
Transcriptomic-Based Quantification of the Epithelial-Hybrid-Mesenchymal Spectrum across Biological Contexts

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Software and Datasets
2.2. Preprocessing of Datasets
2.3. T-Test
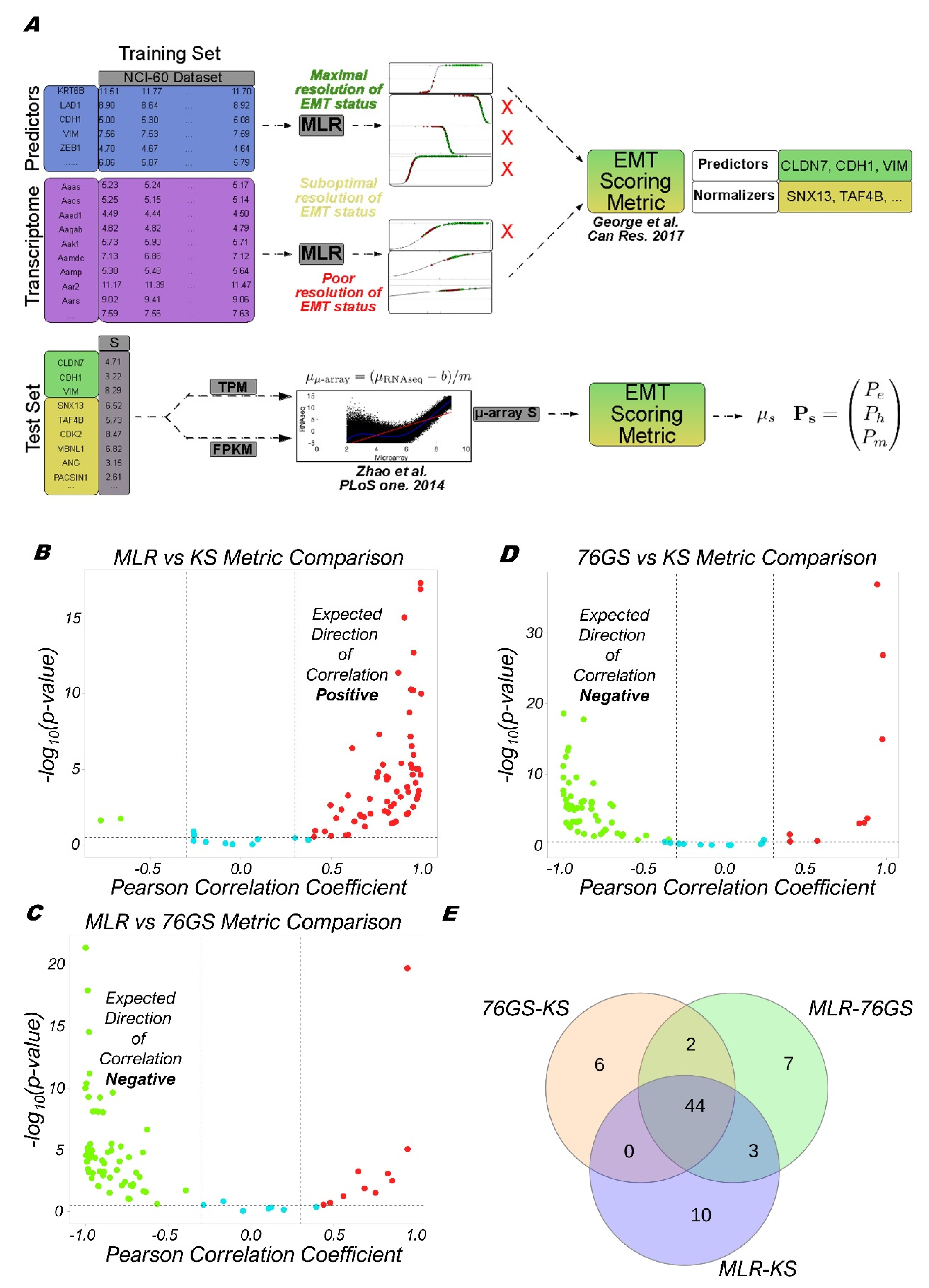
2.4. Calculation of EMT Scores
2.5. MLR Model Applied to RNA-Seq
2.6. Survival Analysis
2.7. ssGSEA
3. Results
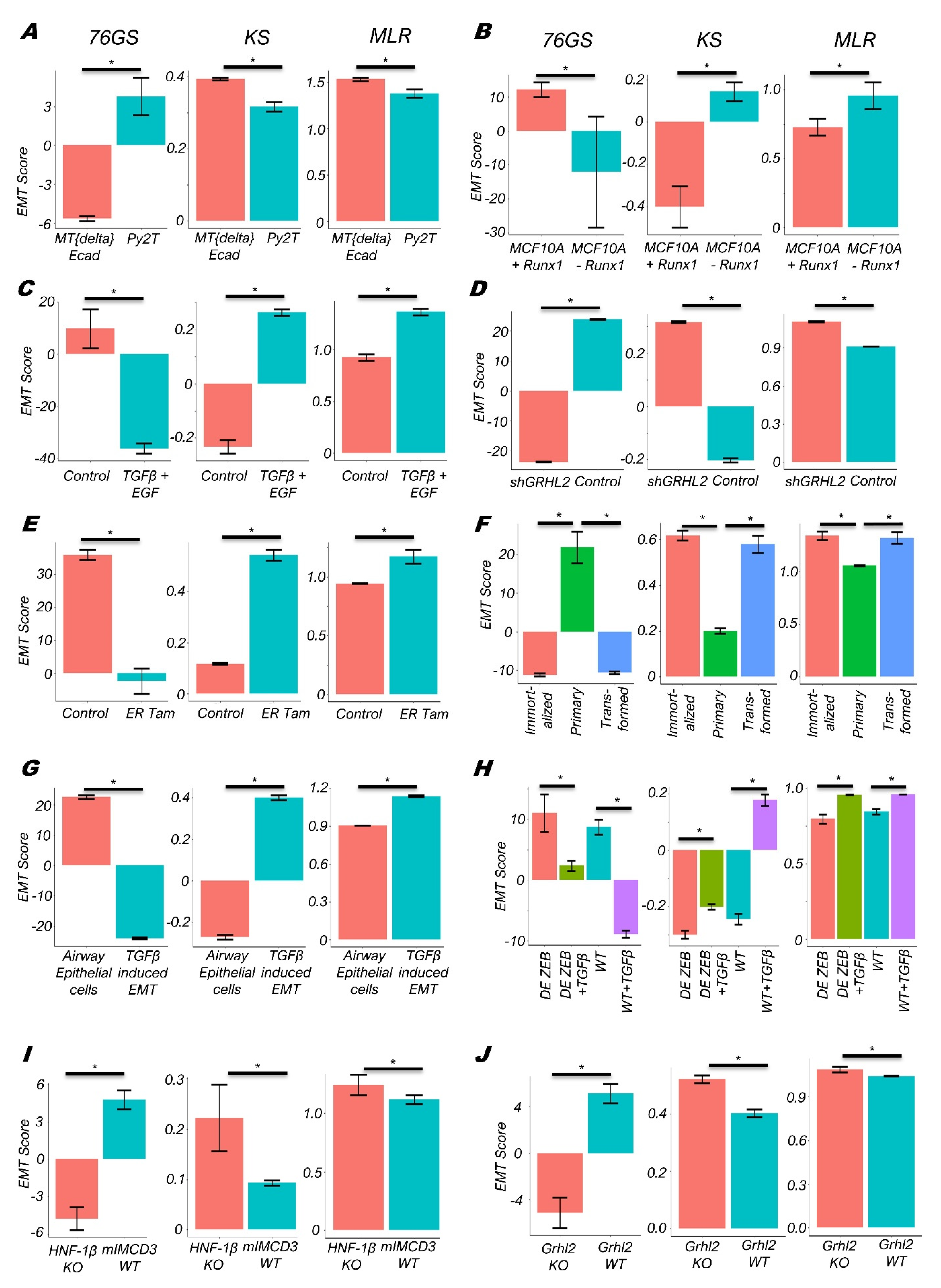
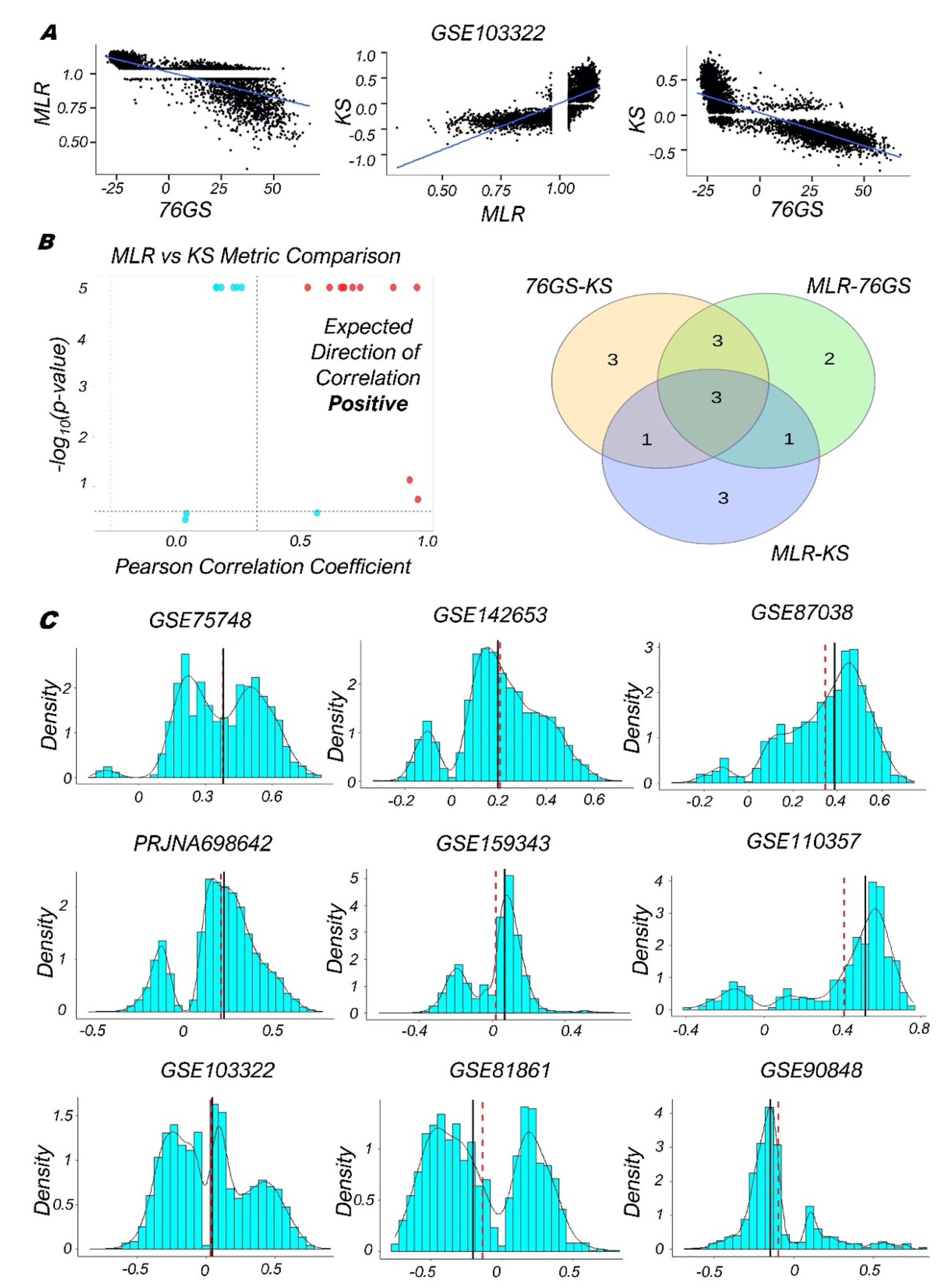
3.1. EMT Scoring Methods Show Concordant Trends across Bulk RNA-Seq Datasets
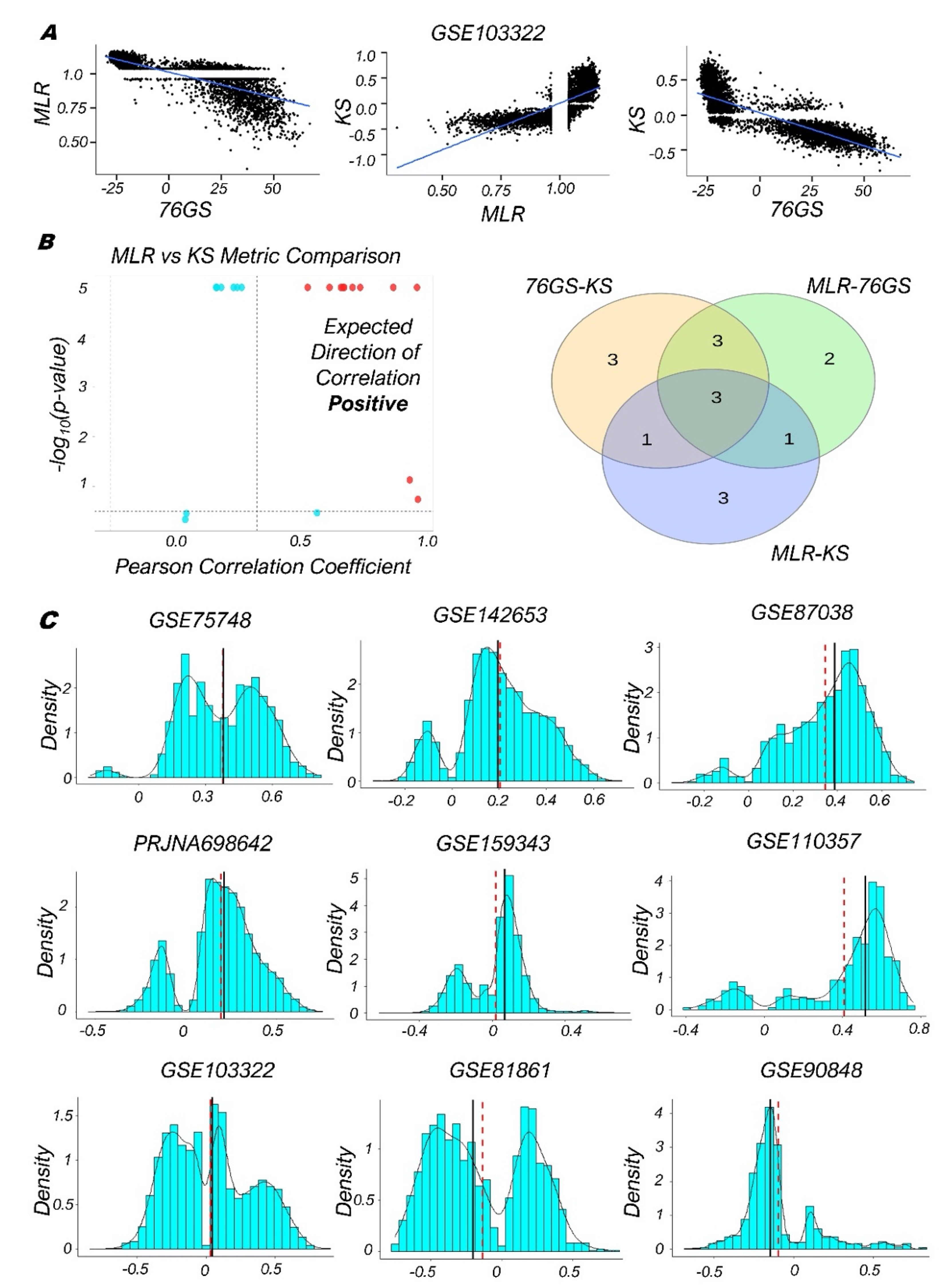
3.2. Single-Cell RNA-Seq Data Analysis Reveals Heterogeneity along the EMP Spectrum
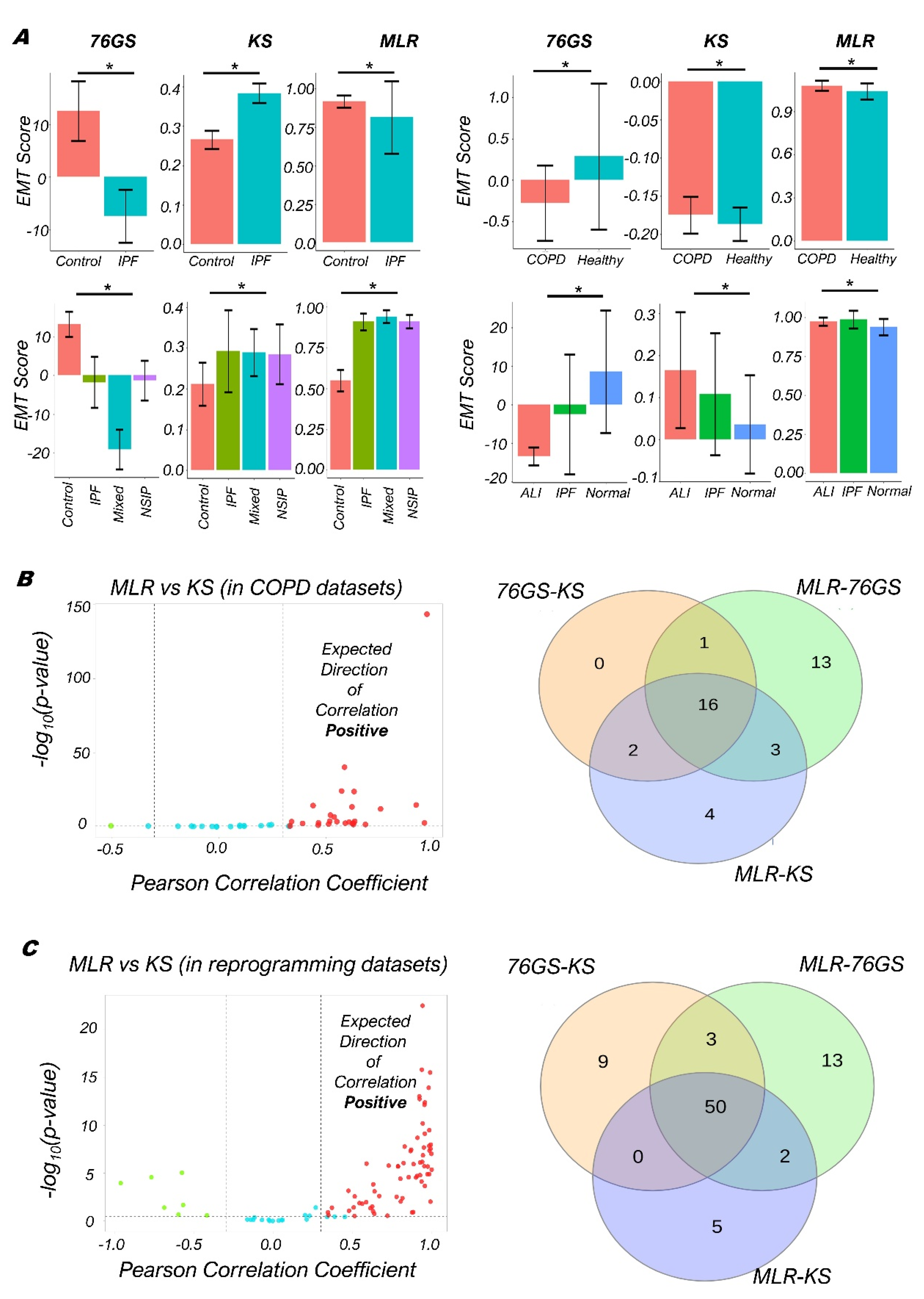
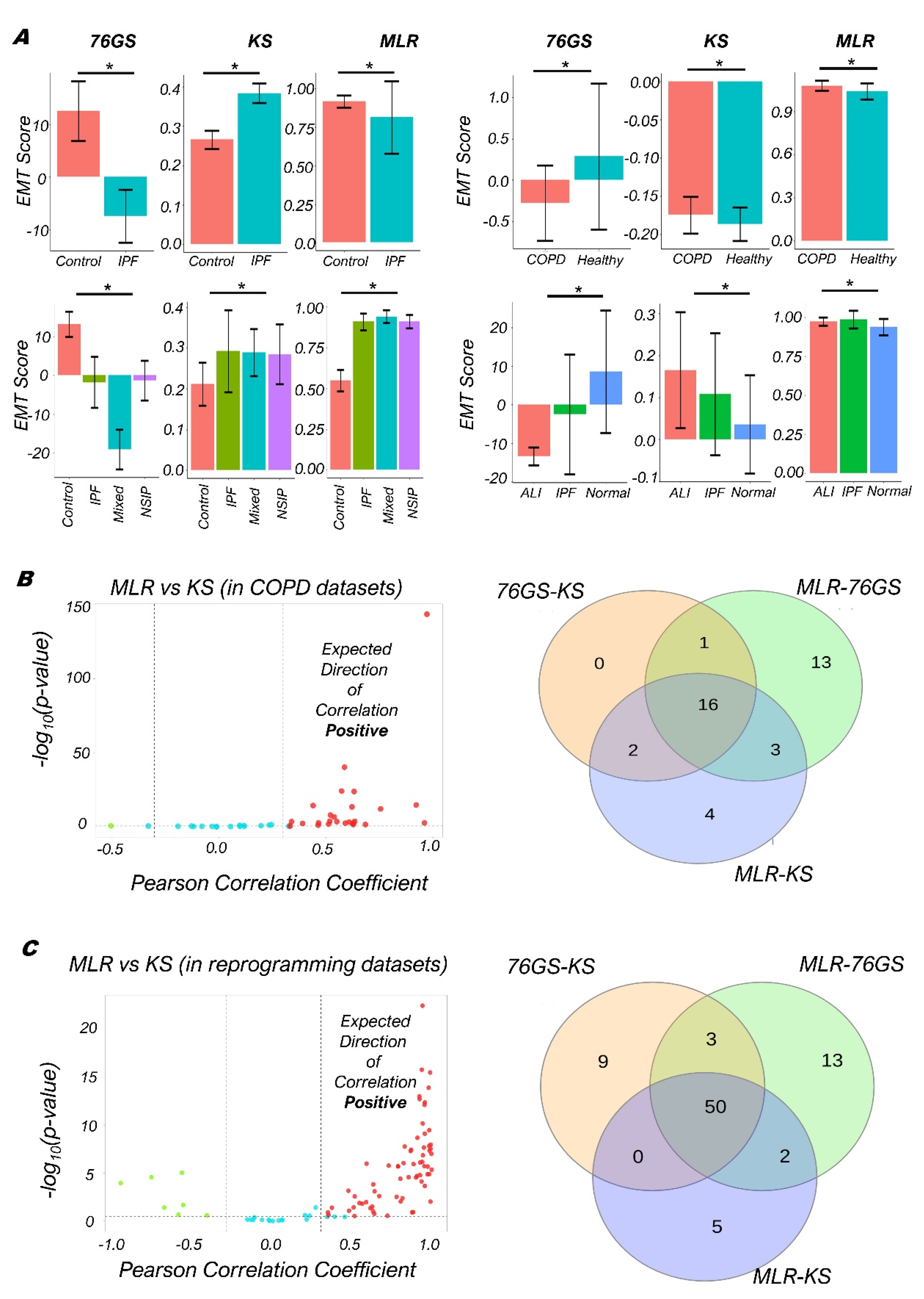
3.3. Quantifying the EMP Spectrum during Lung Diseases and Cellular Reprogramming
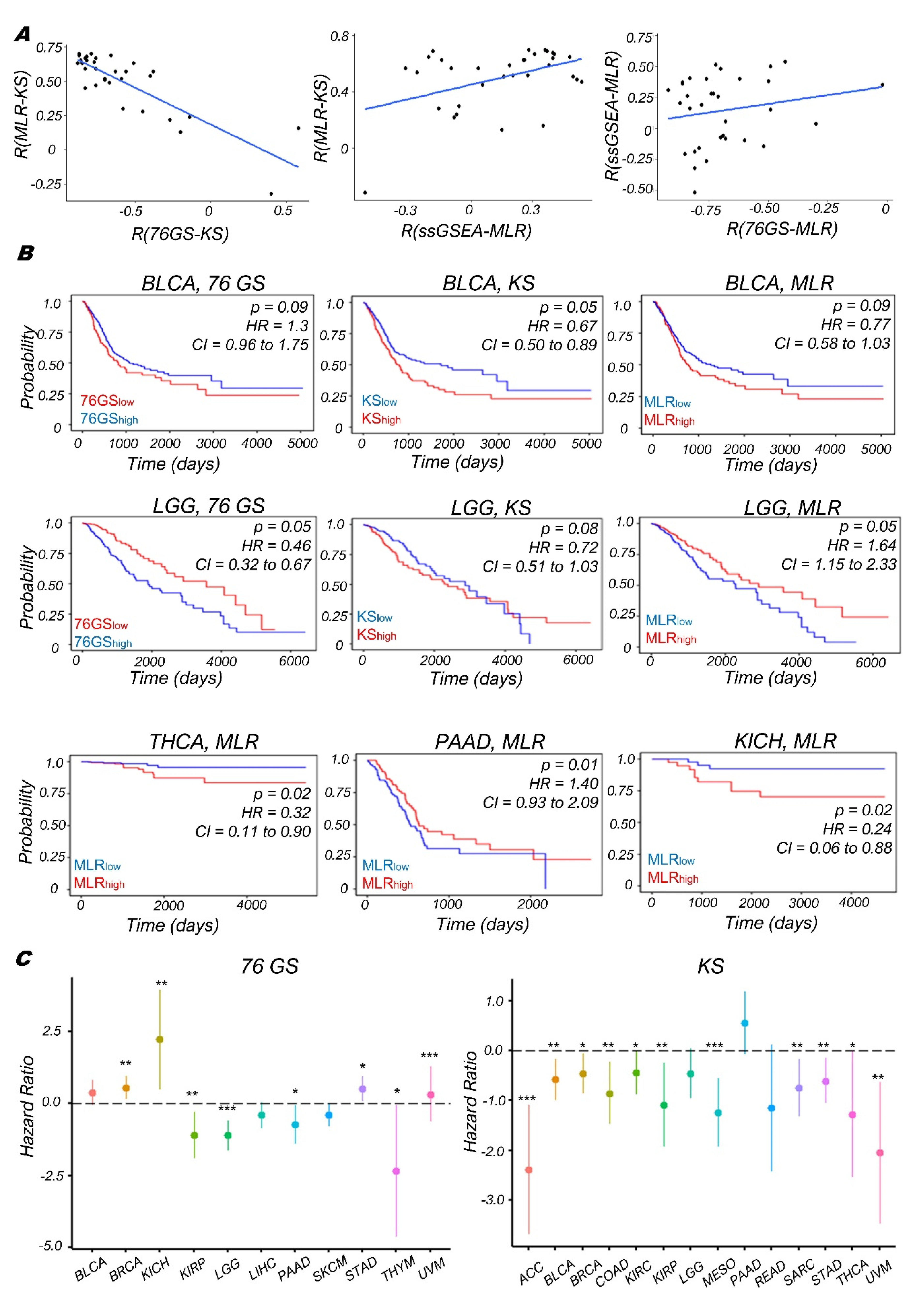
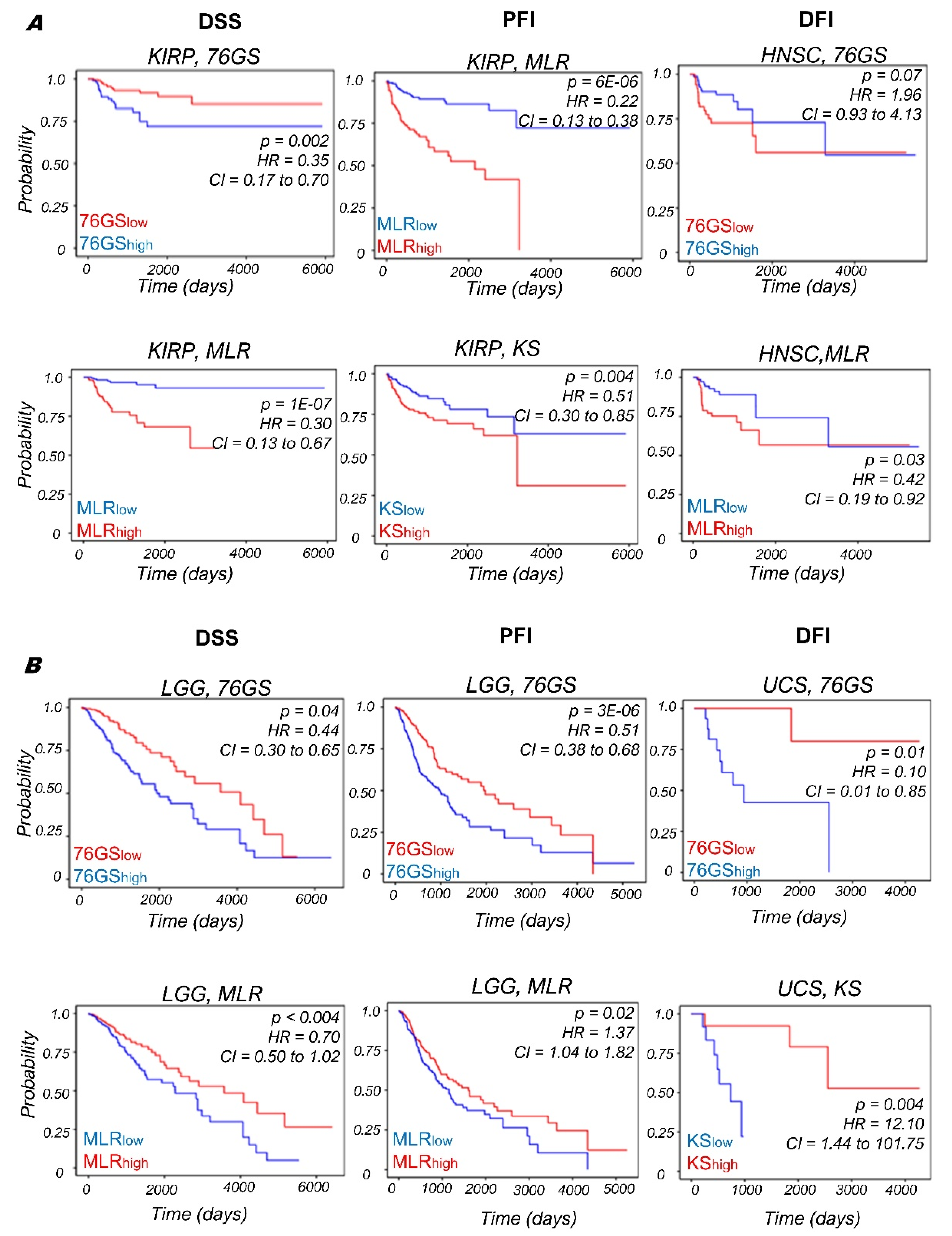
3.4. Context-Specific Association of EMP Status with Patient Survival
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Gupta, G.P.; Massague, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Levine, H. Computational systems biology of epithelial-hybrid-mesenchymal transitions. Curr. Opin. Syst. Biol. 2017, 3, 1–6. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Pal, A.; Barrett, T.F.; Paolini, R.; Parikh, A.; Puram, S.V. Partial EMT in head and neck cancer biology: A spectrum instead of a switch. Oncogene 2021, 40, 5049–5065. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Pasani, S.; Sahoo, S.; Jolly, M.K. Hybrid E/M phenotype(s) and stemness: A mechanistic connection embedded in network topology. J. Clin. Med. 2021, 10, 60. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl. Acad. Sci. USA 2016, 113, E1555–E1564. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, S.; Mishra, A.; Kaur, H.; Hari, K.; Muralidharan, S.; Mandal, S.; Jolly, M.K. A mechanistic model captures the emergence and implications of non-genetic heterogeneity and reversible drug resistance in ER+ breast cancer cells. NAR Cancer 2021, 3, zcab027. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, S.; Nayak, S.P.; Hari, K.; Purkait, P.; Mandal, S.; Kishore, A.; Levine, H.; Jolly, M.K. Immunosuppressive traits of the hybrid epithelial/mesenchymal phenotype. Front. Immunol. 2021, 12, 797261. [Google Scholar] [CrossRef]
- Godin, L.; Balsat, C.; Van Eycke, Y.; Allard, J.; Royer, C.; Remmelink, M.; Pastushenko, I.; Haene, N.D.; Blanpain, C.; Salmon, I.; et al. A Novel Approach for Quantifying Cancer Cells Showing Hybrid Epithelial/Mesenchymal States in Large Series of Tissue Samples: Towards a New Prognostic Marker. Cancers 2020, 12, 906. [Google Scholar] [CrossRef] [Green Version]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef] [Green Version]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.Y.-J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.-S.; Matsumura, N.; Lai, H.-C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [Green Version]
