
Human Parainfluenza Virus 3 Phosphoprotein Is a Tetramer and Shares Structural and Interaction Features with Ebola Phosphoprotein VP35

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construct Design and Protein Purification
2.2. Predictions Disorder, Secondary Structure, and Coiled Coil
2.3. Pulldown Assays
2.4. Circular Dichroism
2.5. ITC
2.6. Analytical Ultracentrifugation
2.7. RNA Production
2.8. Multi-Angle Light Scattering
2.9. Turbidity Assays
2.10. Sequence Alignment and Phylogenetics
3. Results
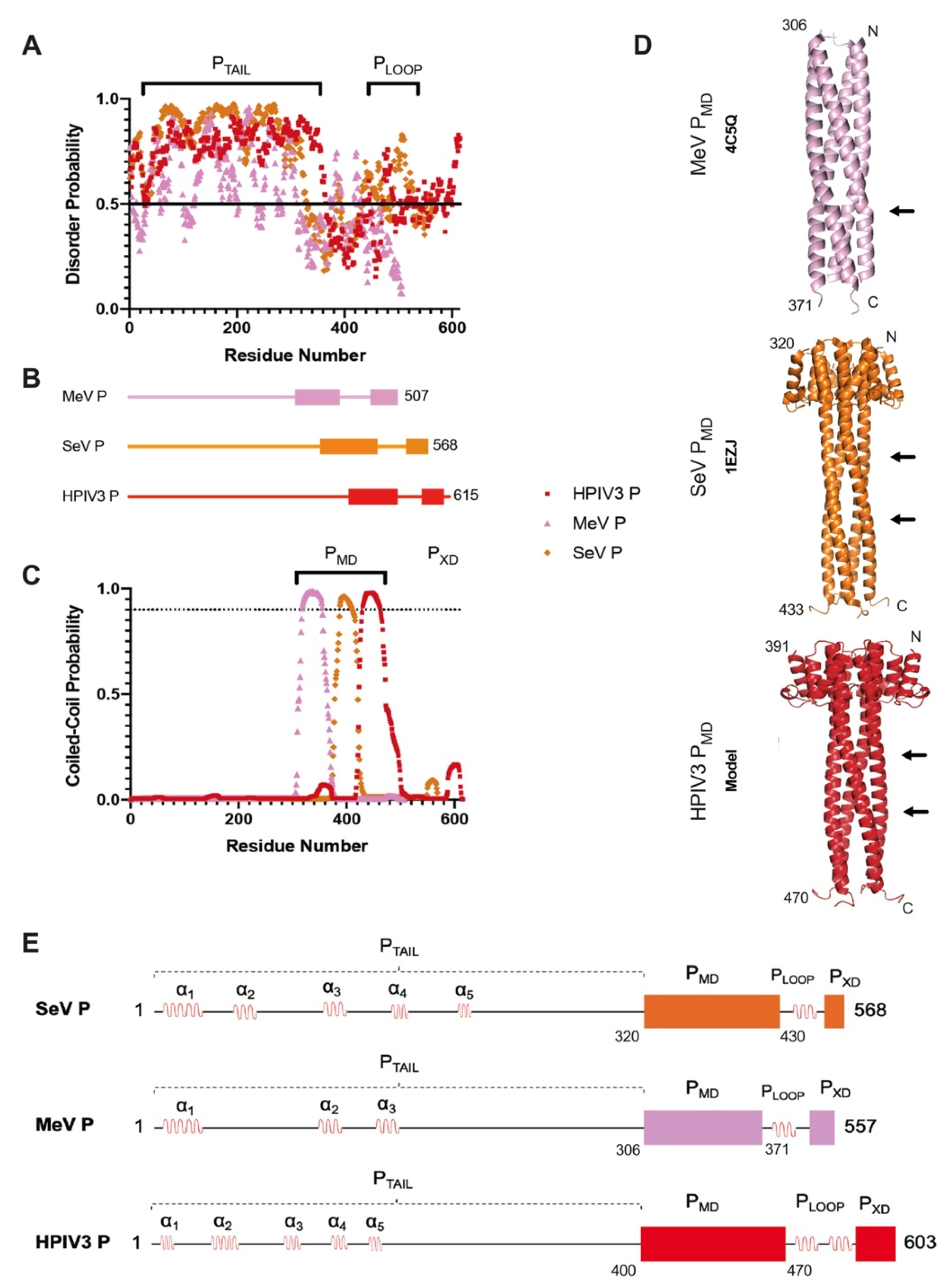
3.1. HPIV3 P Architecture Is Similar to Phosphoproteins from Other Paramyxoviruses
3.2. The HPIV3 P Multimerization Domain Is a Stable Coiled-Coil Tetramer
3.3. The PTAIL Contains an LC8 Binding Motif That Binds LC8 In Vitro
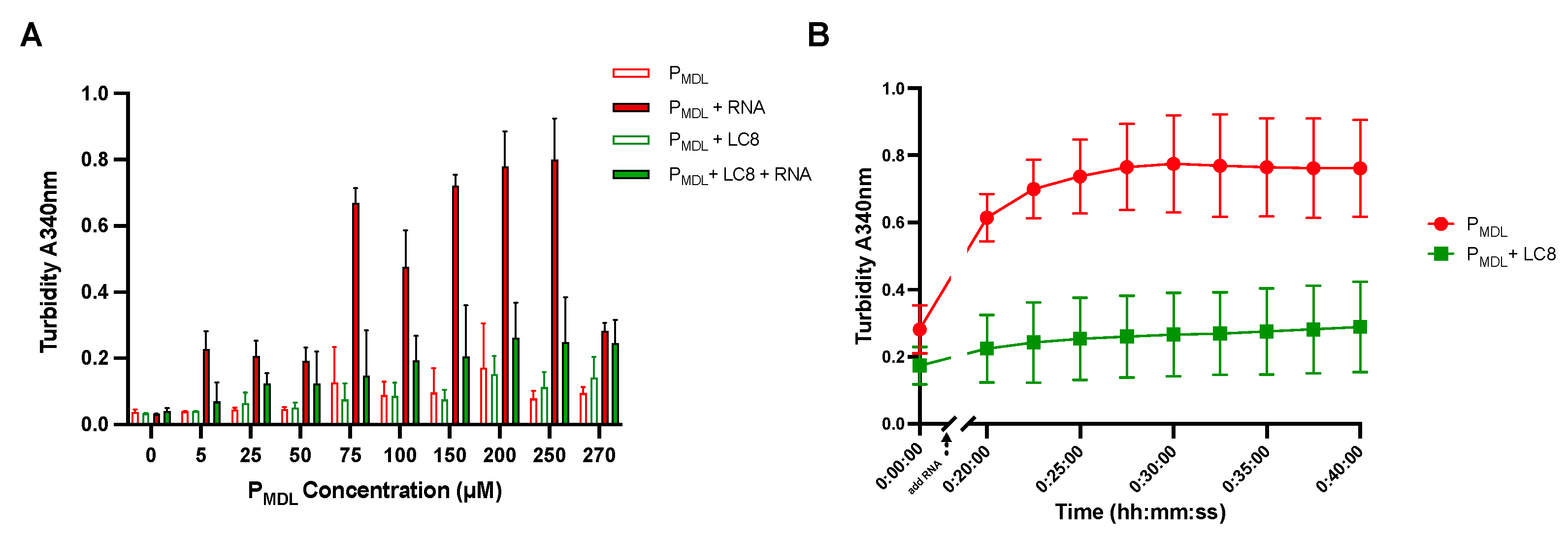
3.4. PMDL Underwent RNA-Induced Aggregation but Was Abrogated by LC8 In Vitro
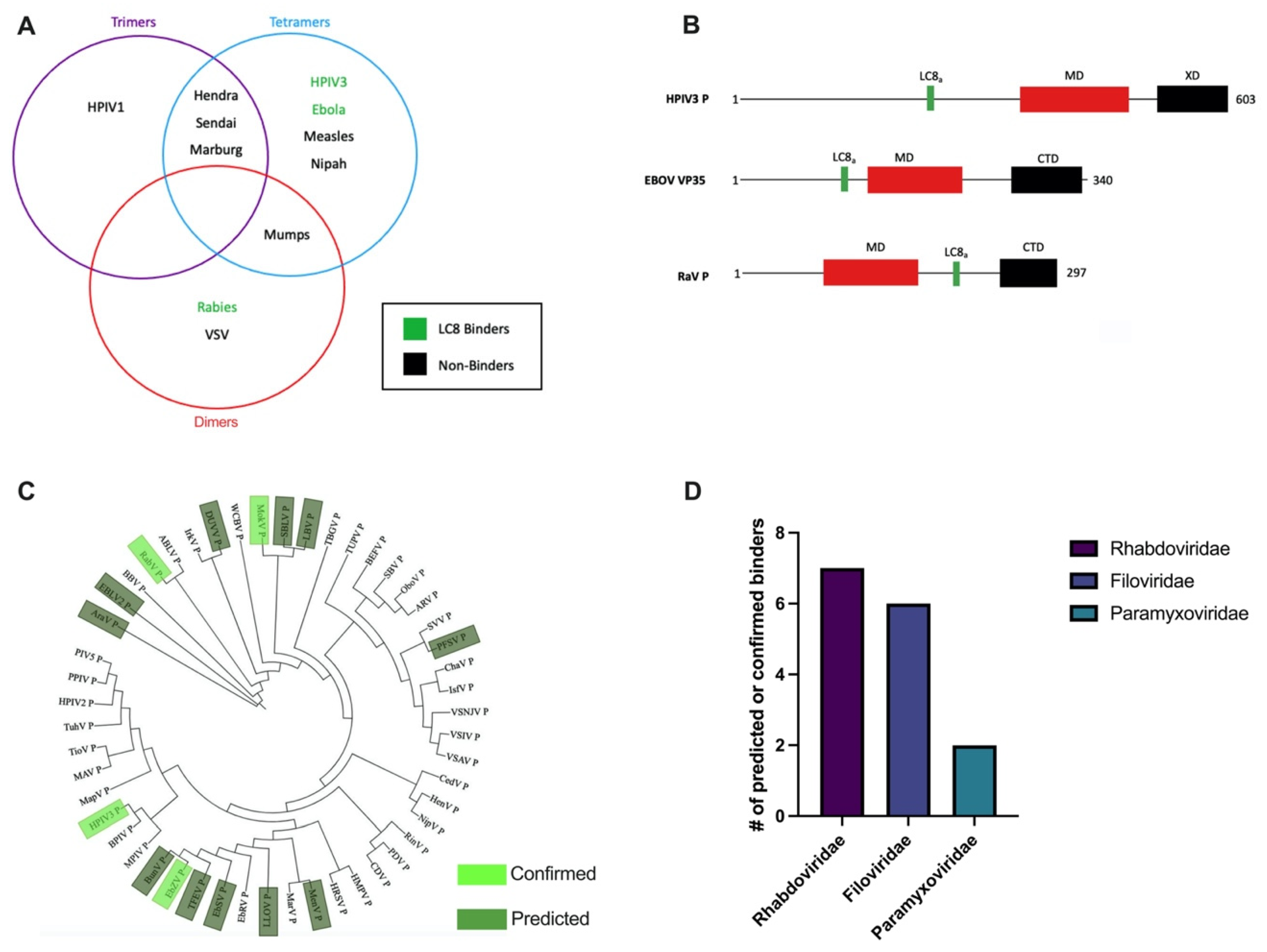
3.5. Association State of the Multimerization Domain of Ebola Zaire Phosphoprotein VP35
4. Discussion
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rhabdoviridae | VSIV | Vesicular Stomatitis Indiana Virus | Paramyxoviridae | PIV5 | Parainfluenza Virus 5 |
| VSNJV | Vesicular Stomatitis New Jersey Virus | HPIV2 | Human Parainfluenza Virus 2 | ||
| VSAV | Vesicular Stomatitis Alagoas Virus | MapV | Mapuera Virus | ||
| IsfV | Isfahan Virus | PPIV | Porcine Rubulavirus | ||
| ChaV | Chandipura Virus | MAV | Menangle Virus | ||
