
TORC1 Signaling Controls the Stability and Function of α-Arrestins Aly1 and Aly2

 ,
, 


Abstract
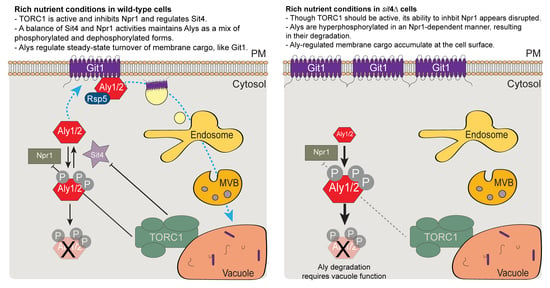
:
1. Introduction
2. Materials and Methods
2.1. Yeast Strains and Growth Conditions
2.2. Serial Dilution Growth Assays
2.3. Plasmids and DNA Manipulations
2.4. KinDel Library Screen
2.5. Yeast Protein Extraction, CIP Treatments, and Immunblot Analyses
2.6. Protein Stability Assays
2.7. Fluorescence Microscopy
2.8. Image Quantification and Statistical Analyses
2.9. RNA Extraction and Relative Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) Analysis
3. Results
3.1. Identifying Kinases and Phosphatases that Influence Aly1- or Aly2-Mediated Growth in Response to Rapamycin or High-Salt Stress
3.2. Some of the Kinases and Phosphatases that Impact Aly1- Or Aly2-Mediated Growth Phenotypes Alter Aly Protein Abundance
3.3. Sit4 and Npr1 Regulate Aly1 and Aly2 Phosphorylation and Abundance
3.4. TORC1 Inhbition or Loss of Sit4 Induces Aly1 and Aly2 Instability and Defective Vacuole Function Restores Aly Levels
3.5. Sit4 and Npr1 Regulate Aly-Mediated Trafficking of the Git1 Transporter
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowers, K.; Stevens, T.H. Protein Transport from the Late Golgi to the Vacuole in the Yeast Saccharomyces Cerevisiae. Biochim. Biophys. Acta Mol. Cell Res. 2005, 1744, 438–454. [Google Scholar] [CrossRef] [Green Version]
- Feyder, S.; de Craene, J.-O.; Bär, S.; Bertazzi, D.; Friant, S. Membrane Trafficking in the Yeast Saccharomyces Cerevisiae Model. Int. J. Mol. Sci. 2015, 16, 1509–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.H.; MacGurn, J.A.; Chu, T.; Stefan, C.J.; Emr, S.D. Arrestin-Related Ubiquitin-Ligase Adaptors Regulate Endocytosis and Protein Turnover at the Cell Surface. Cell 2008, 135, 714–725. [Google Scholar] [CrossRef] [Green Version]
- Nikko, E.; Pelham, H.R.B. Arrestin-Mediated Endocytosis of Yeast Plasma Membrane Transporters. Traffic 2009, 10, 1856–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, A.F.; Apffel, A.; Gardner, R.G.; Cyert, M.S. α-Arrestins Aly1 and Aly2 Regulate Intracellular Trafficking in Response to Nutrient Signaling. Mol. Biol. Cell 2010, 21, 3552–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahlhofer, J.; Leon, S.; Teis, D.; Schmidt, O. The α-Arrestin Family of Ubiquitin Ligase Adaptors Links Metabolism with Selective Endocytosis. Biol. Cell 2021, 113, 183–219. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, A.F.; Schmidt, M.C. AMPK-Mediated Regulation of Alpha-Arrestins and Protein Trafficking. Int. J. Mol. Sci. 2019, 20, 515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacGurn, J.A.; Hsu, P.C.; Smolka, M.B.; Emr, S.D. TORC1 Regulates Endocytosis via Npr1-Mediated Phosphoinhibition of a Ubiquitin Ligase Adaptor. Cell 2011, 147, 1104–1117. [Google Scholar] [CrossRef] [Green Version]
- Ivashov, V.; Zimmer, J.; Schwabl, S.; Kahlhofer, J.; Weys, S.; Gstir, R.; Jakschitz, T.; Kremser, L.; Bonn, G.K.; Lindner, H.; et al. Complementary A-Arrestin-Ubiquitin Ligase Complexes Control Nutrient Transporter Endocytosis in Response to Amino Acids. eLife 2020, 9, e58246. [Google Scholar] [CrossRef]
- Prosser, D.C.; Pannunzio, A.E.; Brodsky, J.L.; Thorner, J.; Wendland, B.; O’Donnell, A.F. α-Arrestins Participate in Cargo Selection for Both Clathrin-Independentand Clathrin-Mediated Endocytosis. J. Cell Sci. 2015, 128, 4220–4234. [Google Scholar] [CrossRef] [Green Version]