- Simeonov, K.P.; Byrns, C.N.; Clark, M.L.; Norgard, R.J.; Martin, B.; Stanger, B.Z.; Shendure, J.; McKenna, A.; Lengner, C.J. Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell 2021, 39, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Saxena, K.; Subbalakshmi, A.R.; Jolly, M.K. Phenotypic heterogeneity in circulating tumor cells and its prognostic value in metastasis and overall survival. EBioMedicine 2019, 46, 4–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecharpentier, A.; Vielh, P.; Perez-Moreno, P.; Planchard, D.; Soria, J.C.; Farace, F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br. J. Cancer 2011, 105, 1338–1341. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Mandal, S.; Tejaswi, T.; Jolly, M.K. Investigating epithelial-mesenchymal heterogeneity of tumors and circulating tumor cells with transcriptomic analysis and biophysical modeling. Comput. Syst. Oncol. 2021, in press. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Debeb, B.G.; Aceto, N.; Farach-Carson, M.C.; Woodward, W.A.; Levine, H. Inflammatory Breast Cancer: A model for investigating cluster-based dissemination. NPJ Breast Cancer 2017, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Murphy, R.; Bhatia, S.; Whitfield, H.J.; Redfern, A.; Davis, M.J.; Thompson, E.W. Measuring and Modelling the Epithelial- Mesenchymal Hybrid State in Cancer: Clinical Implications. Cells Tissues Organs 2021, in press. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.C.; Majewski, T.; Zhang, L.; Yao, H.; Bondaruk, J.; Wang, Y.; Zhang, S.; Wang, Z.; Lee, J.G.; Lee, S.; et al. Dysregulation of EMT Drives the Progression to Clinically Aggressive Sarcomatoid Bladder Cancer. Cell Rep. 2019, 27, 1781–1793. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival outcomes in cancer patients predicted by a partial EMT gene expression scoring metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.; George, J.T.; Tripathi, S.; Levine, H.; Jolly, M.K. Comparative Study of Transcriptomics-Based Scoring Metrics for the Epithelial-Hybrid-Mesenchymal Spectrum. Front. Bioeng. Biotechnol. 2020, 8, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.; Ward, C.; Eapen, M.S.; Myers, S.; Hallgren, O.; Levine, H.; Sohal, S.S. Epithelial–mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease. Dev. Dyn. 2018, 247, 346–358. [Google Scholar] [CrossRef] [Green Version]
