
Epigenetic Therapy Augments Classic Chemotherapy in Suppressing the Growth of 3D High-Grade Serous Ovarian Cancer Spheroids over an Extended Period of Time

 , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Therapeutic Agents
2.2. Tissue Culture
2.2.1. Establishment of Concurrent 2D and 3D Tissue Culture
2.2.2. Measurement of Spheroid Growth in 3D Culture
2.3. Cellular Assays
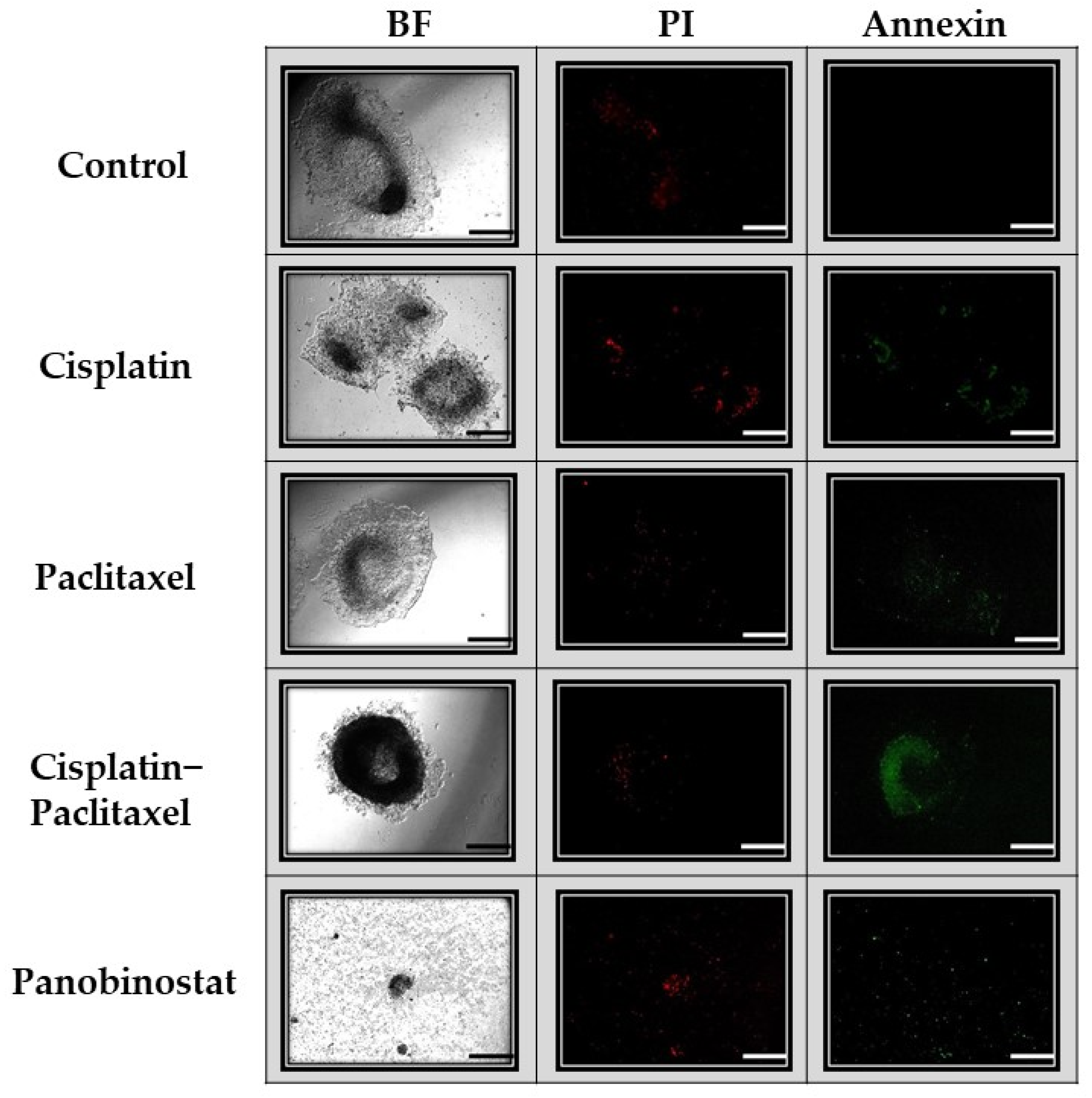
2.3.1. Apoptosis and Necrosis
2.3.2. Viability, Cell Proliferation, Migration and Invasion Assays
2.3.3. Observation of Colony Formation Following Treatment in 3D Culture
2.3.4. Chemoresistance
2.4. Statistical Analysis
3. Results
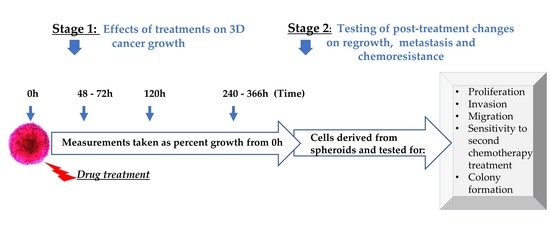
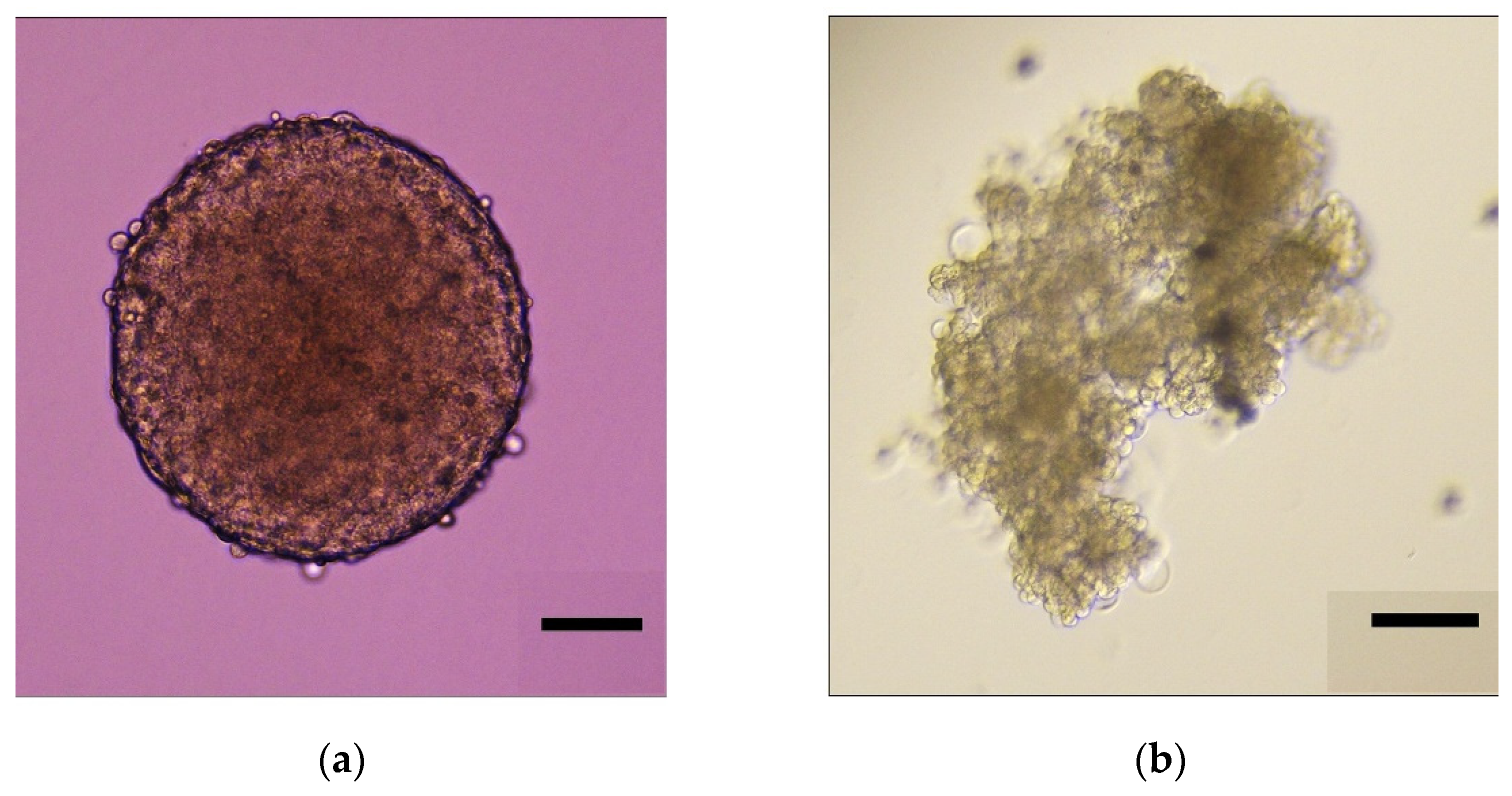
3.1. Spheroid Formation and Experimental Design
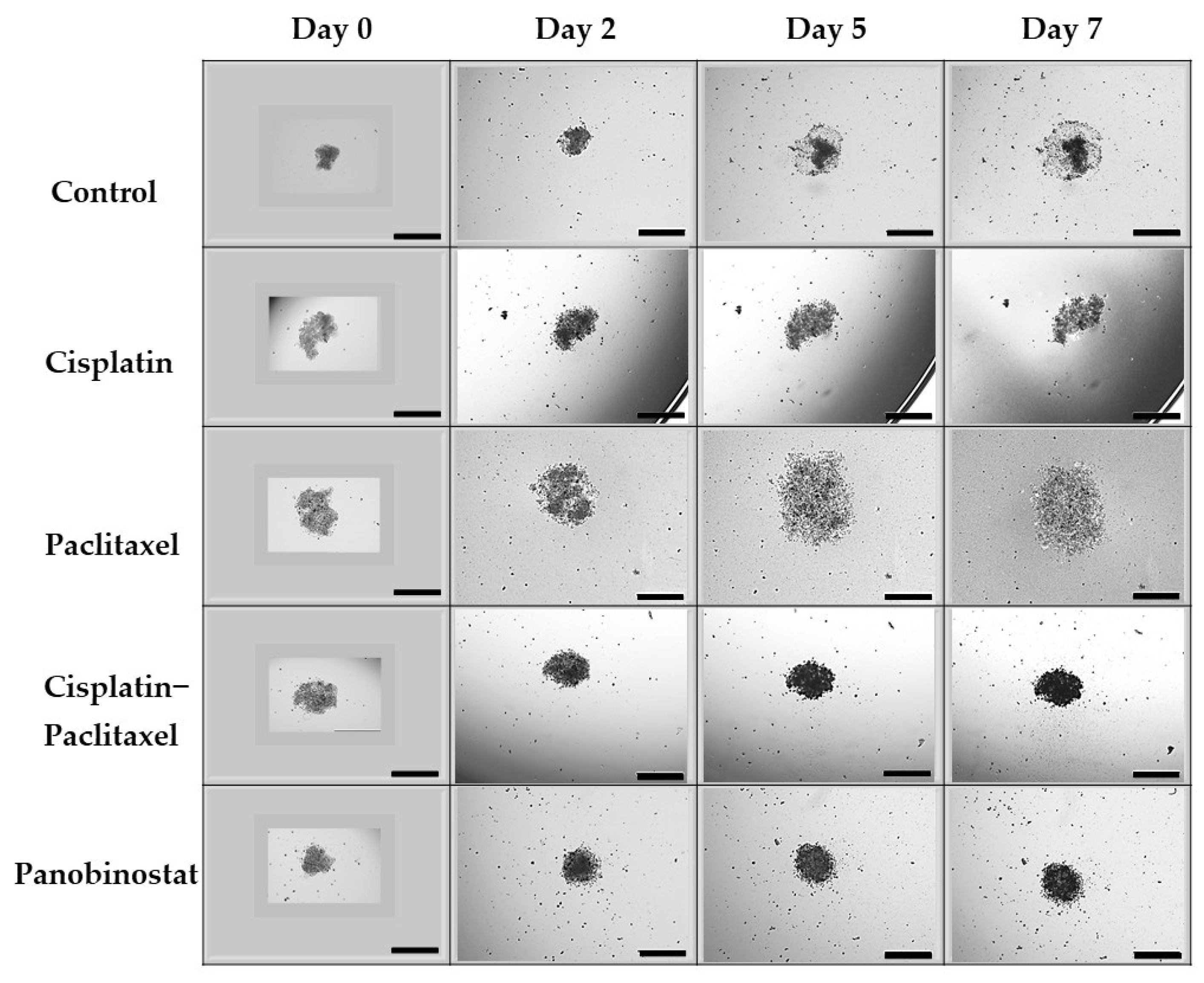
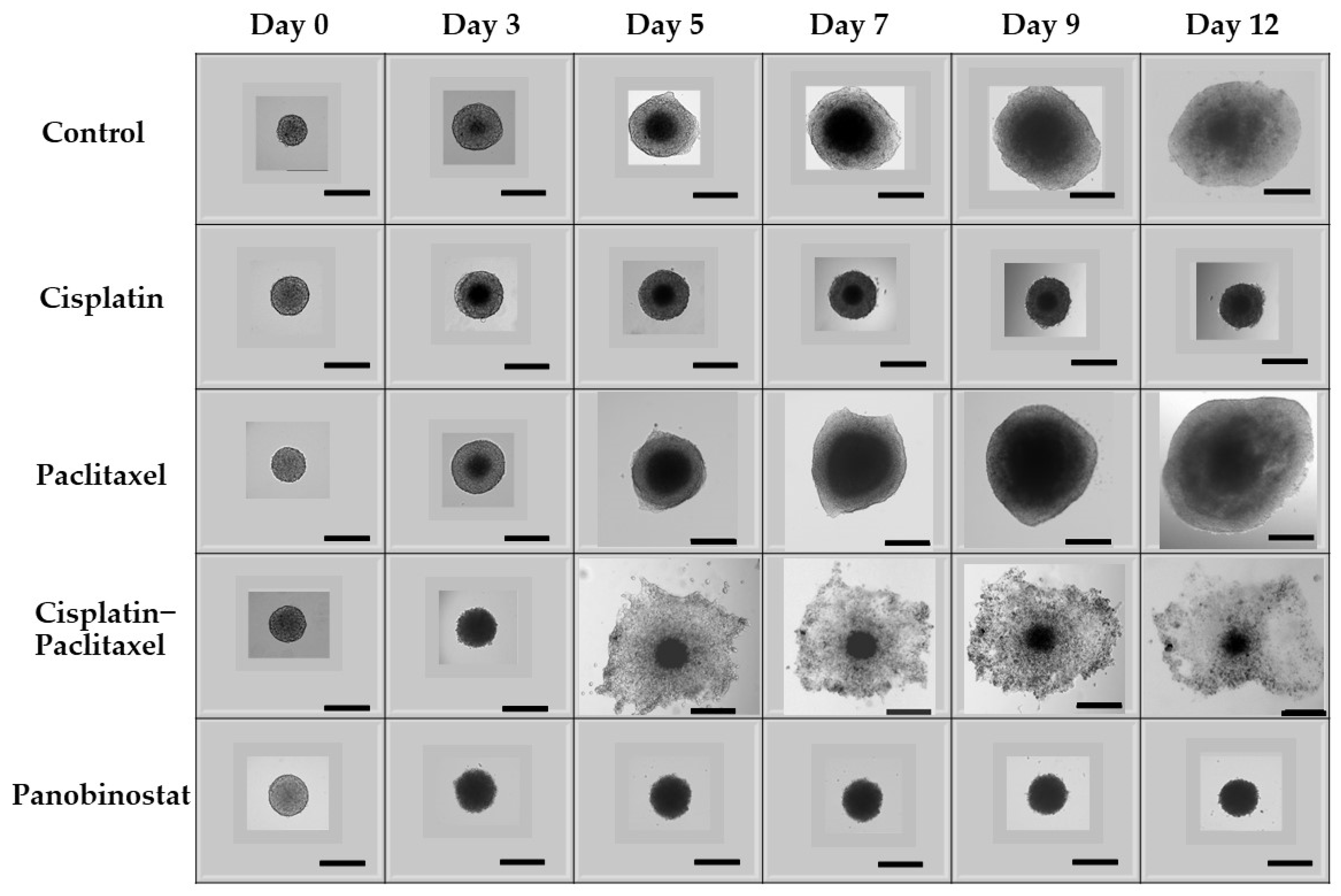
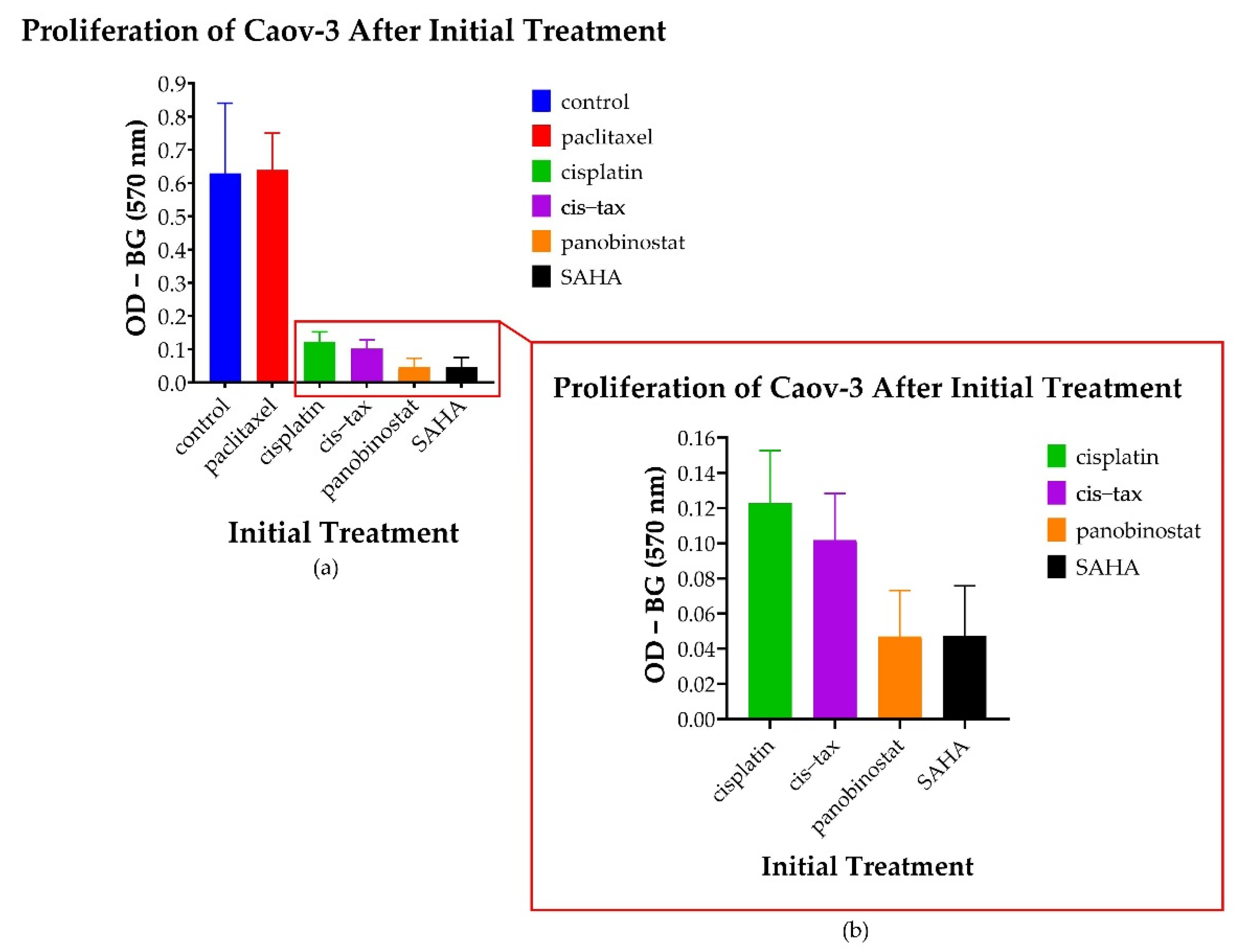
3.2. In Caov-3 Cells, Paclitaxel Alone Does Not Suppress 3D Ovarian Cancer Growth in an Extended Kinetic Assay, Whereas Epigenetic Therapy Causes Spheroid Shrinkage
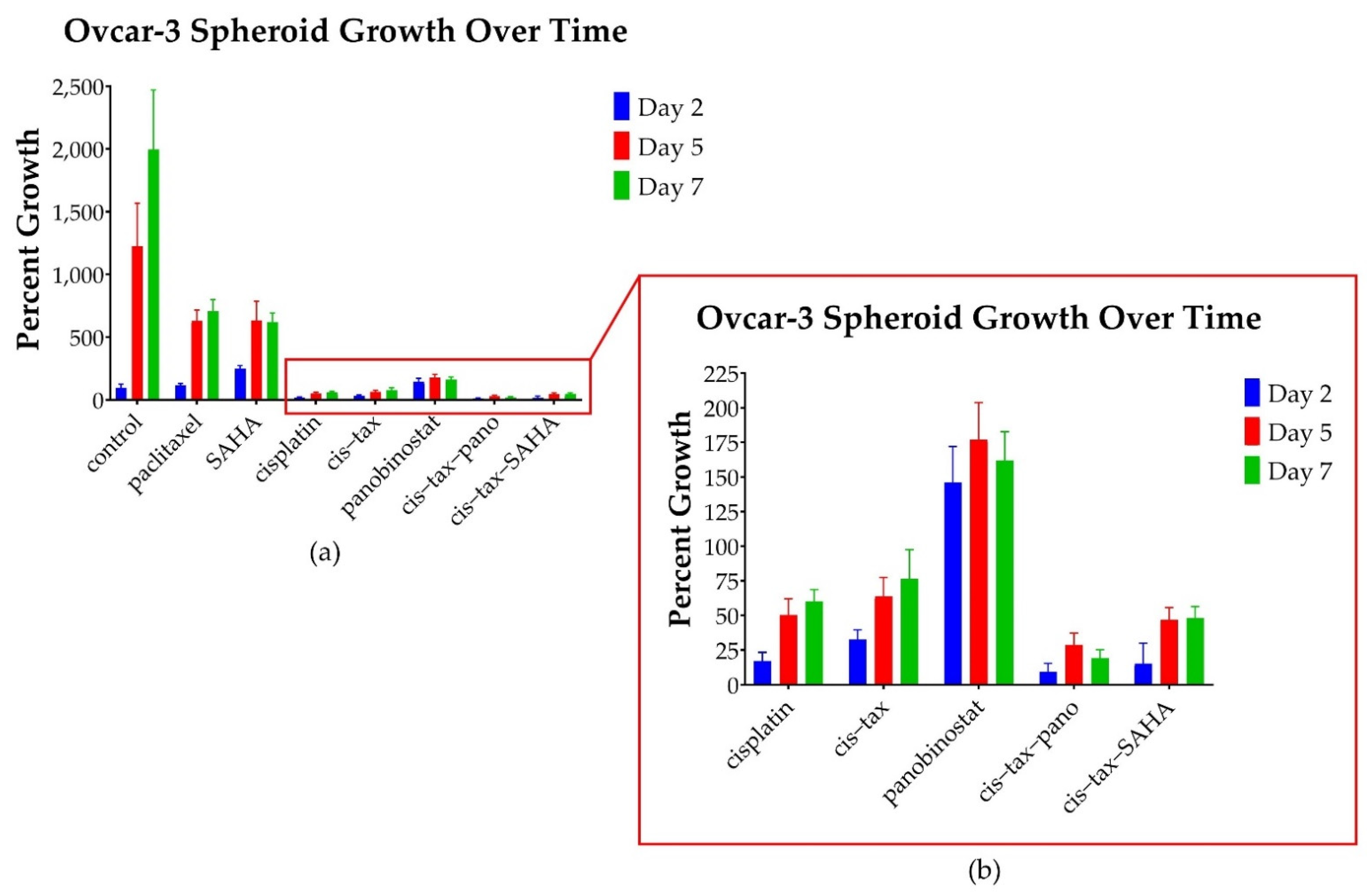
3.3. In Ovcar-3 Cells, the Combination of Classic Chemotherapy with Epigenetic Therapy Is Most Effective
3.4. Cells Observed over an Extended Kinetic in 2D Culture Demonstrate the Same Trends Seen in 3D Culture, However, the Effect Is More Pronounced in 3D as Compared to 2D
3.5. Physiologic Changes in Ovarian Cancer Cells Following Chemotherapy or Epigenetic Treatments in 3D Culture
3.5.1. Spheroid-Derived Cells Exhibit No Change in Proliferation Following Treatment with Paclitaxel; However, There Is a Decreased Proliferative Ability Noted following Treatment with Epigenetic Therapy
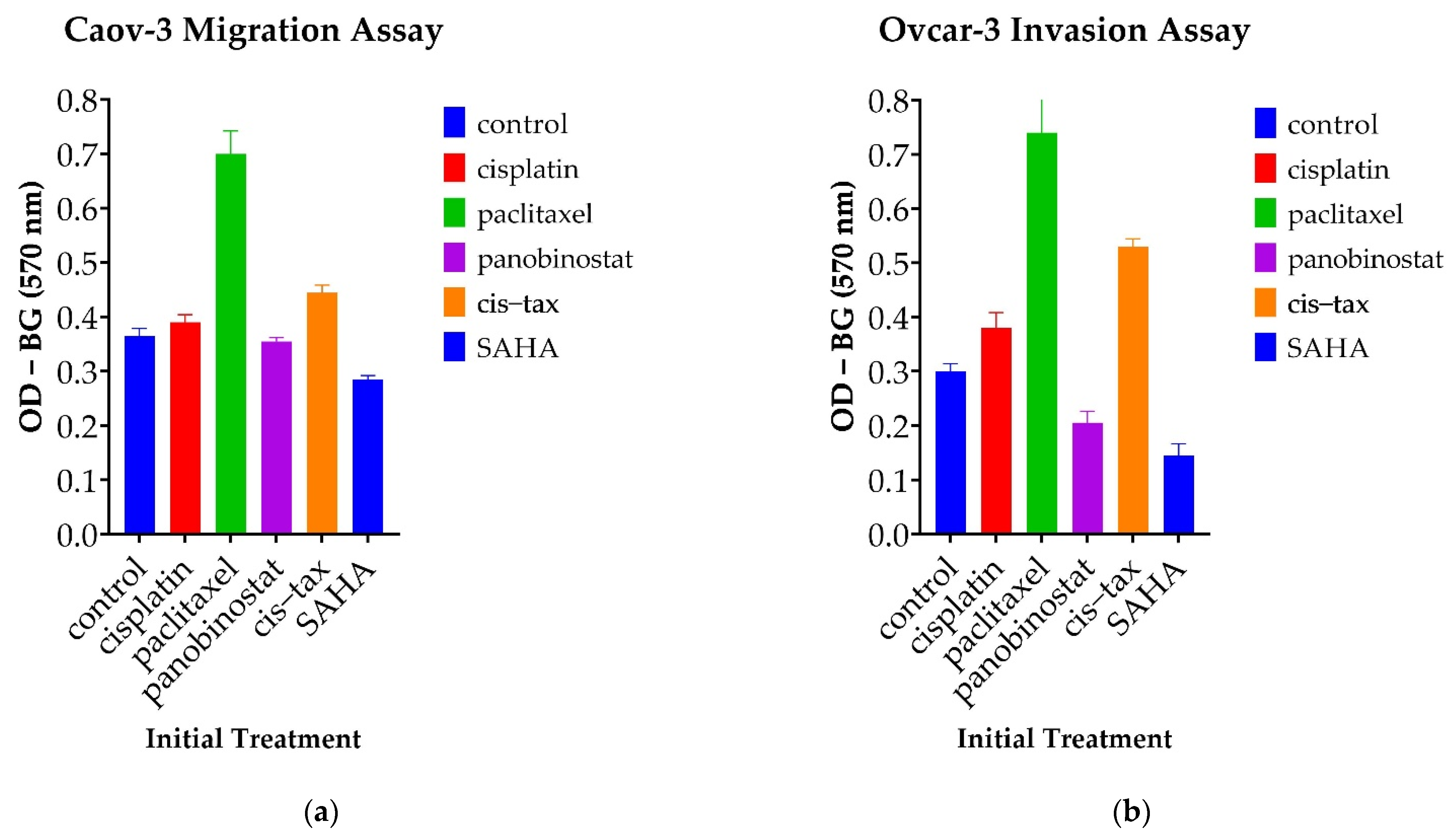
3.5.2. Cells Treated with Paclitaxel Demonstrate Increased Migration Capability, Whereas Those Treated with Epigenetic Therapy Demonstrate Decreased Invasion Capability
