50 mL), dried (Na2SO4), and concentrated under reduced pressure. The product 1 from the deprotection of digallate 6a was obtained as a pure compound in high yield (94%), while the reaction mixture obtained from the deprotection of compound 7 contained the product 1 and gallic acid and after chromatography on silica gel (0.1%CH3COOH in EtOAc) only 56% of product 1 was obtained.
2.4.2. Debenzylation of 9g
Diacylated hamamelofuranose 9g (0.206 g, 0.02 mmol) was dissolved in MeOH (10 mL) and 10% Pd/C (0.06 g) was added. The reaction mixture was intensively stirred at room temperature (25 °C) under hydrogen atmosphere for 18 h and then filtered through Celite 545. After washing the celite cake with MeOH, the filtrate was concentrated to give 0.094 g (97%) of product 1.
2.4.3. Hamamelitannin (1)
Amorphous white solid; α:β = 0.7:1; [α]D20 = +37.9° (c = 1.0, CH3OH). 1H NMR (400 MHz, CD3OD) δ: 7.12 (s, H-Arβ), 7.12 (s, H-Arβ), 7.10 (s, H-Arα), 7.08 (s, H-Arα), 5.34 (s, H-1α), 5.26 (s, H-1β), 4.53 (dd, J = 12.1, 2.6 Hz, H-5aα), 4.52 (dd, J = 11.7, 2.9 Hz, 5aβ), 4.44 (s, H-2′aβ, H-2′bβ), 4.35–4.24 (m, H-5bα, H-5bβ, H-2′aα, H-2′bα, H-4α), 4.20 (ddd, J = 7.8, 4.8, 2.9 Hz, H-4β), 4.17 (d, J = 7.7 Hz, H-3β), 3.90 (d, J = 7.8 Hz, H-3α). 13C NMR (101 MHz, CD3OD) δ 168.6 (COOβ), 168.4 (COOβ), 168.2 (COOα), 168.2 (COOα), 146.5, 146.5, 2 × 146.4 (2 × C-OHβ, 2 × C-OHα), 140.0 (C-OHα), 139.9 (C-OHα), 139.8 (C-OHβ), 139.8 (C-OHβ), 121.5 (C-Arβ), 121.4 (C-Arβ), 121.2 (C-Arα), 121.0 (C-Arα), 110.3, 2 × 110.3, 110.2 (2 × CH-Arβ, 2 × CH-Arα), 102.9 (C-1β), 99.0 (C-1α), 81.4 (C-4β), 80.8 (C-2β), 80.3 (C-4α), 77.7 (C-2α), 74.2 (C-3β), 73.2 (C-3α), 67.4 (C-2′β), 67.1 (C-5β), 66.6 (C-2′α), 65.7 (C-5α). HRMS (ESI): m/z calcd for C20H20O14Na ([M + Na]+) 507,07508; found 507,07464.
3. Results and Discussion
3.1. Synthesis of Hamamelitannin by Acylation of Acceptor 2 Prepared from D-Ribose
D-Ribose was used as the starting material for the preparation of isopropylidenated D-hamamelofuranose 2 (Scheme 1). Treatment of D-ribose with anhydrous acetone in the presence of a catalytic amount of concentrated sulfuric acid yielded the corresponding 2,3-acetonide 2 [46] in 91% yield. The reaction of 2 with aqueous formaldehyde in the presence of potassium carbonate in MeOH introduced the branching hydroxymethyl group through a Ho crossed aldol reaction [47].
In the initial stage of the work, an acetyl-protecting group was selected for the galloyl moiety because the deacetylation of phenols and the removal of the isopropylidene group from the sugar can be performed in one step under acidic conditions. In the first step, standards of the desired triacetylgallates of hamamelofuranose 8 were chemically prepared. After the preparation of 3,4,5-tri-O-acetylgalloyl chloride (3) from gallic acid in two steps, we looked for optimal conditions for the acylation of hamamelose 2. Acetylated phenols are sensitive to both acidic and basic conditions, but deprotected gallates can oligomerize under basic conditions. Therefore, in the synthesis, it was necessary to find slightly basic or neutral conditions that allow 2′,5-di-O-acylation of compound 2 and to which the acetyl groups are inert. Our secondary goal was to regioselectively achieve 2′,5-di-O-acylated hamamelose 6a in maximum yields, using a minimum of equivalents of acyl reagent 3. A high content of monoacylated or triacylated products was undesirable.
Several conventional acylation methods have been used, operating from mildly basic to neutral conditions. The results are summarized in Table 1. In initial experiments, we investigated mild basic reaction conditions working with acetylated galloyl chloride 3, which has been reliably verified in many acylation reactions [53,54,55]. A mixture of two bases—Et3N and DMAP (1 equiv. and 0.25 equiv. relative to 1 equiv. of acyl) was used in dichloromethane (Table 1, Entry 1, 2). Using 2.2 equivs of acyl reagent 3 and appropriate equivalents of bases led to the mixture of per-O-acetylated trigallate 7, digallate 6a, and relatively high content of monogallates (24%), while conditions for theoretical attachment of three acyl groups (3.3 equivs of 3) led to 7 as a major product (81%). Digallate 6a and monogallates were formed in minimal quantities.
As a very effective tool for providing regioselective acylations of sugars, procedures using organotin reagent–dibutyltin oxide (Bu2SnO) [56,57]. Treatment of 2 by acyl donor 3 (2.2 equivs) and Bu2SnO (2.2 equivs) provided digallate 6a in 84% yields and only minor quantities of trigallate and monogallates were isolated (Table 1, Entry 5). Another modification of the method (Bu2SnO quantity, solvent changed to CH3CN) led to an increase in the amount of monogallates (Table 1, Entry 3, 4).