- Sohal, S.S. Epithelial and endothelial cell plasticity in chronic obstructive pulmonary disease (COPD). Respir. Investig. 2017, 55, 104–113. [Google Scholar] [CrossRef]
- Lai, X.; Li, Q.; Wu, F.; Lin, J.; Chen, J.; Zheng, H.; Guo, L. Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons From Somatic Cell Reprogramming. Front. Cell Dev. Biol. 2020, 8, 760. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/ (accessed on 18 November 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef]
- Ishay-Ronen, D.; Diepenbruck, M.; Kalathur, R.K.R.; Sugiyama, N.; Tiede, S.; Ivanek, R.; Bantug, G.; Morini, M.F.; Wang, J.; Hess, C.; et al. Gain Fat—Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell 2019, 35, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.; Messier, T.L.; Tye, C.E.; Dobson, J.R.; Fritz, A.J.; Sikora, K.R.; Browne, G.; Stein, J.L.; Lian, J.B.; Stein, G.S. Runx1 stabilizes the mammary epithelial cell phenotype and prevents epithelial to mesenchymal transition. Oncotarget 2017, 8, 17610–17627. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, V.; Sixt, K.M.; Gao, S.; Xu, X.; Huang, J.; Weigert, R.; Zhou, M.; Zhang, Y.E. Direct Regulation of Alternative Splicing by SMAD3 through PCBP1 Is Essential to the Tumor-Promoting Role of TGF-β. Mol. Cell 2016, 64, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Li, X.; Kalita, M.; Widen, S.G.; Yang, J.; Bhavnani, S.K.; Dang, B.; Kudlicki, A.; Sinha, M.; Kong, F.; et al. Analysis of the TGFβ-induced program in primary airway epithelial cells shows essential role of NF-κB/RelA signaling network in type II epithelial mesenchymal transition. BMC Genom. 2015, 16, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Panchy, N.; Noguchi, S.; Suzuki, H.; Hong, T. Combinatorial perturbation analysis reveals divergent regulations of mesenchymal genes during epithelial-to-mesenchymal transition. NPJ Syst. Biol. Appl. 2019, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Drápela, S.; Bouchal, J.; Jolly, M.K.; Culig, Z. ZEB1: A Critical Regulator of Cell Plasticity, DNA Damage Response, and Therapy Resistance. Front. Mol. Biosci. 2020, 7, 36. [Google Scholar] [CrossRef]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phentoype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieply, B.; Farris, J.; Denvir, J.; Ford, H.L.; Frisch, S.M. Epithelial-Mesenchymal Transition and Tumor Suppression Are Controlled by a Reciprocal Feedback Loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013, 73, 6299–6309. [Google Scholar] [CrossRef] [Green Version]
