
Leptin Receptor Deficiency Results in Hyperphagia and Increased Fatty Acid Mobilization during Fasting in Rainbow Trout (Oncorhynchus mykiss)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. CRISPR Gene Modification
2.3. Microinjection and Early Rearing
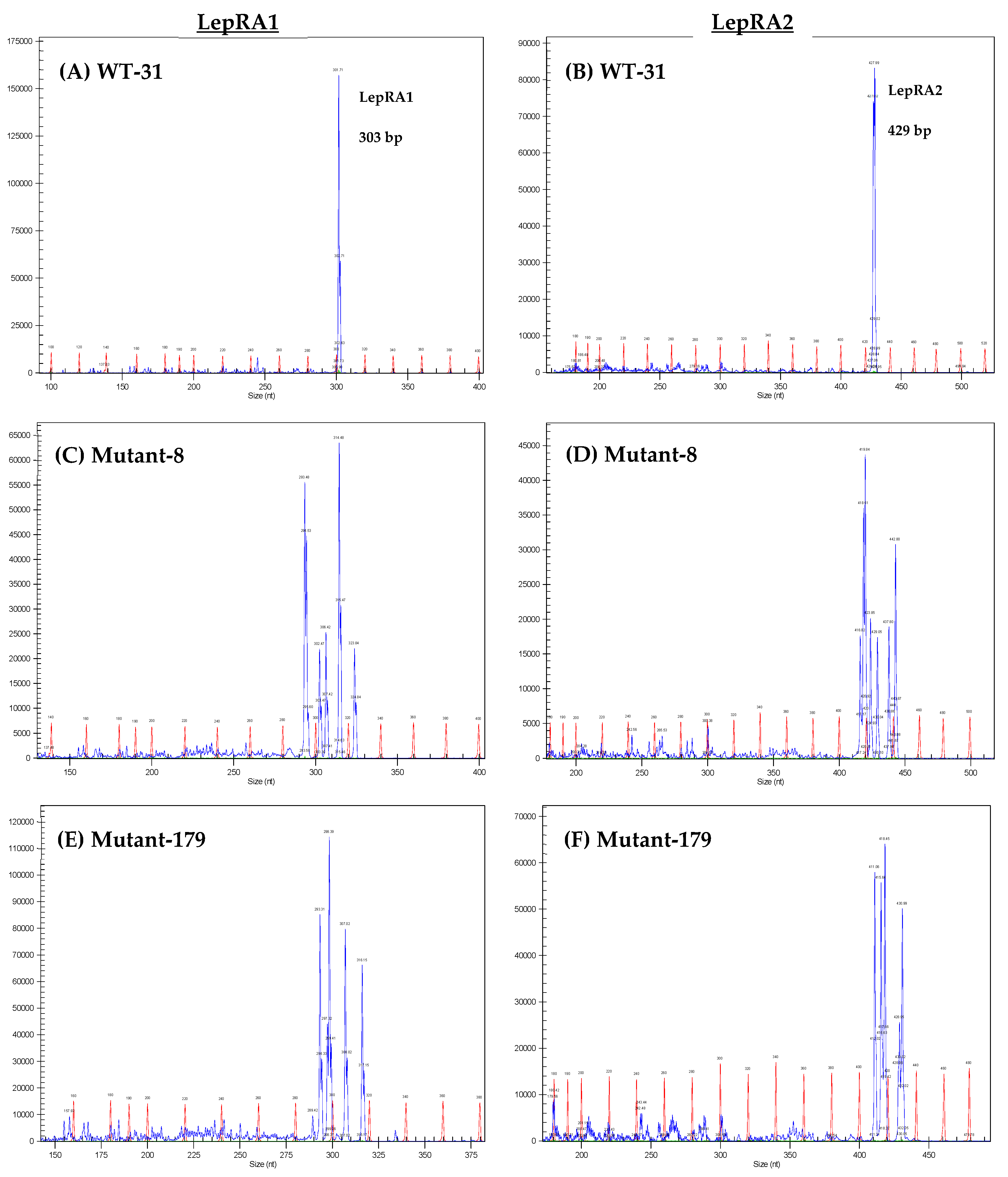
2.4. Genotyping and Mutant Characterization
2.5. Phenotyping Study
2.6. Glycogen Analysis
2.7. Plasma Leptin and Cortisol ELISA
2.8. Fatty Acid Analyses
2.9. RNA Isolation and Quantitative Real-Time PCR
2.10. Statistical Analyses
3. Results
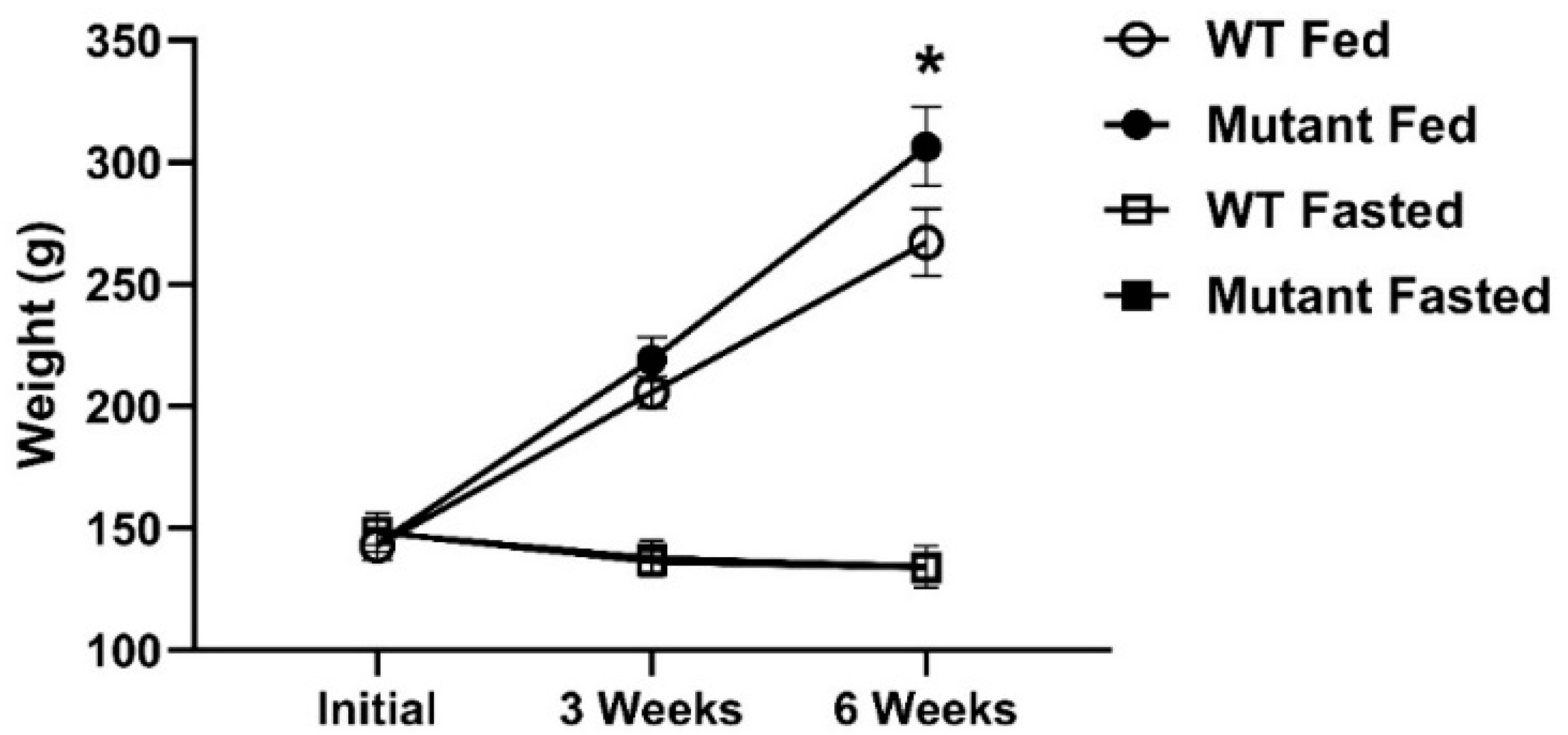
3.1. Growth and Feeding Response to LepR Deficiency
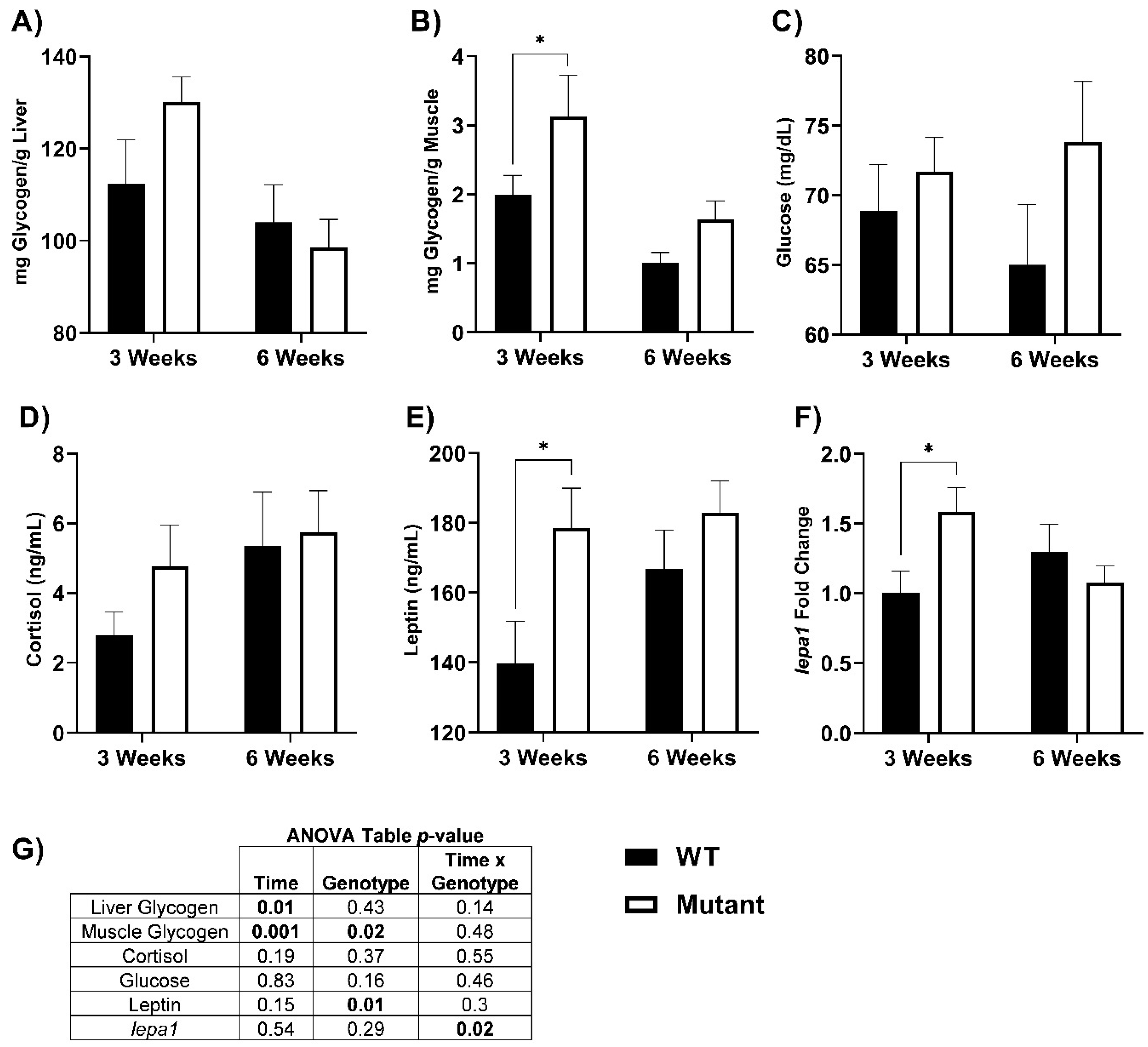
3.2. Glycogen, Cortisol, and Leptin Responses
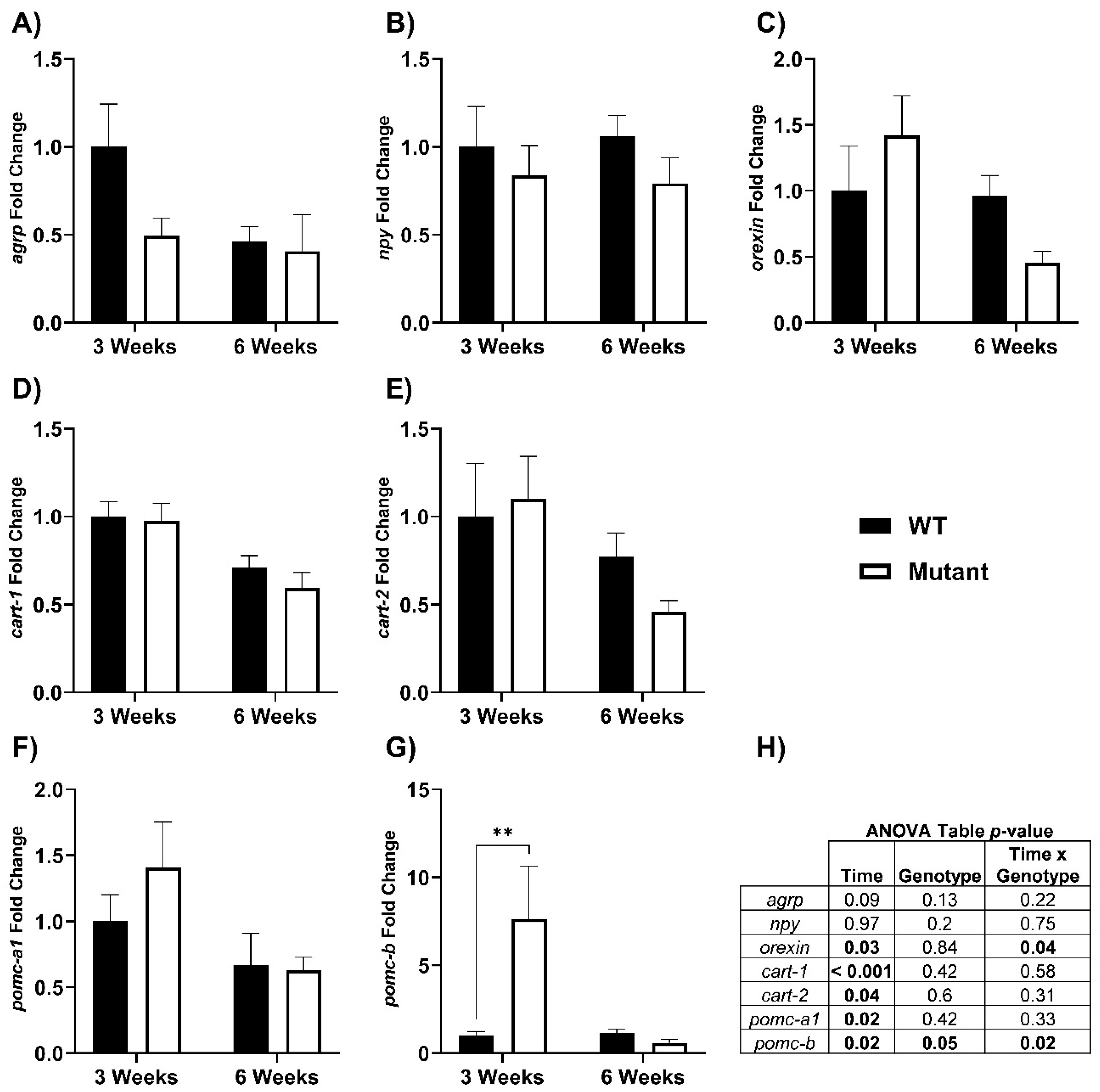
3.3. Hypothalamic mRNA Response
3.4. Fatty Acid Profiles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Londraville, R.L.; Macotela, Y.; Duff, R.J.; Easterling, M.R.; Liu, Q.; Crespi, E.J. Comparative endocrinology of leptin: Assessing function in a phylogenetic context. Gen. Comp. Endocrinol. 2014, 203, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deck, C.A.; Honeycutt, J.L.; Cheung, E.; Reynolds, H.M.; Borski, R.J. Assessing the Functional Role of Leptin in Energy Homeostasis and the Stress Response in Vertebrates. Front. Endocrinol. 2017, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murashita, K.; Uji, S.; Yamamoto, T.; Rønnestad, I.; Kurokawa, T. Production of recombinant leptin and its effects on food intake in rainbow trout (Oncorhynchus mykiss). Comp. Biochem. Physiol. B 2008, 150, 377–384. [Google Scholar] [CrossRef]
- Aguilar, A.J.; Conde-Sieira, M.; Polakof, S.; Míguez, J.M.; Soengas, J.L. Central leptin treatment modulates brain glucosensing function and peripheral energy metabolism of rainbow trout. Peptides 2010, 31, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Baltzegar, D.A.; Reading, B.J.; Douros, J.D.; Borski, R.J. Role for leptin in promoting glucose mobilization during acute hyperosmotic stress in teleost fishes. J. Endocrinol. 2014, 220, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, M.; Page-McCaw, P.S.; Chen, W.; Cone, R.D. Leptin signaling regulates glucose homeostasis, but not adipostasis, in the zebrafish. Proc. Natl. Acad. Sci. USA 2016, 113, 3084–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, A.M.; Soengas, J.L. Leptin signaling in teleost fish with emphasis in food intake regulation. Mol. Cell Endocrinol. 2021, 526, 111209. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.H.; Liang, X.F.; Wang, M.; Zhou, Y.; Bai, X.L.; He, Y. The role of leptin in lipid metabolism in fatty degenerated hepatocytes of the grass carp Ctenopharyngodon idellus. Fish Physiol. Biochem. 2012, 38, 1759–1774. [Google Scholar] [CrossRef] [PubMed]