In one of our previous investigations, we studied ZnO as a convenient catalyst in the 4-O-acetylferuloylation of glycosides [58]. Therefore, we have examined tri-O-acetylgalloylation of 2 in CH2Cl2 (Table 1, Method C, Entry 6), but we isolated a mixture of acetylated monogallates as the main product fraction. Better results were obtained when we used CH3CN as the reaction medium (Table 1, Entry 7, 8). With elevating the reaction temperature to 40 °C, ZnO equivalents and reaction time lead to trigallate 7 a digallate 6a in the summary yield 83% (Table 1, Entry 8). It is interesting that we did not observe compound 7 as the β-anomer and 7α was the exclusive tri-O-acylated product under all conditions; this suggests the neighboring group effect by the C-2′ acyloxymethyl group in the rigid 2,3-isopropylidenated furanose ring, as has been already reported [59].
Recently, we have been intensively dealing with enzymatic acylations of sugars by various phenolic acid donors [43,48,60,61]. The commercial lipase Lipozyme TL IM was shown to be the most effective catalyst in terms of its substrate specificity, reactivity, and stability in these reactions. The disadvantage of its use was the longer reaction time, reaching several days. We have, therefore, galloylated 2 under our optimised conditions for galloylation of methyl β-D-glucopyranoside [61] catalysed by Lipozyme TL IM using 3 equivalents of vinyl gallate 4b as an acyl donor at 37 °C and dry t-butyl alcohol (t-BuOH) as a solvent (Scheme 2). The 2,3-isopropyl-D-hamamelofuranose 2 was surprisingly better accepted by the enzyme than methyl β-D-glucopyranoside. The reaction proceeded regioselectively to the 5-OH position of compound 2 and the maximum yield (66%) of monogallate 5b was obtained after 242 h (Scheme 2). t-BuOH is quite a polar solvent (octanol/water partition coefficient as log Pow: 0.30). According to our experience, the lipase-catalyzed transesterification proceed faster in less polar solvents such as for example t-butyl methyl ether (t-BuMeO) (octanol/water partition coefficient as log Pow: 1.06).
The same reaction of 2 with 3 equiv. of 4b was repeated in t-BuMeO. Under these conditions, after 61 h it gave 5-O-gallate 5b in a yield of 82%. The reaction was significantly faster with a higher yield and the product was again the monoacylated product 5b (Scheme 2). Regarding the increase in reaction rate, we can hypothesize that t-BuMeO opens the hydrophobic lid in the active center of Lipozyme TL IM more efficiently than t-BuOH, and the substrate binding site is more accessible [62].
The lipase specificity for the acyl donor structure can significantly influence the course of the reaction and the degree of acylation, as has been demonstrated for another commercial lipase from Thermomyces lanuginosus—Lipolase 100T [48]. During acylation of α-glucopyranoside with phenolic vinyl esters in CH3CN, Lipolase catalyzed the formation of only 6-O-acylated products. If non-phenolic vinyl esters were used, 2,6-di-O-acylated glucopyranoside was also formed. Therefore, we have used acetylated vinyl gallate 4a (3 equiv.) in the same transesterification reactions catalyzed by Lipozyme TL IM in t-BuMeO, i.e., the phenolic groups were hydrophobized by acetyls. (Scheme 1). After 19 h, the starting compound 2 was consumed and more products (also UV inactive) were visible on the TLC plate. After column chromatography on silica gel, three main UV active products were obtained: 5-monoacylated product 5a (9%), 2′,5-diacyl 6a (6%), and 34% of an unexpected product—2,3-O-isopropylidene-2′-O-acetyl-5-O-(3,4,5-tri-O-acetylgalloyl)-α,β-D-hamamelofuranose (5a-2′-Ac) (Scheme 1, Method D). Lipozyme can probably use the triacetylated vinyl gallate as an activated acetyl donor, and the remaining UV inactive products were partially acetylated derivatives of 2.
3.2. Synthesis of Hamamelitannin by Enzymatic Acylation of D-Hamamelose
Promising results with enzymatic galloylation of compound 2 prompted our increased efforts to prepare hamamelitannin 1 via direct enzymatic acylation of D-hamamelose (8). D-hamamelose, although a rare branched sugar, is commercially available. One of the ways to prepare 8 is molybdic acid-catalyzed isomerization of D-fructose by Bílik reaction [63,64].
At first, we investigated the ability of Lipozyme to acylate 8 with a routine commercial aromatic donor—vinyl benzoate (4c). We were inspired by work [65] in which D-fructose benzoylated with vinyl benzoate (3 equiv.) using lipase from C. antarctica B (CAL) and lipase from Mucor miehei (MML) in t-BuMeO gave after 7 h 1,6-di-O-benzoyl-D-fructofuranoside (80%). In our hands, using 4 equiv. of benzoate 4c, a Lipozyme-catalyzed reaction in t-BuMeO for 50 h afforded di-O-benzoate 9c (37%), traces of monobenzoates 10c and a diverse mixture of tri-O-benzoates 11c, 12c, and 13 in 55% total yield (Scheme 3). The high proportion of variously benzoylated secondary hydroxyl groups, even the existence of tribenzoate 13 in pyranose form, indicated that the reaction with reactive hydrophobic aromatic donors would not be selective.
When proceeding the reaction with 3 equivalents of acetylated vinyl gallate 4a and gallate 4b under similar conditions (Lipozyme TL IM, t-BuMeO, 37 °C), the desired products were obtained in both cases, however, in very low yields. Acetylated 4a gave 13% of diacyl 9a after 41 h and the rest were various UV-inactive acetylated products. The reaction with vinyl gallate 4b was allowed to react for a longer time (292 h), and again obtained only 15% of the acylation product 1 (Scheme 4). The monoacylated product’s content was not visible on TLC. In both cases, we did not observe the presence of aromatic triacylated products. Vinyl gallate 4b was not sufficiently reactive in t-BuOH and no galloylation product with hamamelose 8 was observed even after 10 days.
The structure of the starting compounds in lipase-catalyzed esterifications or transesterifications, especially in the case of phenolic compounds, influences the course of the reaction [48,66]. Therefore, we decided to test other more hydrophobic and stable vinyl esters of gallic acid derivatives in the investigated enzyme reaction. Vinyl esters of syringic acid (4d) and 3,4,5-trimethoxybenzoic acid (4e) were prepared according to our previous work [48]. Two new vinyl esters were also prepared—3,4,5-tri-O-(t-butyldimethylsilyl)gallate (4f) and 3,4,5-tri-O-benzylgallate (4g). The silyl derivative 4f was prepared by silylation of vinyl gallate and the benzylated derivative 4g was prepared by transesterification of 3,4,5-tribenzylgallic acid with vinyl acetate. Silyl and benzyl protective groups are widely used in the syntheses, as they can be effectively removed under mild, relatively neutral conditions [67].