- Chung, V.Y.; Tan, T.Z.; Tan, M.; Wong, M.K.; Kuay, K.T.; Yang, Z.; Ye, J.; Muller, J.; Koh, C.M.; Guccione, E.; et al. GRHL2-miR-200-ZEB1 maintains the epithelial status of ovarian cancer through transcriptional regulation and histone modification. Sci. Rep. 2016, 6, 19943. [Google Scholar] [CrossRef] [Green Version]
- Mooney, S.M.; Talebian, V.; Jolly, M.K.; Jia, D.; Gromala, M.; Levine, H.; McConkey, B.J. The GRHL2/ZEB Feedback Loop—A Key Axis in the Regulation of EMT in Breast Cancer. J. Cell. Biochem. 2017, 118, 2559–2570. [Google Scholar] [CrossRef]
- Chung, V.Y.; Tan, T.Z.; Ye, J.; Huang, R.-L.; Lai, H.-C.; Kappei, D.; Wollmann, H.; Guccione, E.; Huang, R.Y.-J. The role of GRHL2 and epigenetic remodeling in epithelial–mesenchymal plasticity in ovarian cancer cells. Commun. Biol. 2019, 2, 272. [Google Scholar] [CrossRef] [Green Version]
- Aue, A.; Hinze, C.; Walentin, K.; Ruffert, J.; Yurtdas, Y.; Werth, M.; Chen, W.; Rabien, A.; Kilic, E.; Schulzke, J.D.; et al. A grainyhead-like 2/Ovo-like 2 pathway regulates renal epithelial barrier function and lumen expansion. J. Am. Soc. Nephrol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Jolly, M.K.; Boareto, M.; Parsana, P.; Mooney, S.M.; Pienta, K.J.; Levine, H.; Ben-Jacob, E. OVOL guides the epithelial-hybrid-mesenchymal transition. Oncotarget 2015, 6, 15436–15448. [Google Scholar] [CrossRef] [Green Version]
- Carpinelli, M.R.; de Vries, M.E.; Auden, A.; Butt, T.; Deng, Z.; Partridge, D.D.; Miles, L.B.; Georgy, S.R.; Haigh, J.J.; Darido, C.; et al. Inactivation of Zeb1 in GRHL2-deficient mouse embryos rescues mid-gestation viability and secondary palate closure. Dis. Model. Mech. 2020, 13, dmm042218. [Google Scholar] [CrossRef] [Green Version]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, A.; Beltran, M.; Peiro, S.; de Herreros, A.G. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A.; et al. Twist, a master regulator of Morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Harvey, S.E.; Zheng, R.; Lyu, J.; Grzeskowiak, C.L.; Powell, E.; Piwnica-Worms, H.; Scott, K.L.; Cheng, C. The RNA-binding protein AKAP8 suppresses tumor metastasis by antagonizing EMT-associated alternative splicing. Nat. Commun. 2020, 11, 486. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Zhang, Y.; Shao, A.; Avdulov, S.; Herrera, J.; Aboudehen, K.; Pontoglio, M.; Igarashi, P. Mechanism of Fibrosis in HNF1B-Related Autosomal Dominant Tubulointerstitial Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 2493–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.F.; Leng, N.; Zhang, J.; Hou, Z.; Mamott, D.; Vereide, D.T.; Choi, J.; Kendziorski, C.; Stewart, R.; Thomson, J.A. Single-cell RNA-seq reveals novel regulators of human embryonic stem cell differentiation to definitive endoderm. Genome Biol. 2016, 17, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Cui, Y.; Ma, X.; Yong, J.; Yan, L.; Yang, M.; Ren, J.; Tang, F.; Wen, L.; Qiao, J. Single-cell transcriptomics identifies divergent developmental lineage trajectories during human pituitary development. Nat. Commun. 