- De Pedro, N.; Martinez-Alvarez, R.; Delgado, M.J. Acute and chronic leptin reduces food intake and body weight in goldfish (Carassius auratus). J. Endocrinol. 2006, 188, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.; Morgenroth, D.; Einarsdottir, I.E.; Gong, N.; Björnsson, B.T. Energy stores, lipid mobilization and leptin endocrinology of rainbow trout. J. Comp. Physiol. B 2016, 186, 759–773. [Google Scholar] [CrossRef]
- Salmerón, C.; Johansson, M.; Angotzi, A.R.; Rønnestad, I.; Jönsson, E.; Björnsson, B.T.; Gutiérrez, J.; Navarro, I.; Capilla, E. Effects of nutritional status on plasma leptin levels and in vitro regulation of adipocyte leptin expression and secretion in rainbow trout. Gen. Comp. Endocrinol. 2015, 210, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Gorissen, M.; Flik, G. Leptin in teleostean fish, towards the origins of leptin physiology. J. Chem. Neuroanat. 2014, 61, 200–206. [Google Scholar] [CrossRef]
- Prokop, J.W.; Duff, R.J.; Ball, H.C.; Copeland, D.L.; Londraville, R.L. Leptin and leptin receptor: Analysis of a structure to function relationship in interaction and evolution from humans to fish. Peptides 2012, 38, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Londraville, R.L.; Prokop, J.W.; Duff, R.J.; Liu, Q.; Tuttle, M. On the molecular evolution of leptin, leptin receptor, and endospanin. Front. Endocrinol. 2017, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frühbeck, G. Intracellular signalling pathways activated by leptin. Biochem. J. 2006, 393, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabeau, L.; Lavens, D.; Peelman, F.; Eyckerman, S.; Vandekerckhove, J.; Tavernier, J. The ins and outs of leptin receptor activation. FEBS Lett. 2003, 546, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, L.A. The leptin receptor. J. Biol. Chem. 1997, 272, 6093–6096. [Google Scholar] [PubMed] [Green Version]
- Huising, M.O.; Kruiswijk, C.P.; Flik, G. Phylogeny and evolution of class-I helical cytokines. J. Endocrinol. 2006, 189, 1–25. [Google Scholar] [CrossRef]
- Gorska, E.; Popko, K.; Stelmaszczyk-Emmel, A.; Ciepiela, O.; Kucharska, A.; Wasik, M. Leptin receptors. Eur. J. Med. Res. 2010, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Mankiewicz, J.L.; Cleveland, B.M. Characterization of a Leptin Receptor Paralog and Its Response to Fasting in Rainbow Trout (Oncorhynchus mykiss). Int. J. Mol. Sci. 2021, 22, 7732. [Google Scholar] [CrossRef] [PubMed]
- Angotzi, A.R.; Stefansson, S.O.; Nilsen, T.O.; Øvrebø, J.I.; Andersson, E.; Taranger, G.L.; Rønnestad, I. Identification of a novel leptin receptor duplicate in Atlantic salmon: Expression analyses in different life stages and in response to feeding status. Gen. Comp. Endocrinol. 2016, 235, 108–119. [Google Scholar] [CrossRef]