The studied enzymatic acylations were performed according to previously implemented conditions. (1 mmol of 8, 3 equiv. of vinyl ester, 0.2 g of Lipozyme TL IM, 20 mL of solvent, 37 °C). To compare the effect of solvent on the reaction time, the composition of products. and product yields with individual acyl donors; these were carried out in t-BuOH as well as in t-BuMeO. The reaction with the benzylated gallate 4g was also carried out in CH3CN. Acylations were monitored by TLC chromatography. The reactions were stopped when the concentration of the products no longer increased. The lipase and molecular sieves were filtered off and the filtrate was purified by chromatography after concentration.
The results of the reaction (Scheme 5) summarized in Table 2 showed that Lipozyme TL IM catalyzes acylations with all acyl donors except silylated gallate 4f. In the case of 4f, we did not observe any product in both solvents even after hundreds of hours (Table 2, entries 5, 6). Acyl donor 4f is probably too large to interact with the active site of the enzyme. Acylation with syringate 4d proceeded as within the longest reaction times (more than 200 h), while hydrophobic 4e and 4g reacted faster (tens of hours). This is consistent with our previous experience [48], and it appears that the transesterification activity of Lipozyme TL IM, similarly to Lipolase 100T (both are lipases from Thermomyces lanuginosus), corresponds to the hydrolytic activity of type A feruloylesterase [68]. In general, reactions in t-BuOH proceeded slower and a higher quantity of monoacylated products were isolated (Table 2, entries 1, 3, 7). On the contrary, we have observed only negligible amounts of monoacyl-hamameloses in t-BuMeO and mostly 2′,5-di-O-acyls of α,β-D-hamamelofuranose (9d–e, 9g) (Scheme 5) were isolated (Table 2, entries 2, 4, 9). The reactivity of trimethoxybenzoate 4e was similar to that observed for benzoate 4c. Products with acylated secondary hydroxyls were also observed. We isolated a significant proportion of triacyls in t-BuMeO for 4e (Table 2, entry 4), and the highest yields of diacyls for t-BuOH were achieved using 4e (Table 2, entry 3). This suggests that the reaction was directed towards the products that were more soluble in the used solvent.
The acylation of 8 with the bulky benzylated acyl donor 4g had a different reaction course. The reaction in t-BuMeO proceeded with the highest yield (84%) of 2′,5-diacyl 9g (Table 2, entry 9). The desired main product 9g was the only one precipitated from the reaction mixture. After the end of the reaction, it was filtered together with the immobilized enzyme and molecular sieves. It was then washed with hot ethyl acetate from the filter cake. A similar reaction system, in which the starting monosaccharide (D-hamamelose), biocatalyst and the product (9g) were insoluble or almost insoluble in the reaction solvent, which serves as an adjuvant, were known from enzymatic syntheses of sugar fatty acids esters [69]. The low solubility of 9g in the reaction medium probably protects it from unwanted acylations to the secondary hydroxyls. Reactions in more polar solvents (t-BuOH, CH3CN) proceeded more slowly with low product conversion. This could indicate that the lipase does not have a favorable conformation for the bulky acyl 4g or is inactivated by the products.
We also examined the structural composition of our mixtures of monoacylated products isolated from reactions in t-BuOH. Theoretical structures in the mixture of hamamelose monoacyls are shown in Scheme 6. They could not be separated individually, but their mixtures were analyzed by NMR, and the H-1 and C-1 signals for individual anomers and conformations were assigned with the support of literature data. The available literature and experimental values are listed in Table 3. These data demonstrate that the initial acylation is not selective (Entries 9–14). In the mixture, 5-O-monoacylated and predominating 2′-O-acylated hamameloses are visible. Generally, the primary 2′-OH position is sterically less favorable than the primary 5-OH position. It is possible that the acylation takes place first in the 2′-OH position if hamamelose is present in the reaction medium in pyranose form. This acylated pyranose is then transformed into furanose via mutarotation.
3.3. Deprotections for Obtaining Hamamelitannin 1
In the final step, the tri-O-acetylgalloylated compounds 6a, 7 and benzylgalloylated 9g were deprotected. Acidic conditions—3M HCl in CH3CN—were found to be sufficient for simultaneous deisopropylidenation and deacetylation, while the galloyl groups were retained. The product 1 from the deprotection of diacyl 6a was obtained as a pure compound in high yield (94%, conditions (a) in Scheme 7), while the reaction mixture obtained from the deprotection of compound 7 contained 1 and gallic acid. Gallic acid originated from deacylation of anomeric gallate moiety sensitive to acidic conditions. The disadvantage of this method was the long reaction time (3 days at laboratory temperature). Debenzylation of 9g by reductive cleavage with molecular hydrogen over 10% Pd/C proceeded smoothly. After 18 h, the reaction mixture was filtered through Celite 545, and after the concentration of the filtrate, hamamelitannin 1 was obtained with a 97% yield and satisfactory purity (conditions (b), Scheme 3).