3.5.3. Epigenetic Therapy Reduces the Ability for Ovarian Cancer Cells to Form Colonies, Whereas Classic Chemotherapy Does Not Have This Effect
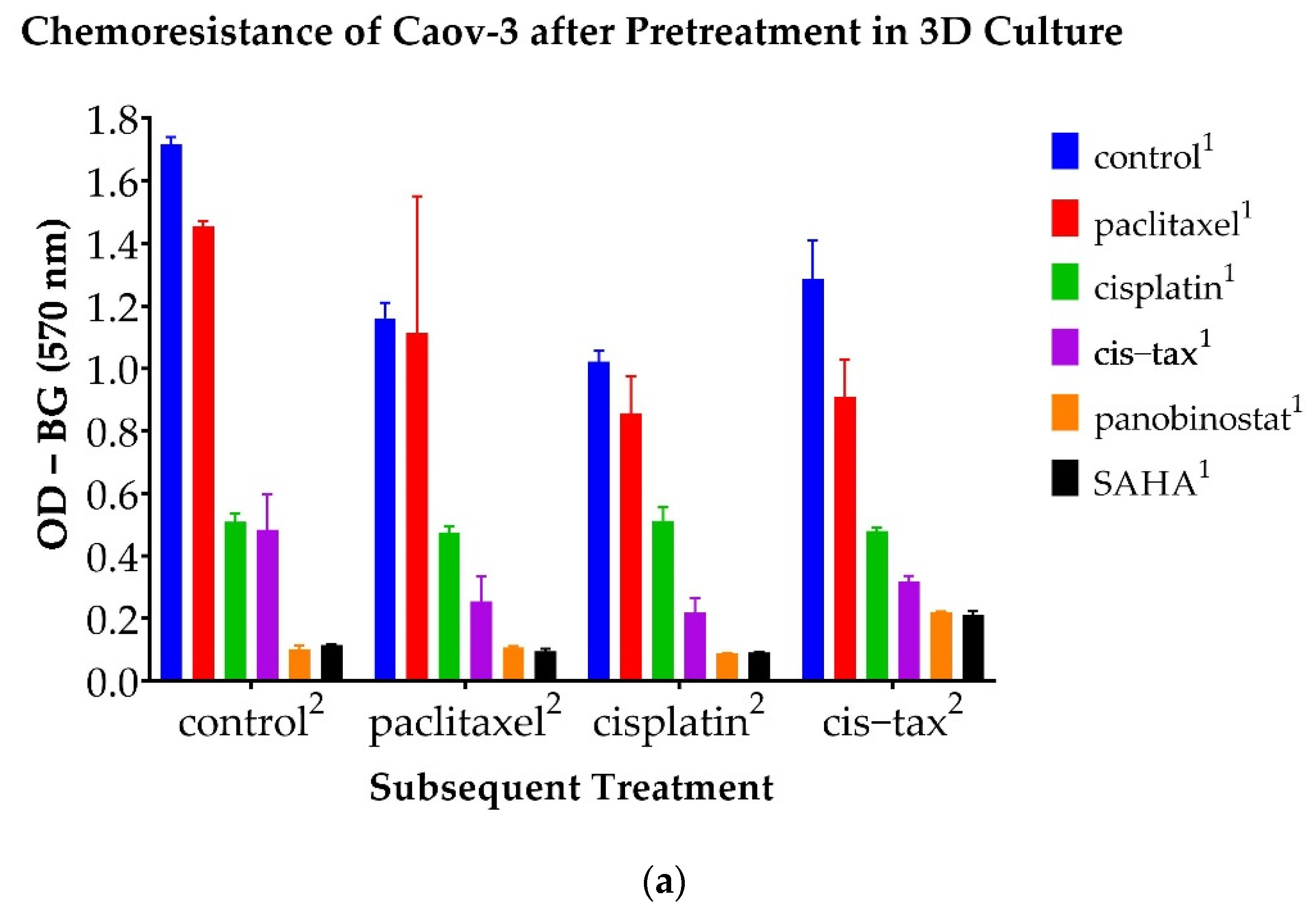
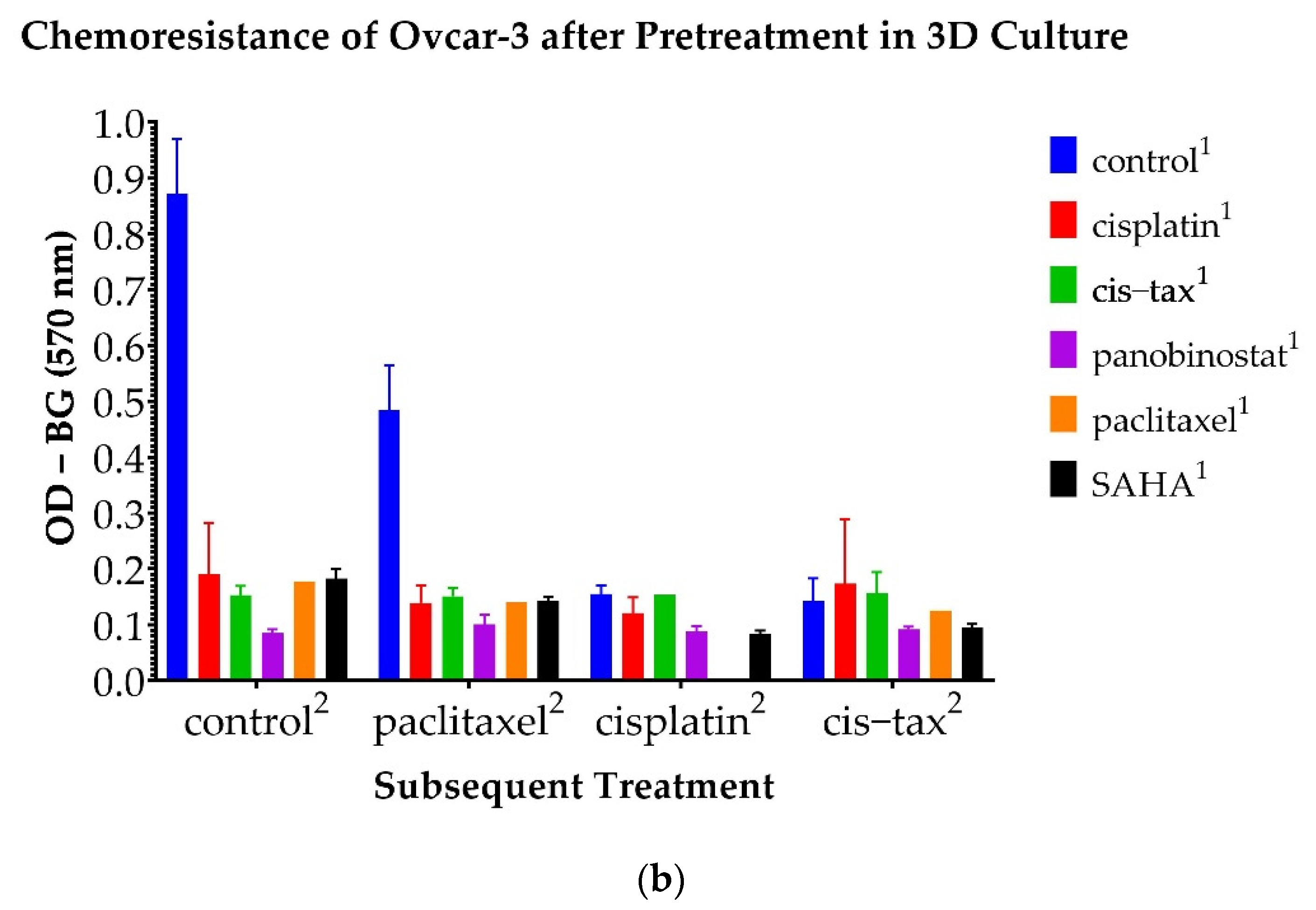
3.6. Chemoresistance Observed in 3D Ovarian Cancer Cells Is Reversible with Epigenetic Therapy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Nik, N.N.; Vang, R.; Shih, I.-M.; Kurman, R.J. Origin and pathogenesis of pelvic (ovarian, tubal, and primary peritoneal) serous carcinoma. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Sopik, V.; Iqbal, J.; Rosen, B.; Narod, S.A. Why have ovarian cancer mortality rates declined? Part II. Case-Fatal. Gynecol. Oncol. 2015, 138, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Chi, D.S.; Berchuck, A.; Dizon, D.S.; Yashar, C. Principles and Practice of Gynecologic Oncology, 7th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2017. [Google Scholar]
- Markman, M.; Rothman, R.; Hakes, T.; Reichman, B.; Hoskins, W.; Rubin, S.; Lewis, J.L., Jr. Second-line platinum therapy in patients with ovarian cancer previously treated with cisplatin. J. Clin. Oncol. 1991, 9, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Freyer, G.; Ray-Coquard, I.; Fischer, D.; Martín, A.G.; Kielhorn, A.; Chia, V.; Taylor, A. Routine clinical practice for patients with recurrent ovarian carcinoma: Results from the TROCADERO study. Int. J. Gynecol. Cancer 2016, 26, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Cooke, S.L.; Brenton, J.D. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol. 2011, 12, 1169–1174. [Google Scholar] [CrossRef]
- Coleman, R.L.; Brady, W.E.; McMeekin, D.S.; Rose, P.G.; Soper, J.T.; Lentz, S.S.; Shahin, M.S. A phase II evaluation of nanoparticle, albumin-bound (nab) paclitaxel in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: A Gynecologic Oncology Group study. Gynecol. Oncol. 2011, 122, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114. [Google Scholar]