| PFV | Pike Fry Rhabdovirus | TioV | Tioman Virus | ||
| SVV | Spring Viraemia of Carp Virus | TuhV | Tuhoko Virus 1 | ||
| TUPV | Tupaia Virus | HenV | Hendra Virus | ||
| TBGV | Tibrogargan Virus | NipV | Nipah Virus | ||
| BEFV | Bovine Ephemeral Fever Virus | CedV | Cedar Henipavirus | ||
| OboV | Obodhiang Virus | CDV | Canine Distemper Virus | ||
| ARV | Adelaide River Virus | PDV | Phocine Distemper Virus | ||
| IrkV | Irkut Virus | RinV | Rinderpest Virus | ||
| WCBV | West Caucasian Bat | HRSV | Human Respiratory Syncytial Virus | ||
| EBLV2 | European Bat Virus 2 | HPIV3 | Human Parainfluenza Virus 3 | ||
| MokV | Mokola Virus | MPIV | Murine Parainfluenza Virus | ||
| LBV | Lagos Bat Virus | BPIV | Bovine Parainfluenza Virus | ||
| RabV | Rabies Virus | HMPV | Human Metapneumovirus | ||
| DUVV | Duvenhage Virus | Filoviridae | EbZV | Zaire Ebola Virus | |
| ABLV | Australian Bat Virus | TFEV | Tai Forest Virus | ||
| SBLV | Shimoni Virus | BunV | Bundibugyo Virus | ||
| AraV | Aravan Virus | MenV | Mengla Virus | ||
| BBV | Bokeloh Bat Virus | EbSV | Sudan Ebola Virus | ||
| SBV | Santa Barbara Virus | ResV | Reston Virus | ||
| LLOV | Lloviu Virus | ||||
| MarV | Marburg Virus |
References
- Amarasinghe, G.K.; Ayllon, M.A.; Bao, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef] [Green Version]
- Fox, T.G.; Christenson, J.C. Influenza and parainfluenza viral infections in children. Pediatr. Rev. 2014, 35, 217–227. [Google Scholar] [CrossRef]