4. Conclusions
2,3-Isopropylhamamelofuranose 2 and D-hamamelose 8 were studied as acceptors in chemoenzymatic galloylations with the aim of developing an efficient preparation of hamamelitannin. The chemical preparation of hamamelitannin from furanose 2 proceeded smoothly. Base-catalyzed acylation of 2 with acetylated galloyl chloride 3 provided 81% of 1,2′,5-trigallate 7. The Bu2SnO-promoted reaction yielded 84% of 2′,5-digallate 6a regioselectively. Enzymatic reactions using vinyl gallate 4b or its acetylated analogue 4a catalyzed by Lipozyme TL IM provided mainly 5-O-galloyl derivatives. Reaction condition using acyl donor 4b in t-BuMeO afforded 82% 5-O-gallate 5b after 61 h. The pilot enzymatic benzoylation of hamamelose 8 using vinyl benzoate and Lipozyme TL IM as a biocatalyst gave mainly benzoylated furanoses (89%), of which mainly tribenzoates (52%). Similar reactions with vinyl gallate 4b and its acetylated analogue 4a gave 2′,5-diacylated hamameloses but in yields below 20%. Acetylated vinyl gallate 4a also appeared as an acetyl donor. The Lipozyme TL IM, in its presence in the reaction mixture, also performed acetylation of acceptors 2 and 8. Hamamelose 8 in t-BuMeO with vinyl gallates, where phenolic groups were hydrophobized with methyl or benzyl moiety, readily afforded 2′,5-diacylated hamamelofuranoses (65–84%), with the exception of the reaction with the silylated gallate 4f. The best results were obtained with tribenzylated gallate 4g, where the desired 2′,5-diacyl 9g precipitated from the reaction mixture. Similar reactions in more polar solvent t-BuOH gave mainly monoacyls and proceeded more slowly. They did not proceed on secondary hydroxyls and were not regioselective on primary hydroxyls. Finally, after deacetylation and deisopropylidenation of compound 6a under acidic conditions, product 1 was obtained in 94% yield (79% after two steps). Similarly, after the reductive debenzylation of compound 9g, hamamelitannin 1 was obtained with a yield of 97% (82% after two steps from hamamelose). The accomplished syntheses (especially the enzymatic method) open the way to multigram preparations of bioactive hamamelitannin and its analogs.
Author Contributions
Conceptualization, M.M.; methodology, M.M.; investigation, M.M. and V.M.; data analysis, M.M.; writing—original draft preparation, M.M.; writing—review and editing, V.M.; supervision, M.M. and V.M.; project administration, M.M. and V.M.; funding acquisition, M.M. and V.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Slovak Research and Development Agency, grant number APVV-18-0188 and by the Slovak Grant Agency for Science VEGA, grant number 2/0111/22.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
The authors are grateful to Veronika Bencová for technical assistance and Iveta Uhliariková, Mária Kopáčová, Michal Šoral, and Filip Pančík for NMR and HRMS measurements. The authors also thank Peter Magdolen and Ján Kozák for providing D-hamamelose. The contribution of COST Action CA17128 “LIGNOCOST—Establishment of a Pan-European Network on the Sustainable Valorisation of Lignin” supported by COST (European Cooperation in Science and Technology), in promoting interaction, exchange of knowledge, and collaborations in the field of chemistry and transformation of natural phenolic substances is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Khanbabaee, K.; van Ree, T. Tannins: Classification and definition. Nat. Prod. Rep. 2001, 18, 641–649. [Google Scholar] [CrossRef]
- Okuda, T.; Ito, H. Tannins of constant structure in medicinal and food plants-hydrolyzable tannins and polyphenols related to tannins. Molecules 2011, 16, 2191–2217. [Google Scholar] [CrossRef]
- Jourdes, M.; Pouységu, L.; Deffieux, D.; Teissedre, P.-L.; Quideau, S. Hydrolyzable Tannins: Gallotannins and Ellagitannins. In Natural Products; Springer: Berlin/Heidelberg, Germany, 2013; Volume 66, pp. 1975–2010. [Google Scholar]
- He, Q.; Shi, B.; Yao, K.; Luo, Y.; Ma, Z. Synthesis of gallotannins. Carbohydr. Res. 2001, 335, 245–250. [Google Scholar] [CrossRef]
- Li, C.W.; Dong, H.J.; Cui, C. Bin The synthesis and antitumor activity of twelve galloyl glucosides. Molecules 2015, 20, 2034–2060. [Google Scholar] [CrossRef] [Green Version]
- Pouységu, L.; Deffieux, D.; Malik, G.; Natangelo, A.; Quideau, S. Synthesis of ellagitannin natural products. Nat. Prod. Rep. 2011, 28, 853–874. [Google Scholar] [CrossRef]
- Mayer, W.; Kunz, W.; Loebich, F. Die Struktur des Hamamelitannins. Justus Liebigs Ann. Chem. 1965, 688, 232–238. [Google Scholar] [CrossRef]
- Hartisch, C.; Kolodziej, H. Galloylhamameloses and proanthocyanidins from Hamamelis virginiana. Phytochemistry 1996, 42, 191–198. [Google Scholar] [CrossRef]
- Grüttner, F. Beiträge zur Chemie der Rinde von Hamamelis virginica L. Arch. Pharm. 1898, 236, 278–320. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, G.; Ishimaru, K.; Tanaka, T.; Nishioka, I. Tannins and related compounds. XVII. Galloylhamameloses from Castanea crenata L. and Sanguisorba officinalis L. Chem. Pharm. Bull. 1984, 32, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, T.; Kobayashi, S.; Seki, R.; Imagawa, H. A New Gallotannin from Bark of Chestnut Tree, Castanea crenata Sieb. et Zucc. Agric. Biol. Chem. 1984, 48, 1411–1416. [Google Scholar] [CrossRef]