- Morini, M.; Pasquier, J.; Dirks, R.; van den Thillart, G.; Tomkiewicz, J.; Rousseau, K.; Dufour, S.; Lafont, A.G. Duplicated leptin receptors in two species of eel bring new insights into the evolution of the leptin system in vertebrates. PLoS ONE 2015, 10, e0126008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasauer, S.M.; Neuhauss, S.C. Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genom. 2014, 289, 1045–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, N.; Einarsdottir, I.E.; Johansson, M.; Björnsson, B.T. Alternative splice variants of the rainbow trout leptin receptor encode multiple circulating leptin-binding proteins. Endocrinology 2013, 154, 2331–2340. [Google Scholar] [CrossRef] [PubMed]
- Rønnestad, I.; Gomes, A.S.; Murashita, K.; Angotzi, R.; Jönsson, E.; Volkoff, H. Appetite-controlling endocrine systems in teleosts. Front. Endocrinol. 2017, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Volkoff, H. The neuroendocrine regulation of food intake in fish: A review of current knowledge. Front. Neurosci. 2016, 10, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.W.; Woods, S.C.; Porte, D.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Murashita, K.; Kurokawa, T. Multiple cocaine-and amphetamine-regulated transcript (CART) genes in medaka, Oryzias latipes: Cloning, tissue distribution and effect of starvation. Gen. Comp. Endocrinol. 2011, 170, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Bonacic, K.; Martínez, A.; Martín-Robles, Á.J.; Muñoz-Cueto, J.A.; Morais, S. Characterization of seven cocaine-and amphetamine-regulated transcripts (CARTs) differentially expressed in the brain and peripheral tissues of Solea senegalensis (Kaup). Gen. Comp. Endocrinol. 2015, 224, 260–272. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Coleman, D.L. Obese and diabetes: Two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 1978, 14, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Ioffe, E.; Fidahusein, N.; Connolly, E.; Friedman, J.M. Absence of Soluble Leptin Receptor in Plasma from dbPas/dbPas and Other db/db Mice. J. Biol. Chem. 1998, 273, 10078–10082. [Google Scholar] [CrossRef] [Green Version]
- Suriano, F.; Vieira-Silva, S.; Falony, G.; Roumain, M.; Paquot, A.; Pelicaen, R.; Régnier, M.; Delzenne, N.M.; Raes, J.; Muccioli, G.G.; et al. Novel insights into the genetically obese (ob/ob) and diabetic (db/db) mice: Two sides of the same coin. Microbiome 2021, 9, 147. [Google Scholar] [CrossRef]
- Chisada, S.I.; Kurokawa, T.; Murashita, K.; Rønnestad, I.; Taniguchi, Y.; Toyoda, A.; Sakaki, Y.; Takeda, S.; Yoshiura, Y. Leptin receptor-deficient (knockout) medaka, Oryzias latipes, show chronical up-regulated levels of orexigenic neuropeptides, elevated food intake and stage specific effects on growth and fat allocation. Gen. Comp. Endocrinol. 2014, 195, 9–20. [Google Scholar] [CrossRef]