2020, 11, 5275. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Hu, Y.; Fan, X.; Wu, X.; Mao, Y.; Hu, B.; Guo, H.; Wen, L.; Tang, F. Single-cell RNA-seq analysis unveils a prevalent epithelial/mesenchymal hybrid state during mouse organogenesis. Genome Biol. 2018, 19, 31. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; Den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef]
- Hendley, A.M.; Rao, A.A.; Leonhardt, L.; Ashe, S.; Smith, J.A.; Giacometti, S.; Peng, X.L.; Jiang, H.; Berrios, D.I.; Pawlak, M.; et al. Single-cell transcriptome analysis defines heterogeneity of the murine pancreatic ductal tree. Elife 2021, 10, e67776. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Yang, H.; Adam, R.C.; Ge, Y.; Hua, Z.L.; Fuchs, E. Epithelial-Mesenchymal Micro-niches Govern Stem Cell Lineage Choices. Cell 2017, 169, 483–496. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Huang, X.; Li, Y.; Xu, X.; Li, S.; Jiang, D.; Liang, J.; Jiang, D.; Wang, C.; Dai, H. Down-regulation of USP13 mediates phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasse, A.; Binder, H.; Schupp, J.C.; Kayser, G.; Bargagli, E.; Jaeger, B.; Hess, M.; Rittinghausen, S.; Vuga, L.; Lynn, H.; et al. BAL Cell Gene Expression Is Indicative of Outcome and Airway Basal Cell Involvement in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, M.J.; Hosein, K.; Howlett, C.J.; Joseph, M.; Mura, M. Comprehensive gene expression profiling identifies distinct and overlapping transcriptional profiles in non-specific interstitial pneumonia and idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 153. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, P.; Thompson, J.R.; Ammar, R.; Porteous, M.; McCoubrey, C.; Cantu, E.; Ravi, K.; Zhang, Y.; Luo, Y.; Streltsov, D.; et al. RNA sequencing of transplant-stage idiopathic pulmonary fibrosis lung reveals unique pathway regulation. ERJ Open Res. 2019, 5, 00117–02019. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-benítez, N.E.; Parotto, M.; Post, M.; Han, B.; Spieth, P.M.; Cheng, W.-E.; Valladares, F.; Villar, J.; Liu, M.; Sato, M.; et al. Mechanical stress induces lung fibrosis by epithelial- mesenchymal transition (EMT). Crit. Care Med. 2012, 40, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouda, M.M.; Shaikh, S.B.; Bhandary, Y.P. Inflammatory and Fibrinolytic System in Acute Respiratory Distress Syndrome. Lung 2018, 196, 609–616. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Deshmukh, A.P.; den Hollander, P.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- McFaline-Figueroa, J.L.; Hill, A.J.; Qiu, X.; Jackson, D.; Shendure, J.; Trapnell, C. A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nat. Genet. 2019, 51, 1389–1398. [Google Scholar] [CrossRef]