- Nonaka, G.; Ageta, M.; Nishioka, I. Tannins and related compounds. XXV. A new class of gallotannins possessing a (-)-shikimic acid core from Castanopsis cuspidata var. sieboldii Nakai. (1). Chem. Pharm. Bull. 1985, 33, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Lampire, O.; Mila, I.; Raminosoa, M.; Michon, V.; Herve Du Penhoat, C.; Faucheur, N.; Laprevote, O.; Scalbert, A. Polyphenols isolated from the bark of Castanea sativa Mill. chemical structures and auto-association. Phytochemistry 1998, 49, 623–631. [Google Scholar] [CrossRef]
- Masaki, H.; Atsumi, T.; Sakurai, H. Protective activity of hamamelitannin on cell damage of murine skin fibroblasts induced by UVB irradiation. J. Dermatol. Sci. 1995, 10, 25–34. [Google Scholar] [CrossRef]
- Korting, H.C.; Schäfer-Korting, M.; Klövekon, W.; Klövekorn, G.; Martin, C.; Laux, P. Comparative efficacy of hamamelis distillate and hydrocortisone cream in atopic eczema. Eur. J. Clin. Pharmacol. 1995, 48, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Piazza, S.; Martinelli, G.; Magnavacca, A.; Fumagalli, M.; Pozzoli, C.; Terno, M.; Canilli, L.; Angarano, M.; Maranta, N.; Dell’Agli, M.; et al. Unveiling the Ability of Witch Hazel (Hamamelis virginiana L.) Bark Extract to Impair Keratinocyte Inflammatory Cascade Typical of Atopic Eczema. Int. J. Mol. Sci. 2022, 23, 9279. [Google Scholar] [CrossRef]
- Piazza, S.; Martinelli, G.; Vrhovsek, U.; Masuero, D.; Fumagalli, M.; Magnavacca, A.; Pozzoli, C.; Canilli, L.; Terno, M.; Angarano, M.; et al. Anti-Inflammatory and Anti-Acne Effects of Hamamelis virginiana Bark in Human Keratinocytes. Antioxidants 2022, 11, 1119. [Google Scholar] [CrossRef]
- Díaz-González, M.; Rocasalbas, G.; Francesko, A.; Touriño, S.; Torres, J.L.; Tzanov, T. Inhibition of deleterious chronic wound enzymes with plant polyphenols. Biocatal. Biotransform. 2012, 30, 102–110. [Google Scholar] [CrossRef]
- Habtemariam, S. Hamamelitannin from Hamamelis virginiana inhibits the tumour necrosis factor-α (TNF)-induced endothelial cell death in vitro. Toxicon 2002, 40, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Nishimura, N.; Okada, K.; Sekido, C.; Yamamichi, S.; Hasumi, K. Inhibitors of autoactivation of plasma hyaluronan-binding protein (factor VII activating protease). Biol. Pharm. Bull. 2011, 34, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.R.; Choi, J.S.; Han, Y.N.; Bae, S.J.; Chung, H.Y. Peroxynitrite scavenging activity of herb extracts. Phyther. Res. 2002, 16, 364–367. [Google Scholar] [CrossRef]
- Dauer, A.; Hensel, A.; Lhoste, E.; Knasmüller, S.; Mersch-Sundermann, V. Genotoxic and antigenotoxic effects of catechin and tannins from the bark of Hamamelis virginiana L. in metabolically competent, human hepatoma cells (Hep G2) using single cell gel electrophoresis. Phytochemistry 2003, 63, 199–207. [Google Scholar] [CrossRef]
- Masaki, H.; Atsumi, T.; Sakurai, H. Hamamelitannin as a new potent active oxygen scavenger. Phytochemistry 1994, 37, 337–343. [Google Scholar] [CrossRef]
- Sánchez-Tena, S.; Fernández-Cachón, M.L.; Carreras, A.; Mateos-Martín, M.L.; Costoya, N.; Moyer, M.P.; Nuñez, M.J.; Torres, J.L.; Cascante, M. Hamamelitannin from witch hazel (Hamamelis virginiana) displays specific cytotoxic activity against colon cancer cells. J. Nat. Prod. 2012, 75, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Kiran, M.D.; Adikesavan, N.V.; Cirioni, O.; Giacometti, A.; Silvestri, C.; Scalise, G.; Ghiselli, R.; Saba, V.; Orlando, F.; Shoham, M.; et al. Discovery of a quorum-sensing inhibitor of drug-resistant staphylococcal infections by structure-based virtual screening. Mol. Pharmacol. 2008, 73, 1578–1586. [Google Scholar] [CrossRef] [Green Version]
- Cobrado, L.; Silva-Dias, A.; Azevedo, M.M.; Pina-Vaz, C.; Rodrigues, A.G. In vivo antibiofilm effect of cerium, chitosan and hamamelitannin against usual agents of catheter-related bloodstream infections. J. Antimicrob. Chemother. 2013, 68, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Brackman, G.; Breyne, K.; De Rycke, R.; Vermote, A.; Van Nieuwerburgh, F.; Meyer, E.; Van Calenbergh, S.; Coenye, T. The Quorum Sensing Inhibitor Hamamelitannin Increases Antibiotic Susceptibility of Staphylococcus aureus Biofilms by Affecting Peptidoglycan Biosynthesis and eDNA Release. Sci. Rep. 2016, 6, 20321. [Google Scholar] [CrossRef] [Green Version]
- Brackman, G.; Garcia-Fernandez, M.J.; Lenoir, J.; De Meyer, L.; Remon, J.P.; De Beer, T.; Concheiro, A.; Alvarez-Lorenzo, C.; Coenye, T. Dressings Loaded with Cyclodextrin–Hamamelitannin Complexes Increase Staphylococcus aureus Susceptibility Toward Antibiotics Both in Single as well as in Mixed Biofilm Communities. Macromol. Biosci. 2016, 16, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Vermote, A.; Brackman, G.; Risseeuw, M.D.P.; Coenye, T.; Van Calenbergh, S. Design, synthesis and biological evaluation of novel hamamelitannin analogues as potentiators for vancomycin in the treatment of biofilm related Staphylococcus aureus infections. Bioorgan. Med. Chem. 2016, 24, 4563–4575. [Google Scholar] [CrossRef]