- Cleveland, B.M.; Yamaguchi, G.; Radler, L.M.; Shimizu, M. Editing the duplicated insulin-like growth factor binding protein-2b gene in rainbow trout (Oncorhynchus mykiss). Sci. Rep. 2018, 8, 16054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleveland, B.M.; Habara, S.; Oikawa, J.; Radler, L.M.; Shimizu, M. Compensatory Response of the Somatotropic Axis from IGFBP-2b Gene Editing in Rainbow Trout (Oncorhynchus mykiss). Genes 2020, 11, 1488. [Google Scholar] [CrossRef]
- Gao, G.; Magadan, S.; Waldbieser, G.C.; Youngblood, R.C.; Wheeler, P.A.; Scheffler, B.E.; Thorgaard, G.H.; Palti, Y. A long reads-based de-novo assembly of the genome of the Arlee homozygous line reveals chromosomal rearrangements in rainbow trout. G3-Genes Genom. Genet. 2021, 11, jkab052. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information (NCBI) [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. 1988. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 30 November 2021).
- Kapustin, Y.; Souvorov, A.; Tatusova, T.; Lipman, D. Splign: Algorithms for computing spliced alignments with identification of paralogs. Biol. Direct 2008, 3, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepage, G.; Roy, C.C. Direct transesterification of all classes of lipids in a one-step reaction. J. Lipid Res. 1986, 27, 114–120. [Google Scholar] [CrossRef]
- Cleveland, B.M.; Weber, G.M.; Raatz, S.K.; Rexroad, C.E.; Picklo, M.J. Fatty acid partitioning varies across fillet regions during sexual maturation in female rainbow trout (Oncorhynchus mykiss). Aquaculture 2017, 475, 52–60. [Google Scholar]
- Picklo, M.J.; Murphy, E.J. A high-fat, high-oleic diet, but not a high-fat, saturated diet, reduces hepatic α-linolenic acid and eicosapentaenoic acid content in mice. Lipids 2016, 51, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wootton, R.J. Feeding. In Ecology of Teleost Fishes; Springer: Dordrecht, The Netherlands, 1990; pp. 32–72. [Google Scholar] [CrossRef]
- Volkoff, H.; Peter, R.E. Feeding behavior of fish and its control. Zebrafish 2006, 3, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Else, P.L.; Hulbert, A.J. Comparison of the “mammal machine” and the “reptile machine”: Energy production. Am. J. Physiol. 1981, 240, R3–R9. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.E. Thermogenic mechanisms and their hormonal regulation. Physiol. Rev. 2006, 86, 435–464. [Google Scholar] [CrossRef]
- Del Vecchio, G.; Murashita, K.; Verri, T.; Gomes, A.S.; Rønnestad, I. Leptin receptor-deficient (knockout) zebrafish: Effects on nutrient acquisition. Gen. Comp. Endocrinol. 2021, 310, 113832. [Google Scholar] [CrossRef]