- Alvaro, C.G.; O’Donnell, A.F.; Prosser, D.C.; Augustine, A.A.; Goldman, A.; Brodsky, J.L.; Cyert, M.S.; Wendland, B.; Thorner, J. Specific α-Arrestins Negatively Regulate Saccharomyces Cerevisiae Pheromone Response by down-Modulating the G-Protein-Coupled Receptor Ste2. Mol. Cell. Biol. 2014, 34, 2660–2681. [Google Scholar] [CrossRef] [Green Version]
- Merhi, A.; André, B. Internal Amino Acids Promote Gap1 Permease Ubiquitylation via TORC1/Npr1/14-3-3-Dependent Control of the Bul Arrestin-Like Adaptors. Mol. Cell. Biol. 2012, 32, 4510–4522. [Google Scholar] [CrossRef] [Green Version]
- Crapeau, M.; Merhi, A.; André, B. Stress Conditions Promote Yeast Gap1 Permease Ubiquitylation and Down-Regulation via the Arrestin-like Bul and Aly Proteins. J. Biol. Chem. 2014, 289, 22103–22116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vina-Vilaseca, A.; Sorkin, A. Lysine 63-Linked Polyubiquitination of the Dopamine Transporter Requires WW3 and WW4 Domains of Nedd4-2 and UBE2D Ubiquitin-Conjugating Enzymes. J. Biol. Chem. 2010, 285, 7645–7656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchese, A.; Railborg, C.; Santini, F.; Keen, J.; Stenmark, H.; Benovic, J.L. The E3 Ubiquitin Ligase AIP4 Mediates Ubiquitination and Sorting of the G Protein-Coupled Receptor CXCR4. Dev. Cell 2003, 5, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Hicke, L.; Zanolari, B.; Riezman, H. Cytoplasmic Tail Phosphorylation of The-Factor Receptor Is Required for Its Ubiquitination and Internalization. J. Cell Biol. 1998, 141, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piper, R.C.; Dikic, I.; Lukacs, G.L. Ubiquitin-Dependent Sorting in Endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a016808. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Lefkowitz, R.J. Seven-Transmembrane Receptor Signaling Through β-Arrestin. Science’s STKE 2005, 2005, cm10. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. β-Arrestins and Cell Signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.; Shenoy, S.K.; Luttrell, L.M.; Lefkowitz, R.J. SnapShot: β-Arrestin Functions. Cell 2020, 182, 1362. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Lin, H.; Grimsey, N.J.; Mendez, F.; Trejo, J. The α-Arrestin ARRDC3 Mediates ALIX Ubiquitination and G Protein–Coupled Receptor Lysosomal Sorting. Mol. Biol. Cell 2015, 26, 4660–4673. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Irannejad, R.; Bowman, S.L.; Du, Y.; Puthenveedu, M.A.; von Zastrow, M.; Benovic, J.L. The α-Arrestin ARRDC3 Regulates the Endosomal Residence Time and Intracellular Signaling of the Β2-Adrenergic Receptor. J. Biol. Chem. 2016, 291, 14510–14525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puca, L.; Chastagner, P.; Meas-Yedid, V.; Israël, A.; Brou, C. α-Arrestin 1 (ARRDC1) and β-Arrestins Cooperate to Mediate Notch Degradation in Mammals. J. Cell Sci. 2013, 126, 4457–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, F.F.; Rowell, J.L.; Li, Y.; Chang, T.H.; Alvarez, C.E.; Means, R.E. Mammalian Alpha Arrestins Link Activated Seven Transmembrane Receptors to Nedd4 Family E3 Ubiquitin Ligases and Interact with Beta Arrestins. PLoS ONE 2012, 7, e50557. [Google Scholar] [CrossRef] [PubMed]