- Vermote, A.; Brackman, G.; Risseeuw, M.D.P.; Vanhoutte, B.; Cos, P.; Van Hecke, K.; Breyne, K.; Meyer, E.; Coenye, T.; Van Calenbergh, S. Hamamelitannin analogues that modulate quorum sensing as potentiators of antibiotics against Staphylococcus aureus. Angew. Chem. Int. Ed. 2016, 55, 6551–6555. [Google Scholar] [CrossRef] [PubMed]
- Vermote, A.; Brackman, G.; Risseeuw, M.D.P.; Coenye, T.; Van Calenbergh, S. Novel hamamelitannin analogues for the treatment of biofilm related MRSA infections–A scaffold hopping approach. Eur. J. Med. Chem. 2017, 127, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Theisen, L.L.; Erdelmeier, C.A.J.; Spoden, G.A.; Boukhallouk, F.; Sausy, A.; Florin, L.; Muller, C.P. Tannins from Hamamelis virginiana bark extract: Characterization and improvement of the antiviral efficacy against influenza a virus and human papillomavirus. PLoS ONE 2014, 9, e88062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.H.; Chen, G.Y.; Tang, C.H.; Huang, W.C.; Yang, J.C.; Wu, Y.C. Drug screening of potential multiple target inhibitors for estrogen receptor-α-positive breast cancer. In Vivo 2021, 35, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xue, J.; Chen, X.; Elsaid, F.G.; Salem, E.T.; Ghanem, R.A.; El-kott, A.F.; Xu, Z. Bioactivity of hamamelitannin, flavokawain A, and triacetyl resveratrol as natural compounds: Molecular docking study, anticolon cancer, and anti-Alzheimer potentials. Biotechnol. Appl. Biochem. 2022. early view. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, J.; Qiu, G.; Alahmadi, T.A.; Alharbi, S.A.; Wainwright, M.; Duan, W. Biological Activities of Some Natural Compounds and Their Cytotoxicity Studies against Breast and Prostate Cancer Cell Lines and Anti-COVID19 Studies. J. Oleo Sci. 2022, 71, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Samdani, M.N.; Morshed, N.; Reza, R.; Asaduzzaman, M.; Islam, A.B.M.M.K. Targeting SARS-CoV-2 non-structural protein 13 via helicase-inhibitor-repurposing and non-structural protein 16 through pharmacophore-based screening. Mol. Divers. 2022. [Google Scholar] [CrossRef]
- Ezekiel, A.D.; Overend, W.G.; Williams, N.R. Branched-chain sugars. Carbohydr. Res. 1969, 11, 233–239. [Google Scholar] [CrossRef]
- Overend, W.G.; Williams, N.R. 622. Branched-chain sugars. Part IV. The synthesis of D-hamamelose and D-epihamamelose. J. Chem. Soc. 1965, 3446–3448. [Google Scholar] [CrossRef]
- Ren, B.; Zhang, L.; Zhang, M. Progress on Selective Acylation of Carbohydrate Hydroxyl Groups. Asian J. Org. Chem. 2019, 8, 1813–1823. [Google Scholar] [CrossRef]
- Kadereit, D.; Waldmann, H. Enzymatic protecting group techniques. Chem. Rev. 2001, 101, 3367–3396. [Google Scholar] [CrossRef]
- Iribarren, A.M.; Iglesias, L.E. An update of biocatalytic selective acylation and deacylation of monosaccharides. RSC Adv. 2016, 6, 16358–16386. [Google Scholar] [CrossRef]
- Godoy, C.A.; Pardo-Tamayo, J.S.; Barbosa, O. Microbial Lipases and Their Potential in the Production of Pharmaceutical Building Blocks. Int. J. Mol. Sci. 2022, 23, 9933. [Google Scholar] [CrossRef]
- Mastihubová, M.; Mastihuba, V.; Bilaničová, D.; Boreková, M. Commercial enzyme preparations catalyse feruloylation of glycosides. J. Mol. Catal. B Enzym. 2006, 38, 54–57. [Google Scholar] [CrossRef]
- Carrea, G.; Riva, S. Medium Engineering of Enzymatic Reactions: E nzyme selectivity in organic solvents can differ from that in water and Properties and Synthetic Applications of Enzymes in Organic Solvents. Angew. Chem. Int. Ed. 2000, 39, 2226–2254. [Google Scholar] [CrossRef]
- Zeuner, B.; Kontogeorgis, G.M.; Riisager, A.; Meyer, A.S. Thermodynamically based solvent design for enzymatic saccharide acylation with hydroxycinnamic acids in non-conventional media. New Biotechnol. 2012, 29, 255–270. [Google Scholar] [CrossRef]
- Kim, W.H.; Kang, J.A.; Lee, H.R.; Park, A.Y.; Chun, P.; Lee, B.; Kim, J.; Kim, J.A.; Jeong, L.S.; Moon, H.R. Efficient and practical synthesis of L-hamamelose. Carbohydr. Res. 2009, 344, 2317–2321. [Google Scholar] [CrossRef]
- Ho, P.-T. Branched-chain sugars. I. reaction between furanoses and formaldehyde: A synthesis of D-hamamelose. Tetrahedron Lett. 1978, 19, 1623–1626. [Google Scholar] [CrossRef]
- Mastihubová, M.; Mastihuba, V. Donor specificity and regioselectivity in Lipolase mediated acylations of methyl α-D-glucopyranoside by vinyl esters of phenolic acids and their analogues. Bioorgan. Med. Chem. Lett. 2013, 23, 5389–5392. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhao, Y. Synthesis of 7-O-galloyl-D-sedoheptulose. Carbohydr. Res. 2007, 342, 1510–1513. [Google Scholar] [CrossRef]
- Schilling, G.; Keller, A. Zusammensetzug und Konformation von Hamamelose in Lösung. Liebigs Ann. Chem. 1977, 232, 1475–1479. [Google Scholar] [CrossRef]
- Hricovíniová, Z.; Lamba, D.; Hricovíni, M. Structure of 2-C-(hydroxymethyl)-D-ribose (hamamelose) in the solid-state analyzed by CP MAS NMR and X-ray crystallography. Carbohydr. Res. 2005, 340, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Schilling, G.; Keller, A. Monogalloylhamamelose aus Hamamelis virginiana/Monogalloylhamamelose from Hamamelis virginiana. Z. Naturforsch. C 1986, 41, 253–257. [Google Scholar] [CrossRef]