- North, B.P.; Turnbull, J.F.; Ellis, T.; Porter, M.J.; Migaud, H.; Bron, J.; Bromage, N.R. The impact of stocking density on the welfare of rainbow trout (Oncorhynchus mykiss). Aquaculture 2006, 255, 466–479. [Google Scholar] [CrossRef]
- Vivas, Y.; Azpeleta, C.; Feliciano, A.; Velarde, E.; Isorna, E.; Delgado, M.J.; De Pedro, N. Time-dependent effects of leptin on food intake and locomotor activity in goldfish. Peptides 2011, 32, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Z.; He, A.Y.; Ning, L.J.; Luo, Y.; Li, D.L.; Zhang, M.L.; Chen, L.Q.; Du, Z.Y. Leptin selectively regulates nutrients metabolism in Nile tilapia fed on high carbohydrate or high fat diet. Front. Endocrinol. 2018, 9, 574. [Google Scholar] [CrossRef] [PubMed]
- Busacker, G.P.; Adelman, I.R.; Goolish, E.M. Methods for Fish Biology: Growth; Schreck, C.B., Moyle, P.B., Eds.; American Fisheries Society: Bethesda, MD, USA, 1990; pp. 363–387. [Google Scholar]
- Thornton, J.E.; Cheung, C.C.; Clifton, D.K.; Steiner, R.A. Regulation of hypothalamic proopiomelanocortin mRNA by leptin in ob/ob mice. Endocrinology 1997, 138, 5063–5066. [Google Scholar] [PubMed]
- Ahi, E.P.; Brunel, M.; Tsakoumis, E.; Schmitz, M. Transcriptional study of appetite regulating genes in the brain of zebrafish (Danio rerio) with impaired leptin signaling. Sci. Rep. 2019, 9, 20166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leder, E.H.; Silverstein, J.T. The pro-opiomelanocortin genes in rainbow trout (Oncorhynchus mykiss): Duplications, splice variants, and differential expression. J. Endocrinol. 2006, 188, 355–363. [Google Scholar] [CrossRef]
- Dores, R.M.; Baron, A.J. Evolution of POMC: Origin, phylogeny, posttranslational processing, and the melanocortins. Ann. N. Y. Acad. Sci. 2011, 1220, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Amano, M.; Itoh, T.; Yasuda, A.; Yamanome, T.; Amemiya, Y.; Sasaki, K.; Sakai, M.; Yamamori, K.; Kawauchi, H. Nucleotide sequence and expression of three subtypes of proopiomelanocortin mRNA in barfin flounder. Gen. Comp. Endocrinol. 2005, 141, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Valen, R.; Jordal, A.E.; Murashita, K.; Rønnestad, I. Postprandial effects on appetite-related neuropeptide expression in the brain of Atlantic salmon, Salmo salar. Gen. Comp. Endocrinol. 2011, 171, 359–366. [Google Scholar] [CrossRef]
- Gong, N.; Johansson, M.; Björnsson, B.T. Impaired central leptin signaling and sensitivity in rainbow trout with high muscle adiposity. Gen. Comp. Endocrinol. 2016, 235, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.C.; Liang, X.F.; Cai, W.J.; Li, A.X.; Huang, D.; He, S. Differential roles of two leptin gene paralogues on food intake and hepatic metabolism regulation in mandarin fish. Front. Endocrinol. 2020, 11, 438. [Google Scholar] [CrossRef] [PubMed]