- Lee, J.M.; Mhawech-Fauceglia, P.; Lee, N.; Parsanian, L.C.; Lin, Y.G.; Gayther, S.A.; Lawrenson, K. A three-dimensional microenvironment alters protein expression and chemosensitivity of epithelial ovarian cancer cells in vitro. Lab. Investig. 2013, 93, 528–542. [Google Scholar]
- Shield, K.; Ackland, M.L.; Ahmed, N.; Rice, G.E. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol. Oncol. 2009, 113, 143–148. [Google Scholar] [CrossRef]
- Kim, J.B. Three-dimensional tissue culture models in cancer biology. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 15, pp. 365–377. [Google Scholar]
- Wang, C.; Tang, Z.; Zhao, Y.; Yao, R.; Li, L.; Sun, W. Three-dimensional in vitro cancer models: A short review. Biofabrication 2014, 6, 022001. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Hippo, Y. Current status of patient-derived ovarian cancer models. Cell 2019, 8, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.; Eustace, A.; Busschots, S.; Breen, L.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro development of chemotherapy and targeted therapy drug-resistant cancer cell lines: A practical guide with case studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, B.; Łuczewski, Ł.; Lamperska, K.; et al. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. AMS 2018, 14, 910. [Google Scholar] [PubMed]
- Tanenbaum, L.M.; Mantzavinou, A.; Subramanyam, K.S.; del Carmen, M.G.; Cima, M.J. Ovarian cancer spheroid shrinkage following continuous exposure to cisplatin is a function of spheroid diameter. Gynecol. Oncol. 2017, 146, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-H.; Chen, Y.-D.; Huang, S.-F.; Wang, H.-M.; Wu, M.-H. The effect of primary cancer cell culture models on the results of drug chemosensitivity assays: The application of perfusion microbioreactor system as cell culture vessel. BioMed Res. Int. 2015, 2015, 470283. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.M.; Chu, C.S.; Rubin, S.C. Chemotherapy for Gynecologic Cancers: Society of Gynecologic Oncology Handbook, 3rd ed.; Society of Gynecologic Oncology: Chicago, IL, USA, 2017. [Google Scholar]
- Brouwers, E.E.; Huitema, A.D.; Beijnen, J.H.; Schellens, J.H. Long-term platinum retention after treatment with cisplatin and oxaliplatin. BMC Clin. Pharmacol. 2008, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Balch, C.; Huang, T.H.-M.; Brown, R.; Nephew, K.P. The epigenetics of ovarian cancer drug resistance and resensitization. Am. J. Obstet. Gynecol. 2004, 191, 1552–1572. [Google Scholar] [CrossRef]
- Gujar, H.; Weisenberger, D.J.; Liang, G. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes 2019, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, R.L.; Nemeth, E.; Tran, H.; Shvartsman, H.; Cass, I.; Narod, S.; Karlan, B.Y. BRCA1 promoter region hypermethylation in ovarian carcinoma: A population-based study. Cancer Res. 2000, 60, 5329–5333. [Google Scholar]
- Smith, H.J.; Straughn, J.M.; Buchsbaum, D.J.; Arend, R.C. Epigenetic therapy for the treatment of epithelial ovarian cancer: A clinical review. Gynecol. Oncol. Rep. 2017, 20, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, A.Z.; Afroze, S.H.; Osuji, G.A.; Kayani, S.Y.; Colon, N.; Pantho, A.F.; Kuehl, T.J.; Pilkinton, K.A.; Uddin, M.N. Epigenetic Modifications in Ovarian Cancer: A Review. J. Cancer Treat. Diagn. 2020, 4, doi. [Google Scholar] [CrossRef]
- Matei, D.E.; Nephew, K.P. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol. Oncol. 2010, 116, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Garrett, L.A.; Growdon, W.B.; Rueda, B.R.; Foster, R. Influence of a novel histone deacetylase inhibitor panobinostat (LBH589) on the growth of ovarian cancer. J. Ovarian Res. 2016, 9, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, C.S., 3rd; Greenberg, V.L.; DeSimone, C.P.; Modesitt, S.C.; van Nagell, J.R.; Craven, R.; Zimmer, S.G. Suberoylanilide hydroxamic acid (SAHA) potentiates paclitaxel-induced apoptosis in ovarian cancer cell lines. Gynecol. Oncol. 