- Lu, F.; Ralph, J. Facile Synthesis of 4-Hydroxycinnamyl p-Coumarates. J. Agric. Food Chem. 1998, 46, 2911–2913. [Google Scholar] [CrossRef]
- Mastihubová, M.; Mastihuba, V.; Kremnicky, L.; Willet, J.L.; Côté, G.L. Chemoenzymatic Preparation of Novel Substrates for Feruloyl Esterases. Synlett 2001, 2001, 1559–1560. [Google Scholar] [CrossRef]
- Mastihubová, M.; Szemesová, J.; Biely, P. Two efficient ways to 2-O- and 5-O-feruloylated 4-nitrophenyl α-L-arabinofuranosides as substrates for differentiation of feruloyl esterases. Tetrahedron Lett. 2003, 44, 1671–1673. [Google Scholar] [CrossRef]
- Zhang, Z.; Wong, C.-H. Regioselective benzoylation of sugars mediated by excessive Bu2SnO: Observation of temperature promoted migration. Tetrahedron 2002, 58, 6513–6519. [Google Scholar] [CrossRef]
- Dong, H.; Pei, Z.; Byström, S.; Ramström, O. Reagent-Dependent Regioselective Control in Multiple Carbohydrate Esterifications. J. Org. Chem. 2007, 72, 1499–1502. [Google Scholar] [CrossRef]
- Mastihubová, M.; Biely, P. Preparation of regioselectively feruloylated p-nitrophenyl α-L-arabinofuranosides and β-D-xylopyranosides-convenient substrates for study of feruloyl esterase specificity. Carbohydr. Res. 2010, 345, 1094–1098. [Google Scholar] [CrossRef]
- Yoo, B.N.; Kim, H.O.; Moon, H.R.; Seol, S.K.; Jang, S.K.; Lee, K.M.; Jeong, L.S. Synthesis of 2-C-hydroxymethylribofuranosylpurines as potent anti-hepatitis C virus (HCV) agents. Bioorgan. Med. Chem. Lett. 2006, 16, 4190–4194. [Google Scholar] [CrossRef]
- Chyba, A.; Mastihuba, V.; Mastihubová, M. Effective enzymatic caffeoylation of natural glucopyranosides. Bioorgan. Med. Chem. Lett. 2016, 26, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Chyba, A.; Mastihubová, M.; Mastihuba, V. Regioselective galloylation of methyl β-D-glucopyranoside by a lipase. Mon. Chem. Chem. Mon. 2016, 147, 1137–1142. [Google Scholar] [CrossRef]
- Rehm, S.; Trodler, P.; Pleiss, J. Solvent-induced lid opening in lipases: A molecular dynamics study. Protein Sci. 2010, 19, 2122–2130. [Google Scholar] [CrossRef] [Green Version]
- Hricovíniová, Z.; Hricovíni, M.; Petruš, L. Stereospecific molybdic acid-catalyzed isomerization of D-fructose to branched-chain aldose. The synthesis of D-hamamelose. Chem. Pap. 1998, 52, 692–698. [Google Scholar]
- Hricovíniová-Bíliková, Z.; Hricovíni, M.; Petrušová, M.; Serianni, A.S.; Petruš, L. Stereospecific molybdic acid-catalyzed isomerization of 2-hexuloses to branched-chain aldoses. Carbohydr. Res. 1999, 319, 38–46. [Google Scholar] [CrossRef]
- D’Antona, N.; El-Idrissi, M.; Ittobane, N.; Nicolosi, G. Enzymatic procedures in the preparation of regioprotected D-fructose derivatives. Carbohydr. Res. 2005, 340, 319–323. [Google Scholar] [CrossRef]
- Stamatis, H.; Sereti, V.; Kolisis, F.N. Enzymatic synthesis of hydrophilic and hydrophobic derivatives of natural phenolic acids in organic media. J. Mol. Catal. B Enzym. 2001, 11, 323–328. [Google Scholar] [CrossRef]
- Greene, T.W.; Wuts, P.G.M. Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols. In Greene’s Protective Groups in Organic Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; Volume 9, pp. 17–471. ISBN 0471160199. [Google Scholar]
- Crepin, V.F.; Faulds, C.B.; Connerton, I.F. Functional classification of the microbial feruloyl esterases. Appl. Microbiol. Biotechnol. 2004, 63, 647–652. [Google Scholar] [CrossRef]
- Cao, L.; Fischer, A.; Bornscheuer, U.T.; Schmid, R.D. Lipase-Catalyzed Solid Phase Synthesis of Sugar Fatty Acid Esters. Biocatal. Biotransform. 1996, 14, 269–283. [Google Scholar] [CrossRef]
Figure 1.
Representation of the structure of hamamelitannin.

Scheme 1.
Conventional and enzymatic acylation of acceptor 2 using acetylated galloyl donors. Reagents and conditions are shown in Table 1.
Scheme 1.
Conventional and enzymatic acylation of acceptor 2 using acetylated galloyl donors. Reagents and conditions are shown in Table 1.

Scheme 2.
Enzymatic acylation of acceptor 2 using vinyl gallate. Reagents and conditions: (a) vinyl gallate 4b (3 equiv.), Lipozyme TL IM, t-BuOH, 37 °C, 242 h, 5b (66%); (b) 4b (3 equiv.), Lipozyme TL IM, t-BuMeO, 37 °C, 61 h, 5b (82%).
Scheme 2.
Enzymatic acylation of acceptor 2 using vinyl gallate. Reagents and conditions: (a) vinyl gallate 4b (3 equiv.), Lipozyme TL IM, t-BuOH, 37 °C, 242 h, 5b (66%); (b) 4b (3 equiv.), Lipozyme TL IM, t-BuMeO, 37 °C, 61 h, 5b (82%).

Scheme 3.
Enzymatic benzoylation of hamamelose 8. Reagents and conditions: (a) vinyl benzoate 4c (4 equiv.), Lipozyme TL IM, t-BuMeO, 37 °C, 50 h, 9c (37%), 10c (1%), 11c (35%), 12c (17%), and 13 (3%).
Scheme 3.
Enzymatic benzoylation of hamamelose 8. Reagents and conditions: (a) vinyl benzoate 4c (4 equiv.), Lipozyme TL IM, t-BuMeO, 37 °C, 50 h, 9c (37%), 10c (1%), 11c (35%), 12c (17%), and 13 (3%).

Scheme 4.
Enzymatic galloylation of hamamelose 8. Reagents and conditions: (a) vinyl 3,4,5-tri-O-acetylgallate 4a (3 equiv.), Lipozyme TL IM, t-BuMeO, 37 °C, 41 h, 9a (13%); (b) vinyl gallate 4b (3 equiv.), Lipozyme TL IM, t-BuMeO, 37 °C, 292 h, 1 (15%).
Scheme 4.