- Yada, T.; Azuma, T.; Takahashi, A.; Suzuki, Y.; Hirose, S. Effects of desacetyl-α-MSH on lipid mobilization in the rainbow trout, Oncorhynchus mykiss. Zool. Sci. 2000, 17, 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Copeland, D.L.; Duff, R.J.; Liu, Q.; Prokop, J.; Londraville, R.L. Leptin in teleost fishes: An argument for comparative study. Front. Physiol. 2011, 2, 26. [Google Scholar] [CrossRef] [Green Version]
- Murashita, K.; Jordal, A.E.O.; Nilsen, T.O.; Stefansson, S.O.; Kurokawa, T.; Björnsson, B.T.; Moen, A.G.G.; Rønnestad, I. Leptin reduces Atlantic salmon growth through the central pro-opiomelanocortin pathway. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2011, 158, 79–86. [Google Scholar]
- Unger, R.H.; Zhou, Y.T.; Orci, L. Regulation of fatty acid homeostasis in cells: Novel role of leptin. Proc. Natl. Acad. Sci. USA 1999, 96, 2327–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reidy, S.P.; Weber, J.M. Leptin: An essential regulator of lipid metabolism. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2000, 125, 285–298. [Google Scholar] [CrossRef]
- Bakshi, A.; Singh, R.; Rai, U. Trajectory of leptin and leptin receptor in vertebrates: Structure, function and their regulation. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2022, 257, 110652. [Google Scholar] [CrossRef]
- Salmerón, C.; Johansson, M.; Asaad, M.; Angotzi, A.R.; Rønnestad, I.; Stefansson, S.O.; Jönsson, E.; Björnsson, B.T.; Gutiérrez, J.; Navarro, I.; et al. Roles of leptin and ghrelin in adipogenesis and lipid metabolism of rainbow trout adipocytes in vitro. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2015, 188, 40–48. [Google Scholar] [CrossRef]
- Muoio, D.M.; Dohn, G.L.; Fiedorek, F.T.; Tapscott, E.B.; Coleman, R.A. Leptin directly alters lipid partitioning in skeletal muscle. Diabetes 1997, 46, 1360–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muoio, D.M.; Dohm, G.L.; Tapscott, E.B.; Coleman, R.A. Leptin opposes insulin’s effects on fatty acid partitioning in muscles isolated from obese ob/ob mice. Am. J. Physiol. Endocrinol. Metab. 1999, 276, E913–E921. [Google Scholar] [CrossRef]
- Mankiewicz, J.L.; Deck, C.A.; Taylor, J.D.; Douros, J.D.; Borski, R.J. Epinephrine and glucose regulation of leptin synthesis and secretion in a teleost fish, the tilapia (Oreochromis Mossambicus). Gen. Comp. Endocrinol. 2021, 302, 113669. [Google Scholar] [CrossRef] [PubMed]
- Deck, C.A.; Mankiewicz, J.L.; Borski, R.J. Evidence for a leptin-insulin axis in a teleost fish. J. Endocrinol. 2022, 253, 13–25. [Google Scholar] [CrossRef]