- Patwari, P.; Emilsson, V.; Schadt, E.E.; Chutkow, W.A.; Lee, S.; Marsili, A.; Zhang, Y.; Dobrin, R.; Cohen, D.E.; Larsen, P.R.; et al. The Arrestin Domain-Containing 3 Protein Regulates Body Mass and Energy Expenditure. Cell Metab. 2011, 14, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Ho, H.C.; Tumolo, J.M.; Hsu, P.C.; MacGurn, J.A. Methionine Triggers Ppz-Mediated Dephosphorylation of Art1 to Promote Cargo-Specific Endocytosis. J. Cell Biol. 2019, 218, 977–992. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, A.F. The Running of the Buls: Control of Permease Trafficking by α-Arrestins Bul1 and Bul2. Mol. Cell. Biol. 2012, 32, 4506–4509. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, A.F.; Huang, L.; Thorner, J.; Cyert, M.S. A Calcineurin-Dependent Switch Controls the Trafficking Function of α-Arrestin Aly1/Art6. J. Biol. Chem. 2013, 288, 24063–24080. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, A.F.; McCartney, R.R.; Chandrashekarappa, D.G.; Zhang, B.B.; Thorner, J.; Schmidt, M.C. 2-Deoxyglucose Impairs Saccharomyces Cerevisiae Growth by Stimulating Snf1-Regulated and α-Arrestin-Mediated Trafficking of Hexose Transporters 1 and 3. Mol. Cell. Biol. 2015, 35, 939–955. [Google Scholar] [CrossRef] [Green Version]
- Herrador, A.; Livas, D.; Soletto, L.; Becuwe, M.; Léon, S.; Vincent, O. Casein Kinase 1 Controls the Activation Threshold of an α-Arrestin by Multisite Phosphorylation of the Interdomain Hinge. Mol. Biol. Cell 2015, 26, 2128–2138. [Google Scholar] [CrossRef] [Green Version]
- Becuwe, M.; Vieira, N.; Lara, D.; Gomes-Rezende, J.; Soares-Cunha, C.; Casal, M.; Haguenauer-Tsapis, R.; Vincent, O.; Paiva, S.; Léon, S. A Molecular Switch on an Arrestin-like Protein Relays Glucose Signaling to Transporter Endocytosis. J. Cell Biol. 2012, 196, 247–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savocco, J.; Nootens, S.; Afokpa, W.; Bausart, M.; Chen, X.; Villers, J.; Renard, H.F.; Prévost, M.; Wattiez, R.; Morsomme, P. Yeast α-Arrestin Art2 Is the Key Regulator of Ubiquitylation-Dependent Endocytosis of Plasma Membrane Vitamin B1 Transporters. PLoS Biol. 2019, 17, e3000512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.C.; MacGurn, J.A.; Emr, S.D. Deubiquitinating Enzymes Ubp2 and Ubp15 Regulate Endocytosis by Limiting Ubiquitination and Degradation of ARTs. Mol. Biol. Cell 2017, 28, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Hovsepian, J.; Defenouillère, Q.; Albanèse, V.; Váchová, L.; Garcia, C.; Palková, Z.; Léon, S. Multilevel Regulation of an α-Arrestin by Glucose Depletion Controls Hexose Transporter Endocytosis. J. Cell Biol. 2017, 216, 1811–1831. [Google Scholar] [CrossRef]
- Breitkreutz, A.; Choi, H.; Sharom, J.R.; Boucher, L.; Neduva, V.; Larsen, B.; Lin, Z.-Y.; Breitkreutz, B.-J.; Stark, C.; Liu, G.; et al. A Global Protein Kinase and Phosphatase Interaction Network in Yeast. Science 2010, 328, 1043–1046. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.; Gruhler, A.; Heilbut, A.; Bader, G.D.; Moore, L.; Adams, S.-L.; Millar, A.; Taylor, P.; Bennett, K.; Boutilier, K.; et al. Systematic Identification of Protein Complexes in Saccharomyces Cerevisiae by Mass Spectrometry. Nature 2002, 415, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Dokládal, L.; Stumpe, M.; Hu, Z.; Jaquenoud, M.; Dengjel, J.; de Virgilio, C. Phosphoproteomic Responses of TORC1 Target Kinases Reveal Discrete and Convergent Mechanisms That Orchestrate the Quiescence Program in Yeast. Cell Rep. 2021, 37, 110149. [Google Scholar] [CrossRef]
- Holt, L.J.; Tuch, B.B.; Villén, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global Analysis of Cdk1 Substrate Phosphorylation Sites Provides Insights into Evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef] [Green Version]
- Albuquerque, C.P.; Smolka, M.B.; Payne, S.H.; Bafna, V.; Eng, J.; Zhou, H. A Multidimensional Chromatography Technology for In-Depth Phosphoproteome Analysis. Mol. Cell. Proteom. 2008, 7, 1389–1396. [Google Scholar] [CrossRef] [Green Version]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villén, J. Global Analysis of Phosphorylation and Ubiquitylation Cross-Talk in Protein Degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef]