2010, 116, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Sookram, J.; Zheng, A.; Linden, K.M.; Morgan, A.B.; Brown, S.A.; Ostrovsky, O. Epigenetic therapy can inhibit growth of ovarian cancer cells and reverse chemoresistant properties acquired from metastatic omentum. Int. J. Gynecol. Obstet. 2019, 145, 225–232. [Google Scholar] [CrossRef]
- Sukovas, A.; Cesna, V.; Jasukaitiene, A.; Barauskas, G.; Nadisauskiene, R.J.; Dambrauskas, Z.; Paskauskas, S.; Gulbinas, A. Response of OVCAR-3 Cells to Cisplatin and Hyperthermia: Does Hyperthermia Really Matter? Anticancer Res. 2017, 37, 5011–5018. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.M.; Ho, Y.P.; Chang, W.C.; Dai, Y.C.; Chen, L.; Hung, Y.C.; Ma, W.L. Increase Paclitaxel Sensitivity to Better Suppress Serous Epithelial Ovarian Cancer via Ablating Androgen Receptor/Aryl Hydrocarbon Receptor-ABCG2 Axis. Cancers 2019, 11, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues Moita, A.J.; Bandolik, J.J.; Hansen, F.K.; Kurz, T.; Hamacher, A.; Kassack, M.U. Priming with HDAC Inhibitors Sensitizes Ovarian Cancer Cells to Treatment with Cisplatin and HSP90 Inhibitors. Int. J. Mol. Sci. 2020, 21, 8300. [Google Scholar] [CrossRef]
- Ong, P.S.; Wang, X.Q.; Lin, H.S.; Chan, S.Y.; Ho, P.C. Synergistic effects of suberoylanilide hydroxamic acid combined with cisplatin causing cell cycle arrest independent apoptosis in platinum-resistant ovarian cancer cells. Int. J. Oncol. 2012, 40, 1705–1713. [Google Scholar] [CrossRef] [Green Version]