- Johnson, K.S.; Hussein, S.; Chakraborty, P.; Muruganantham, A.; Mikhail, S.; Gonzalez, G.; Song, S.; Jolly, M.K.; Toneff, M.J.; Benton, M.L.; et al. Epithelial-mesenchymal plasticity through loss of CTCF motif accessibility and protein expression. bioRxiv 2021, 447526. [Google Scholar] [CrossRef]
- Stylianou, N.; Lehman, M.L.; Wang, C.; Fard, A.T.; Rockstroh, A.; Fazli, L.; Jovanovic, L.; Ward, M.; Sadowski, M.C.; Kashyap, A.S.; et al. A molecular portrait of epithelial–mesenchymal plasticity in prostate cancer associated with clinical outcome. Oncogene 2019, 38, 913–934. [Google Scholar] [CrossRef]
- Jia, W.; Deshmukh, A.; Mani, S.A.; Jolly, M.K.; Levine, H. A possible role for epigenetic feedback regulation in the dynamics of the Epithelial-Mesenchymal Transition (EMT). Phys. Biol. 2019, 16, 066004. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.P.; Vanderhyden, B.C. Context specificity of the EMT transcriptional response. Nat. Commun. 2020, 11, 2142. [Google Scholar] [CrossRef]
- Serresi, M.; Kertalli, S.; Li, L.; Schmitt, M.J.; Dramaretska, Y.; Wierikx, J.; Hulsman, D.; Gargiulo, G. Functional antagonism of chromatin modulators regulates epithelial-mesenchymal transition. Sci. Adv. 2021, 7, eabd7974. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Douglas, D.; Zhang, J.; Kumari, S.; Enuameh, M.S.; Dai, Y.; Wallace, C.T.; Watkins, S.C.; Shu, W.; Xing, J. Live-cell imaging and analysis reveal cell phenotypic transition dynamics inherently missing in snapshot data. Sci. Adv. 2020, 6, eaba9319. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, V.; Bose, B. Morphological State Transition Dynamics in EGF-Induced Epithelial to Mesenchymal Transition. J. Clin. Med. 2019, 8, 911. [Google Scholar] [CrossRef] [Green Version]
- Karacosta, L.G.; Anchang, B.; Ignatiadis, N.; Kimmey, S.C.; Benson, J.A.; Shrager, J.B.; Tibshirani, R.; Bendall, S.C.; Plevritis, S.K. Mapping Lung Cancer Epithelial-Mesenchymal Transition States and Trajectories with Single-Cell Resolution. Nat. Commun. 2019, 10, 5587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Riberio, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.S.; Abdollahi, B.; Wilkins, O.M.; Chakraborty, P.; Ognjenovic, N.B.; Muller, K.E.; Kumar Jolly, M.; Hassanpour, S.; Pattabiraman, D.R. Dynamic plasticity within the EMT spectrum, rather than static mesenchymal traits, drives tumor heterogeneity and metastatic progression of breast cancers. bioRxiv 2021, 434993. [Google Scholar] [CrossRef]