- Perry, R.J.; Wang, Y.; Cline, G.W.; Rabin-Court, A.; Song, J.D.; Dufour, S.; Zhang, X.M.; Petersen, K.F.; Shulman, G.I. Leptin mediates a glucose-fatty acid cycle to maintain glucose homeostasis in starvation. Cell 2018, 172, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Haks, M.C.; Forn-Cuní, G.; He, J.; Nowik, N.; Harms, A.C.; Hankemeier, T.; Eeza, M.N.H.; Matysik, J.; Alia, A.; et al. Metabolomic and transcriptomic profiling of adult mice and larval zebrafish leptin mutants reveal a common pattern of changes in metabolites and signaling pathways. Cell Biosci. 2021, 11, 126. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCR Primers | ||||
|---|---|---|---|---|
| Gene | Forward (5′-3′) | Reverse (5′-3′) | Accession # | Product Size (bp) |
| lepra1 | TGTGATGTGGTTATTAGCCTGT | ACATCTCCCCAAAAGGCTGG | JX878485 | 303 |
| lepra2 | CAACTGCCAGTCAGGTGATCTT | TGCATATTACTGTTGACCCATCC | XM_021599667 | 429 |
| qPCR Primers | ||||
| Gene | Forward (5′-3′) | Reverse (5′-3′) | Accession # | Reference |
| lepa1 | GGTGATTAGGATCAAAAAGCTGGA | GACGAGCAGTAGGTCCTGGTAGAA | AB354909 | Salmerón et al., 2015 |
| agrp | ACTCACCGACGATTCCTTCC | CAGCTGCATCACCTCAGCAA | XM_036964549 | − |
| npy | TGAAGGAGAGCACAGACACG | TGAGATCAGTTGCTCGTCGC | AF203902 | − |
| orexin | ATGACGCAGCCACTGGAATC | TGACCCGTGGAGTAGCTGAT | KR080508 | Gong et al., 2016 |
| cart-1 | GCTCCCCTGAAGACCCCATA | CATCAGCATCACACATGGGAA | XM_021564309 | − |
| cart-2 | CGCTGCTGTCCATCCTATGT | TCGTGTAGAGCTCCAAGAAGAC | XM_021573503 | − |
| pomc-a1 | CTCGCTGTCAAGACCTCAACTCT | GAGTTGGGTTGGAGATGGACCTC | X69808 | Leder and Silverstein, 2006 |
| pomc-b | CCAGAACCCTCACTGTGACGG | CCTGCTGCCCTCCTCTACTGC | X69809 | Leder and Silverstein, 2006 |
| ef-1α | CATTGACAAGAGAACCATTGA | CCTTCAGCTTGTCCAGCAC | AF498320 | Cleveland and Weber, 2014 |
| Muscle | ||||||
|---|---|---|---|---|---|---|
| Fatty Acid | WT Fed | Mutant Fed | p-Value | WT Fasted | Mutant Fasted | p-Value |
| 14:0 | 467.1 ± 46.5 | 548.2 ± 54.4 | 0.270 | 334.9 ± 31.6 | 221.2 ± 23.7 * | 0.010 |
| 16:0 | 4703.4 ± 415.9 | 5309.5 ± 443.3 | 0.329 | 3855.8 ± 240.9 | 2811.7 ± 185.0 * | 0.002 |
| 17:0 | 91.8 ± 5.7 | 97.9 ± 7.6 | 0.529 | 88.5 ± 6.1 | 76.2 ± 6.3 | 0.173 |
| 18:0 | 1287.9 ± 102.3 | 1493.9 ± 129.3 | 0.226 | 1240.6 ± 76.9 | 919.3 ± 58.7 * | 0.003 |
| 20:0 | 51.0 ± 4.1 | 58.4 ± 4.8 | 0.255 | 45.1 ± 3.1 | 33.6 ± 3.0 * | 0.013 |
| 21:0 | 40.4 ± 3.6 | 41.8 ± 4.4 | 0.807 | 41.0 ± 4.6 | 39.7 ± 4.2 | 0.830 |
| 22:0 | 36.3 ± 3.2 | 43.6 ± 4.1 | 0.175 | 28.8 ± 2.1 | 20.5 ± 1.8 * | 0.006 |
| SFA | 6814.6 ± 577.7 | 7731.7 ± 645.8 | 0.301 | 5772.0 ± 358.2 | 4243.7 ± 278.7 * | 0.003 |
| 16:1n-7, Z | 1244.9 ± 148.5 | 1479.2 ± 150.2 | 0.278 | 867.7 ± 79.0 | 571.4 ± 64.5 * | 0.009 |
| 18:1n-9, E | 48.5 ± 3.8 | 55.5 ± 4.9 | 0.268 | 43.3 ± 2.8 | 30.3 ± 2.3 * | 0.002 |
| 18:1n-9, Z | 5953.5 ± 666.9 | 7134.2 ± 705.4 | 0.236 | 4971.6 ± 425.2 | 3336.8 ± 341.4 * | 0.007 |