- Paullin, T.; Powell, C.; Menzie, C.; Hill, R.; Cheng, F.; Martyniuk, C.J.; Westerheide, S.D. Spheroid growth in ovarian cancer alters transcriptome responses for stress pathways and epigenetic responses. PLoS ONE 2017, 12, e0182930. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Perzyński, A.; Salomon-Perzyńska, M.; Michalski, B.; Skrzypulec-Plinta, V. High-grade serous ovarian cancer: The clone wars. Arch. Gynecol. Obstet. 2017, 295, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Brenton, J.D. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashashati, A.; Ha, G.; Tone, A.; Ding, J.; Prentice, L.M.; Roth, A.; Shah, S.P. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J. Pathol. 2013, 231, 21–34. [Google Scholar] [CrossRef]
- al Habyan, S.; Kalos, C.; Szymborski, J.; McCaffrey, L. Multicellular detachment generates metastatic spheroids during intra-abdominal dissemination in epithelial ovarian cancer. Oncogene 2018, 37, 5127–5135. [Google Scholar] [CrossRef]
- Motohara, T.; Masuda, K.; Morotti, M.; Zheng, Y.; El-Sahhar, S.; Chong, K.Y.; Wietek, N.; Alsaadi, A.; Karaminejadranjbar, M.; Ahmed, A. An evolving story of the metastatic voyage of ovarian cancer cells: Cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene 2019, 38, 2885–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Wang, Q.; Lau, W.B.; Lau, B.; Xu, L.; Zhao, L.; Zhou, S. Tumor microenvironment: The culprit for ovarian cancer metastasis? Cancer Lett. 2016, 377, 174–182. [Google Scholar] [CrossRef]
- McKenzie, A.J.; Hicks, S.R.; Svec, K.V.; Naughton, H.; Edmunds, Z.L.; Howe, A.K. The mechanical microenvironment regulates ovarian cancer cell morphology, migration, and spheroid disaggregation. Sci. Rep. 2018, 8, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Miow, Q.H.; Tan, T.Z.; Ye, J.; Lau, J.A.; Yokomizo, T.; Thiery, J.P.; Mori, S. Epithelial–mesenchymal status renders differential responses to cisplatin in ovarian cancer. Oncogene 2015, 34, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Nephew, K.P. Ovarian Cancer Stem Cells: Role in Metastasis and Opportunity for Therapeutic Targeting. Cancers 2019, 11, 934. [Google Scholar] [CrossRef] [Green Version]