- Chakraborty, P.; George, J.T.; Woodward, W.A.; Levine, H.; Jolly, M.K. Gene expression profiles of inflammatory breast cancer reveal high heterogeneity across the epithelial-hybrid-mesenchymal spectrum. Transl. Oncol. 2021, 14, 101026. [Google Scholar] [CrossRef]
- Tsuji, T.; Ibaragi, S.; Shima, K.; Hu, M.G.; Katsurano, M.; Sasaki, A.; Hu, G.F. Epithelial-mesenchymal transition induced by growth suppressor p12 CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008, 68, 10377–10386. [Google Scholar] [CrossRef] [Green Version]
- Neelakantan, D.; Zhou, H.; Oliphant, M.U.J.; Zhang, X.; Simon, L.M.; Henke, D.M.; Shaw, C.A.; Wu, M.F.; Hilsenbeck, S.G.; White, L.D.; et al. EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells. Nat. Commun. 2017, 8, 15773. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi-Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Aban, C.; Lombardi, A.; Neiman, G.; Biani, M.C.; La Greca, A.; Walsman, A.; Moro, L.N.; Sevlever, G.; Miriuka, S.; Luzzani, C. Downregulation of E-cadherin in pluripotent stem cells triggers partial EMT. Sci. Rep. 2021, 11, 2048. [Google Scholar] [CrossRef] [PubMed]
- Leroy, P.; Mostov, K.E. Slug Is Required for Cell Survival during Partial Epithelial-Mesenchymal Transition of HGF-induced tubulogenesis. J. Cell Sci. 2007, 18, 1943–1952. [Google Scholar] [CrossRef]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Horan, K.; Cursons, J.; Davis, M.J. Single sample scoring of molecular phenotypes. BMC Bioinformatics 2018, 19, 404. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Jolly, M.K.; George, J.T.; Levine, H.; Onuchic, J.N. A mechanism-based computational model to capture the interconnections among epithelial-mesenchymal transition, cancer stem cells and Notch-Jagged signaling. Oncotarget 2018, 9, 29906–29920. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jolly, M.K.; George, J.T.; Pienta, K.J.; Levine, H. Computational Modeling of the Crosstalk Between Macrophage Polarization and Tumor Cell Plasticity in the Tumor Microenvironment. Front. Oncol. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Wang, P.; Zhou, R.; Thomas, P.; Zhao, L.; Zhou, R.; Mandal, S.; Jolly, M.K.; Richard, D.J.; Rehm, B.H.A.; Ostrikov, K.; et al. Epithelial-to-mesenchymal transition enhances cancer cell sensitivity to cytotoxic effects of zcold atmospheric plasmas in breast and bladder cancer systems. Cancers 2021, 13, 2889. [Google Scholar] [CrossRef]