- Maykowski, P.; Smithgall, M.; Zachariah, P.; Oberhardt, M.; Vargas, C.; Reed, C.; Demmer, R.T.; Stockwell, M.S.; Saiman, L. Seasonality and clinical impact of human parainfluenza viruses. Influenza Other Respir. Viruses 2018, 12, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Cortez, K.J.; Erdman, D.D.; Peret, T.C.; Gill, V.J.; Childs, R.; Barrett, A.J.; Bennett, J.E. Outbreak of human parainfluenza virus 3 infections in a hematopoietic stem cell transplant population. J. Infect. Dis. 2001, 184, 1093–1097. [Google Scholar] [CrossRef]
- Liang, B. Structures of the Mononegavirales Polymerases. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Henrickson, K.J. Parainfluenza viruses. Clin. Microbiol. Rev. 2003, 16, 242–264. [Google Scholar] [CrossRef] [Green Version]
- Habchi, J.; Longhi, S. Structural Disorder within Paramyxoviral Nucleoproteins and Phosphoproteins in Their Free and Bound Forms: From Predictions to Experimental Assessment. Int. J. Mol. Sci. 2015, 16, 15688–15726. [Google Scholar] [CrossRef]
- Choudhary, S.K.; Malur, A.G.; Huo, Y.; De, B.P.; Banerjee, A.K. Characterization of the oligomerization domain of the phosphoprotein of human parainfluenza virus type 3. Virology 2002, 302, 373–382. [Google Scholar] [CrossRef] [Green Version]
- De, B.P.; Gupta, S.; Banerjee, A.K. Cellular protein kinase C isoform zeta regulates human parainfluenza virus type 3 replication. Proc. Natl. Acad. Sci. USA 1995, 92, 5204–5208. [Google Scholar] [CrossRef] [Green Version]