- Modesitt, S.C.; Parsons, S.J. In vitro and in vivo histone deacetylase inhibitor therapy with vorinostat and paclitaxel in ovarian cancer models: Does timing matter? Gynecol. Oncol. 2010, 119, 351–357. [Google Scholar] [CrossRef]
- Chen, M.Y.; Liao, W.S.L.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Bast, R.C., Jr. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef] [Green Version]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.; Berlin, S.; Lee, H.; Whalen, C.; Obermayer, E.; Penson, R.; Liu, J.; Campos, S.; Krasner, C.; Horowitz, N. Phase I study of combination of vorinostat, carboplatin, and gemcitabine in women with recurrent, platinum-sensitive epithelial ovarian, fallopian tube, or peritoneal cancer. Cancer Chemother. Pharmacol. 2015, 76, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Moufarrij, S.; Dandapani, M.; Arthofer, E.; Gomez, S.; Srivastava, A.; Lopez-Acevedo, M.; Chiappinelli, K.B. Epigenetic therapy for ovarian cancer: Promise and progress. Clin. Epigenetics 2019, 11, 7. [Google Scholar] [CrossRef]
- Jones, S.F.; Bendell, J.C.; Infante, J.R.; Spigel, D.R.; Thompson, D.S.; Yardley, D.A.; Greco, F.A.; Murphy, P.B.; Burris, H.A., 3rd. A phase I study of panobinostat in combination with gemcitabine in the treatment of solid tumors. Clin. Adv. Hematol. Oncol. 2011, 9, 225–230. [Google Scholar] [PubMed]
- Wilson, A.; Sarfo-Kantanka, K.; Barrack, T.; Steck, A.; Saskowski, J.; Crispens, M.; Khabele, D. Panobinostat sensitizes cyclen E high, homologous reombination-proficient ovarian cancer to olaparib. Gynecol. Oncol. 2016, 143, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Mechanism | Relevance |
|---|---|---|
| cisplatin | classic chemotherapy; platinum alkylating agent | commonly used in HGSC alone or in combination with paclitaxel |