- Ling, J.; Nie, Q.; Li, C. Landscape and kinetic path quantify critical transitions in epithelial-mesenchymal transition. Biophys. J. 2021, 120, 4484–4500. [Google Scholar] [CrossRef]
- Tripathi, S.; Levine, H.; Jolly, M.K. The Physics of Cellular Decision-Making during Epithelial-Mesenchymal Transition. Annu. Rev. Biophys. 2020, 49, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celià-Terrassa, T.; Bastian, C.; Liu, D.D.; Ell, B.; Aiello, N.M.; Wei, Y.; Zamalloa, J.; Blanco, A.M.; Hang, X.; Kunisky, D.; et al. Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat. Commun. 2018, 9, 5005. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Park, J.H.; Kaur, H.; Jung, K.H.; Yang, S.; Tripathi, S.; Galbraith, M.; Deng, Y.; Jolly, M.K.; Kaipparettu, B.A.; et al. Towards decoding the coupled decision-making of metabolism and epithelial-to-mesenchymal transition in cancer. Br. J. Cancer 2021, 124, 1902–1911. [Google Scholar] [CrossRef]
- Dumont, N.; Wilson, M.B.; Crawford, Y.G.; Reynolds, P.A.; Sigaroudinia, M.; Tlsty, T.D. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 14867–14872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C.; et al. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Epithelial-Mesenchymal Transition Is the Main Driver of Intrinsic Metabolism in Cancer Cell Lines. Available online: https://www.biorxiv.org/content/10.1101/2021.11.02.466992v1.full (accessed on 18 December 2021).
- Quantifying the Patterns of Metabolic Plasticity and Heterogeneity along the Epithelial-Hybrid-Mesenchymal Spectrum in Cancer. Available online: https://www.biorxiv.org/content/10.1101/2021.12.18.473275v1 (accessed on 18 December 2021).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandal, S.; Tejaswi, T.; Janivara, R.; Srikrishnan, S.; Thakur, P.; Sahoo, S.; Chakraborty, P.; Sohal, S.S.; Levine, H.; George, J.T.; et al. Transcriptomic-Based Quantification of the Epithelial-Hybrid-Mesenchymal Spectrum across Biological Contexts. Biomolecules 2022, 12, 29. https://doi.org/10.3390/biom12010029
Mandal S, Tejaswi T, Janivara R, Srikrishnan S, Thakur P, Sahoo S, Chakraborty P, Sohal SS, Levine H, George JT, et al. Transcriptomic-Based Quantification of the Epithelial-Hybrid-Mesenchymal Spectrum across Biological Contexts. Biomolecules. 2022; 12(1):29. https://doi.org/10.3390/biom12010029
Chicago/Turabian StyleMandal, Susmita, Tanishq Tejaswi, Rohini Janivara, Syamanthak Srikrishnan, Pradipti Thakur, Sarthak Sahoo, Priyanka Chakraborty, Sukhwinder Singh Sohal, Herbert Levine, Jason T. George, and et al. 2022. "Transcriptomic-Based Quantification of the Epithelial-Hybrid-Mesenchymal Spectrum across Biological Contexts" Biomolecules 12, no. 1: 29. https://doi.org/10.3390/biom12010029