- MacGilvray, M.E.; Shishkova, E.; Place, M.; Wagner, E.R.; Coon, J.J.; Gasch, A.P. Phosphoproteome Response to Dithiothreitol Reveals Unique Versus Shared Features of Saccharomyces Cerevisiae Stress Responses. J. Proteome Res. 2020, 19, 3405–3417. [Google Scholar] [CrossRef] [PubMed]
- Cyert, M.S. Calcineurin Signaling in Saccharomyces Cerevisiae: How Yeast Go Crazy in Response to Stress. Biochem. Biophys. Res. Commun. 2003, 311, 1143–1150. [Google Scholar] [CrossRef]
- Nakamura, T.; Liu, Y.; Hirata, D.; Namba, H.; Harada, S.; Hirokawa, T.; Miyakawa, T. Protein Phosphatase Type 2B (Calcineurin)-Mediated, FK506-Sensitive Regulation of Intracellular Ions in Yeast Is an Important Determinant for Adaptation to High Salt Stress Conditions. EMBO J. 1993, 12, 4063–4071. [Google Scholar] [CrossRef]
- Westfall, P.J.; Thorner, J. Analysis of Mitogen-Activated Protein Kinase Signaling Specificity in Response to Hyperosmotic Stress: Use of an Analog-Sensitive HOG1 Allele. Eukaryotic Cell 2006, 5, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohmann, S. Control of High Osmolarity Signalling in the Yeast Saccharomyces Cerevisiae. FEBS Lett. 2009, 583, 4025–4029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szopinska, A.; Degand, H.; Hochstenbach, J.F.; Nader, J.; Morsomme, P. Rapid Response of the Yeast Plasma Membrane Proteome to Salt Stress. Mol. Cell. Proteom. 2011, 10, M111. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Guo, B.; Arndt, K.T.; Schmelzle, T.; Hall, M.N. TIP41 Interacts with TAP42 and Negatively Regulates the TOR Signaling Pathway. Mol. Cell 2001, 8, 1017–1026. [Google Scholar] [CrossRef]
- Jiang, Y.; Broach, J.R. Tor Proteins and Protein Phosphatase 2A Reciprocally Regulate Tap42 in Controlling Cell Growth in Yeast. EMBO J. 1999, 18, 2782–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo, J.L.; Helliwell, S.B.; Wiederkehr, C.; Demougin, P.; Fowler, B.; Primig, M.; Hall, M.N. NPR1 Kinase and RSP5-BUL1/2 Ubiquitin Ligase Control GLN3-Dependent Transcription in Saccharomyces Cerevisiae. J. Biol. Chem. 2004, 279, 37512–37517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckstaens, M.; Llinares, E.; van Vooren, P.; Marini, A.M. The TORC1 Effector Kinase Npr1 Fine Tunes the Inherent Activity of the Mep2 Ammonium Transport Protein. Nat. Commun. 2014, 5, 3101. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Beck, T.; Koller, A.; Kunz, J.; Hall, M.N.; Schmidt, A.; Beck, T. The TOR Nutrient Signalling Pathway Phosphorylates NPR1 and Inhibits Turnover of the Tryptophan Permease. EMBO J. 1998, 17, 6924–6931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.J.; Tolley, E.A.; Cooper, T.G. Sit4 and PP2A Dephosphorylate Nitrogen Catabolite Repression-Sensitive Gln3 When TorC1 Is up- as Well as Downregulated. Genetics 2019, 212, 1205–1225. [Google Scholar] [CrossRef]
- Tate, J.J.; Rai, R.; de Virgilio, C.; Cooper, T.G. N- and C-Terminal Gln3-Tor1 Interaction Sites: One Acting Negatively and the Other Positively to Regulate Nuclear Gln3 Localization. Genetics 2021, 217, iyab017. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.B.; Ott, E.M.; Keener, J.M.; Curtiss, M.; Sandrin, V.; Babst, M. Regulation of Membrane Protein Degradation by Starvation-Response Pathways. Traffic 2012, 13, 468–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, B.P.; Hawbaker, S.; Chiang, A.; Jordahl, E.M.; Anaokar, S.; Nikiforov, A.; Bowman, R.W.; Ziegler, P.; McAtee, C.K.; Patton-Vogt, J.; et al. Alpha-Arrestins Aly1/Art6 and Aly2/Art3 Regulate Trafficking of the Glycerophosphoinositol Transporter Git1 and Impact Phospholipid Homeostasis. Biol. Cell 2022, 114, 3–31. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.; Pringle, G.; Hartwell, G. Coordination of Growth with Cell Division in the Yeast. Exp. Cell Res. 1977, 105, 79–98. [Google Scholar] [CrossRef]