Enzymatic galloylation of hamamelose 8. Reagents and conditions: (a) vinyl 3,4,5-tri-O-acetylgallate 4a (3 equiv.), Lipozyme TL IM, t-BuMeO, 37 °C, 41 h, 9a (13%); (b) vinyl gallate 4b (3 equiv.), Lipozyme TL IM, t-BuMeO, 37 °C, 292 h, 1 (15%).

Scheme 5.
Enzymatic acylation of 8 by variously hydrophobized vinyl gallates.

Scheme 6.
Theoretical products of non-regioselective monoacylation of D-hamamelose.

Scheme 7.
Preparation of 1 by deprotection of suitable starting materials. Reagents and conditions: (a) 3M HCl, CH3CN, 25 °C, 72 h (94% from 6a, 56% from 7); (b) 10% Pd/C, MeOH, H2, 25 °C, 18 h, 1 (97%).
Scheme 7.
Preparation of 1 by deprotection of suitable starting materials. Reagents and conditions: (a) 3M HCl, CH3CN, 25 °C, 72 h (94% from 6a, 56% from 7); (b) 10% Pd/C, MeOH, H2, 25 °C, 18 h, 1 (97%).

Table 2.
Results of the enzymatic acylation of 8 by various acyl donors using Lipozyme TL IM at 37 °C.
Table 2.
Results of the enzymatic acylation of 8 by various acyl donors using Lipozyme TL IM at 37 °C.
| Entry | Acyl Donor (3 Equiv.) | Solvent | Time (h) | Diacyls 9d–g 1 (%) | Monoacyls 10d–g 3 (%) | Triacyls 11d–g, 12d–g |
|---|---|---|---|---|---|---|
| 1 | 4d | t-BuOH | 272 | 22 | 67 | n.d. |
| 2 | 4d | t-BuMeO | 212 | 68 | 4 | n.d. |
| 3 | 4e | t-BuOH | 98 | 41 | 51 | n.d. |
| 4 | 4e | t-BuMeO | 46 | 65 | n.d. | 8 4 |
| 5 | 4f | t-BuOH | 102 | n.d. 2 | n.d. | n.d. |
| 6 | 4f | t-BuMeO | 231 | n.d. | n.d. | n.d. |
| 7 | 4g | t-BuOH | 198 | 2 | 15 | n.d. |
| 8 | 4g | CH3CN | 154 | 17 | 16 | n.d. |
| 9 | 4g | t-BuMeO | 96 | 84 | n.d. | n.d. |
1 Isolated yields. 2 Not detected. 3 The mixture of α,β anomers of 5-O- and/or 2′-O-monoacylated furanoses and/or 2′-O-monoacylated pyranose was isolated. 4 The mixture of α,β anomers of 2′,3, 5-O- and 1,2′,5-O-triacylated furanose regioisomers was isolated.
Table 3.
Assignment of selected NMR signals from mixtures of various monoacylated D-hamamelose (entries 9–14). Comparison with data from the literature on D-hamamelose (entries 1–4) and D-hamamelose monogallates (entries 5–8).
Table 3.
Assignment of selected NMR signals from mixtures of various monoacylated D-hamamelose (entries 9–14). Comparison with data from the literature on D-hamamelose (entries 1–4) and D-hamamelose monogallates (entries 5–8).
| Entry | Compound | Atom | Solvent | 5αF | 2′αF | 5βF | 2′βF | 2′αP | 2′βP |
|---|---|---|---|---|---|---|---|---|---|
| 1 1 | Ham 6 | H-1 | DMSO-d6 | 5.14 | 4.89 | 4.48 | 4.85 | ||
| 2 1 | Ham | H-1 | D2O | 5.24 | 5.18 | 5.09 | 4.76 | ||
| 3 1 | Ham | C-1 | DMSO-d6 | 96.6 | 101.4 | 94.5 | 95.0 | ||
| 4 1,2 | Ham | C-1 | D2O | 97.8 | 101.5 | 94.8 | 95.3 | ||
| 5 3 | HG 7 | C-1 | DMSO-d6 | 96.9 | 96.9 | 101.9 | 100.8 | - | 94.3 |
| 6 4 | HG | C-1 | Acetone-d6 | 98.2 | 103.0 | ||||
| 7 5 | HG | C-1 | Acetone-d6 | 96.6 | 101.8 | ||||
| 8 5 | HG | H-1 | Acetone-d6 | 5.43 | 5.31 | ||||
| 9 | 10d | H-1 | CD3OD | 5.31 (0.8) | 5.23 (1) | 5.16 (0.3) | 5.21 (1) | n.d | 4.75 (0.5) |
| 10 | 10d | C-1 | CD3OD | 99.0 | 96.1 | 103.5 | 102.7 | n.d | 96.3 |
| 11 | 10e | H-1 | CD3OD | 5.31 (0.7) | 5.22 (1) | 5.16 (0.1) | 5.20 (0.8) | n.d | 4.74 (0.4) |
| 12 | 10e | C-1 | CD3OD | 99.0 | 96.1 | 102.7 | 102.7 | n.d | 96.2 |
| 13 | 10g | H-1 | CD3OD | 5.30 (0.6) | 5.20 (1) | n.d. | 5.20 (1) | n.d | 4.62 (0.3) |
| 14 | 10g | C-1 | CD3OD | 99.0 | 96.0 | n.d. | 102.7 | n.d | 96.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mastihubová, M.; Mastihuba, V. From Hamamelitannin Synthesis to the Study of Enzymatic Acylations of D-Hamamelose. Biomolecules 2023, 13, 519. https://doi.org/10.3390/biom13030519
AMA Style
Mastihubová M, Mastihuba V. From Hamamelitannin Synthesis to the Study of Enzymatic Acylations of D-Hamamelose. Biomolecules. 2023; 13(3):519. https://doi.org/10.3390/biom13030519
Chicago/Turabian StyleMastihubová, Mária, and Vladimír Mastihuba. 2023. "From Hamamelitannin Synthesis to the Study of Enzymatic Acylations of D-Hamamelose" Biomolecules 13, no. 3: 519. https://doi.org/10.3390/biom13030519
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.