| paclitaxel | classic chemotherapy; | commonly used in HGSC alone or in combination with cisplatin |
| taxane antimicrotubule agent | ||
| panobinostat | epigenetic therapy; histone deacetylase inhibitor | improves efficacy of the cisplatin–paclitaxel combination in preclinical models [27] |
| suberoylanilide hydroxamic acid/vorinostat | epigenetic therapy; histone deacetylase inhibitor | improves efficacy of paclitaxel combination in preclinical models [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilbao, M.; Katz, C.; Kass, S.L.; Smith, D.; Hunter, K.; Warshal, D.; Aikins, J.K.; Ostrovsky, O. Epigenetic Therapy Augments Classic Chemotherapy in Suppressing the Growth of 3D High-Grade Serous Ovarian Cancer Spheroids over an Extended Period of Time. Biomolecules 2021, 11, 1711. https://doi.org/10.3390/biom11111711
Bilbao M, Katz C, Kass SL, Smith D, Hunter K, Warshal D, Aikins JK, Ostrovsky O. Epigenetic Therapy Augments Classic Chemotherapy in Suppressing the Growth of 3D High-Grade Serous Ovarian Cancer Spheroids over an Extended Period of Time. Biomolecules. 2021; 11(11):1711. https://doi.org/10.3390/biom11111711
Chicago/Turabian StyleBilbao, Michelle, Chelsea Katz, Stephanie L. Kass, Devon Smith, Krystal Hunter, David Warshal, James K. Aikins, and Olga Ostrovsky. 2021. "Epigenetic Therapy Augments Classic Chemotherapy in Suppressing the Growth of 3D High-Grade Serous Ovarian Cancer Spheroids over an Extended Period of Time" Biomolecules 11, no. 11: 1711. https://doi.org/10.3390/biom11111711
APA StyleBilbao, M., Katz, C., Kass, S. L., Smith, D., Hunter, K., Warshal, D., Aikins, J. K., & Ostrovsky, O. (2021). Epigenetic Therapy Augments Classic Chemotherapy in Suppressing the Growth of 3D High-Grade Serous Ovarian Cancer Spheroids over an Extended Period of Time. Biomolecules, 11(11), 1711. https://doi.org/10.3390/biom11111711