- Gietz, R.D.; Schiestl, R.H. High-Efficiency Yeast Transformation Using the LiAc/SS Carrier DNA/PEG Method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Hager, N.A.; Krasowski, C.J.; Mackie, T.D.; Kolb, A.R.; Needham, P.G.; Augustine, A.A.; Dempsey, A.; Szent-Gyorgyi, C.; Bruchez, M.P.; Bain, D.J.; et al. Select α-Arrestins Control Cell-Surface Abundance of the Mammalian Kir2.1 Potassium Channel in a Yeast Model. J. Biol. Chem. 2018, 293, 11006–11021. [Google Scholar] [CrossRef] [Green Version]
- Volland, C.; Urban-Grimal, D.; Géraud, G.; Haguenauer-Tsapis, R. Endocytosis and Degradation of the Yeast Uracil Permease under Adverse Conditions. J. Biol. Chem. 1994, 269, 9833–9841. [Google Scholar] [CrossRef]
- Liesche, J.; Marek, M.; Günther-Pomorski, T. Cell Wall Staining with Trypan Blue Enables Quantitative Analysis of Morphological Changes in Yeast Cells. Front. Microbiol. 2015, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.E.; Brown, T.A.; Trumpower, B.L. A Rapid and Simple Method for Preparation of RNA from Saccharomyces Cerevisiae. Nucleic Acids Res. 1990, 18, 3091–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, C.G.; Thorner, J. Heterotrimeric G Protein-Coupled Receptor Signaling in Yeast Mating Pheromone Response. J. Biol. Chem. 2016, 291, 7788–7795. [Google Scholar] [CrossRef] [Green Version]
- Dohlman, H.G.; Thorner, J. Regulation of G Protein–Initiated Signal Transduction in Yeast: Paradigms and Principles. Annu. Rev. Biochem. 2001, 70, 703–754. [Google Scholar] [CrossRef]
- Raught, B.; Gingras, A.-C.; Sonenberg, N. The Target of Rapamycin (TOR) Proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 7037–7044. [Google Scholar] [CrossRef] [Green Version]
- Urban, J.; Soulard, A.; Huber, A.; Lippman, S.; Mukhopadhyay, D.; Deloche, O.; Wanke, V.; Anrather, D.; Ammerer, G.; Riezman, H.; et al. Sch9 Is a Major Target of TORC1 in Saccharomyces Cerevisiae. Mol. Cell 2007, 26, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Binda, M.; Péli-Gulli, M.-P.; Bonfils, G.; Panchaud, N.; Urban, J.; Sturgill, T.W.; Loewith, R.; de Virgilio, C. The Vam6 GEF Controls TORC1 by Activating the EGO Complex. Mol. Cell 2009, 35, 563–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, G.A.; Gomez, T.A.; Deshaies, R.J.; Tansey, W.P. Combined Chemical and Genetic Approach to Inhibit Proteolysis by the Proteasome. Yeast 2010, 27, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Woolford, C.A.; Daniels, L.B.; Park, F.J.; Jones, E.W.; van Arsdell, J.N.; Innis, M.A. The PEP4 Gene Encodes an Aspartyl Protease Implicated in the Posttranslational Regulation of Saccharomyces Cerevisiae Vacuolar Hydrolases. Mol. Cell. Biol. 1986, 6, 2500–2510. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.U.; Kane, T.; Tipper, C.; Spatrick, P.; Jenness, D.D. Yeast Mutants Affecting Possible Quality Control of Plasma Membrane Proteins. Mol. Cell. Biol. 1999, 19, 3588–3599. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Guerriero, C.J.; Brodsky, J.L. Substrate Ubiquitination Retains Misfolded Membrane Proteins in the Endoplasmic Reticulum for Degradation. Cell Rep. 2021, 36, 109717. [Google Scholar] [CrossRef] [PubMed]