| 18:1n-7, Z | 708.8 ± 71.9 | 809.3 ± 78.3 | 0.355 | 573.9 ± 46.4 | 394.6 ± 36.7 * | 0.006 |
| 20:1n-9 | 406.3 ± 45.8 | 479.2 ± 45.4 | 0.269 | 381.6 ± 35.6 | 269.8 ± 30.0 * | 0.026 |
| 22:1n-9 | 61.1 ± 6.5 | 71.1 ± 7.1 | 0.308 | 62.7 ± 5.4 | 43.3 ± 4.5 * | 0.012 |
| 24:1n-9 | 60.7 ± 4.8 | 69.6 ± 5.9 | 0.251 | 62.9 ± 4.7 | 48.3 ± 3.2 * | 0.019 |
| MUFA | 8534.7 ± 949.8 | 10,155.9 ± 999.0 | 0.251 | 7002.2 ± 595.7 | 4722.0 ± 481.4 * | 0.007 |
| 18:2n-6, Z, Z | 4693.1 ± 463.9 | 5237.4 ± 488.2 | 0.427 | 3843.6 ± 313.0 | 2696.0 ± 235.0 * | 0.008 |
| 18:3n-6 | 94.7 ± 8.2 | 119.0 ± 9.7 | 0.068 | 67.7 ± 4.9 | 49.4 ± 4.8 * | 0.013 |
| 18:3n-3 | 492.3 ± 45.3 | 549.3 ± 50.1 | 0.409 | 393.2 ± 28.1 | 277.4 ± 23.0 * | 0.004 |
| 20:2n-6 | 363.5 ± 29.4 | 408.6 ± 36.9 | 0.352 | 293.1 ± 23.3 | 212.3 ± 17.6 * | 0.012 |
| 20:3n-6 | 236.2 ± 11.9 | 257.1 ± 23.1 | 0.438 | 206.6 ± 11.3 | 162.5 ± 9.3 * | 0.007 |
| 20:3n-3 | 41.9 ± 3.5 | 45.9 ± 4.1 | 0.475 | 34.4 ± 2.4 | 25.3 ± 1.8 * | 0.007 |
| 20:4n-6 | 308.8 ± 15.4 | 349.3 ± 22.9 | 0.159 | 339.2 ± 13.0 | 298.2 ± 11.7 * | 0.029 |
| 22:2n-6 | 32.4 ± 3.1 | 37.3 ± 3.7 | 0.326 | 28.5 ± 2.6 | 20.5 ± 2.1 * | 0.024 |
| 20:5n-3 | 462.8 ± 22.0 | 491.1 ± 30.3 | 0.462 | 513.5 ± 17.1 | 457.8 ± 16.1 * | 0.027 |
| 22:4n-6 | 42.0 ± 2.4 | 48.9 ± 3.7 | 0.134 | 36.6 ± 1.8 | 28.1 ± 1.8 * | 0.003 |
| 22:5n-6 | 111.6 ± 5.4 | 128.6 ± 7.7 | 0.086 | 99.3 ± 4.5 | 85.2 ± 4.4 * | 0.035 |
| 22:5n-3 | 142.6 ± 6.4 | 152.6 ± 10.8 | 0.445 | 161.1 ± 7.0 | 127.4 ± 7.0 * | 0.002 |
| 22:6n-3 | 2778.0 ± 96.9 | 2904.4 ± 146.2 | 0.484 | 3368.6 ± 125.2 | 2965.2 ± 68.6 * | 0.012 |
| PUFA | 9800.1 ± 693.5 | 10,729.4 ± 807.1 | 0.394 | 9385.5 ± 496.7 | 7405.4 ± 379.0 * | 0.005 |
| n-3 | 3917.7 ± 167.4 | 4143.3 ± 233.3 | 0.447 | 4470.8 ± 167.5 | 3853.1 ± 108.1 * | 0.006 |
| Total | 25,149.5 ± 2216.3 | 28,617.0 ± 2441.6 | 0.304 | 22,159.7 ± 1426.4 | 16,371.1 ± 1134.2 * | 0.005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mankiewicz, J.L.; Picklo, M.J.; Idso, J.; Cleveland, B.M. Leptin Receptor Deficiency Results in Hyperphagia and Increased Fatty Acid Mobilization during Fasting in Rainbow Trout (Oncorhynchus mykiss). Biomolecules 2022, 12, 516. https://doi.org/10.3390/biom12040516
Mankiewicz JL, Picklo MJ, Idso J, Cleveland BM. Leptin Receptor Deficiency Results in Hyperphagia and Increased Fatty Acid Mobilization during Fasting in Rainbow Trout (Oncorhynchus mykiss). Biomolecules. 2022; 12(4):516. https://doi.org/10.3390/biom12040516
Chicago/Turabian StyleMankiewicz, Jamie L., Matthew J. Picklo, Joseph Idso, and Beth M. Cleveland. 2022. "Leptin Receptor Deficiency Results in Hyperphagia and Increased Fatty Acid Mobilization during Fasting in Rainbow Trout (Oncorhynchus mykiss)" Biomolecules 12, no. 4: 516. https://doi.org/10.3390/biom12040516