- de Craene, J.O.; Soetens, O.; Andre, B. The Npr1 Kinase Controls Biosynthetic and Endocytic Sorting of the Yeast Gap1 Permease. J. Biol. Chem. 2001, 276, 43939–43948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, C.; André, B. A C-Terminal Di-Leucine Motif and Nearby Sequences Are Required for NH4(+)-Induced Inactivation and Degradation of the General Amino Acid Permease, Gap1p, of Saccharomyces Cerevisiae. Mol. Microbiol. 1997, 24, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Jauniaux, J.C.; Grenson, M. GAP1, the General Amino Acid Permease Gene of Saccharomyces Cerevisiae. Nucleotide Sequence, Protein Similarity with the Other Bakers Yeast Amino Acid Permeases, and Nitrogen Catabolite Repression. Eur. J. Biochem. 1990, 190, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Springael, J.Y.; André, B. Nitrogen-Regulated Ubiquitination of the Gap1 Permease of Saccharomyces Cerevisiae. Mol. Biol. Cell 1998, 9, 1253–1263. [Google Scholar] [CrossRef] [Green Version]
- Leskoske, K.L.; Roelants, F.M.; Emmerstorfer-Augustin, A.; Augustin, C.M.; Si, E.P.; Hill, J.M.; Thorner, J. Phosphorylation by the Stress-Activated MAPK Slt2 down-Regulates the Yeast TOR Complex 2. Genes Dev. 2018, 32, 1576–1590. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pRS415-TEF1pr-Aly1-GFP | pRS415-TEF1pr-Aly2-GFP | |||||||
|---|---|---|---|---|---|---|---|---|
| Gene | NaCl | Rapa | Protein Levels | Mobility Shift? | NaCl | Rapa | Protein Levels | Mobility Shift? |
| bub1∆ | Sens | Sens | Equal | No | Sens | Sens | Equal | No |
| ctk1∆ | Sens | Sens | Lower | No | Sens | Sens | Lower | Maybe |
| fus3∆ | Sens | Res | Hi/Equal | No | Sens | Equal | Equal | No |
| gip2∆ | Sens | Res | Equal | No | Sens | Res | Equal | Yes |
| kin82∆ | Equal | Equal | Equal | No | Equal | Res | Equal | No |
| nem1∆ | Equal | Sens | Lower | No | Equal | Sens | Lower | No |
| ptc4∆ | Equal | Equal | Equal/Lower | No | Equal | Equal | Equal/Lower | No |
| rck2∆ | Equal | Equal | Equal/Lower | No | Equal | Equal | Equal/Lower | No |
| sip2∆ | Res | Sens | Lower | Yes | Res | Sens | Lower | No |
| sit4∆ | Sens | Sens | Lower | Yes | Sens | Res | Lower | Yes |
| slt2∆ | Sens | Sens | Equal | No | Equal | Sens | Lower | No |
| spo7∆ | Equal | Sens | Lower | No | Res | Sens | Equal/Lower | No |
| ste7∆ | Res | Res | Equal/Lower | No | Res | Res | Equal/Lower | No |
| ste20∆ | Sens | Sens | Lower | No | Sens | Sens | Equal/Lower | Yes |
| tip41∆ | Equal | Res | Lower | No | Equal | Res | Equal/Lower | No |
| tor1∆ | Equal | Sens | Equal/Lower | No | Equal | Sens | Equal/Lower | No |
| ymr1∆ | Equal | Res | Equal/Lower | No | Equal | Equal | Equal | No |
| yvh1∆ | Sens | Sens | Lower | Yes | Sens | Sens | Lower | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowman, R.W., II; Jordahl, E.M.; Davis, S.; Hedayati, S.; Barsouk, H.; Ozbaki-Yagan, N.; Chiang, A.; Li, Y.; O’Donnell, A.F. TORC1 Signaling Controls the Stability and Function of α-Arrestins Aly1 and Aly2. Biomolecules 2022, 12, 533. https://doi.org/10.3390/biom12040533
Bowman RW II, Jordahl EM, Davis S, Hedayati S, Barsouk H, Ozbaki-Yagan N, Chiang A, Li Y, O’Donnell AF. TORC1 Signaling Controls the Stability and Function of α-Arrestins Aly1 and Aly2. Biomolecules. 2022; 12(4):533. https://doi.org/10.3390/biom12040533
Chicago/Turabian StyleBowman, Ray W., II, Eric M. Jordahl, Sydnie Davis, Stefanie Hedayati, Hannah Barsouk, Nejla Ozbaki-Yagan, Annette Chiang, Yang Li, and Allyson F. O’Donnell. 2022. "TORC1 Signaling Controls the Stability and Function of α-Arrestins Aly1 and Aly2" Biomolecules 12, no. 4: 533. https://doi.org/10.3390/biom12040533