- Huntley, C.C.; De, B.P.; Murray, N.R.; Fields, A.P.; Banerjee, A.K. Human parainfluenza virus type 3 phosphoprotein: Identification of serine 333 as the major site for PKC zeta phosphorylation. Virology 1995, 211, 561–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugai, A.; Sato, H.; Yoneda, M.; Kai, C. Phosphorylation of measles virus phosphoprotein at S86 and/or S151 downregulates viral transcriptional activity. FEBS Lett. 2012, 586, 3900–3907. [Google Scholar] [CrossRef]
- Curran, J. A role for the Sendai virus P protein trimer in RNA synthesis. J. Virol. 1998, 72, 4274–4280. [Google Scholar] [CrossRef] [Green Version]
- Curran, J.; Boeck, R.; Lin-Marq, N.; Lupas, A.; Kolakofsky, D. Paramyxovirus phosphoproteins form homotrimers as determined by an epitope dilution assay, via predicted coiled coils. Virology 1995, 214, 139–149. [Google Scholar] [CrossRef]
- Bloyet, L.M.; Schramm, A.; Lazert, C.; Raynal, B.; Hologne, M.; Walker, O.; Longhi, S.; Gerlier, D. Regulation of measles virus gene expression by P protein coiled-coil properties. Sci. Adv. 2019, 5, eaaw3702. [Google Scholar] [CrossRef] [Green Version]
- Pickar, A.; Elson, A.; Yang, Y.; Xu, P.; Luo, M.; He, B. Oligomerization of Mumps Virus Phosphoprotein. J. Virol. 2015, 89, 11002–11010. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Green, T.J.; Lu, S.; Luo, M. Crystal structure of the oligomerization domain of the phosphoprotein of vesicular stomatitis virus. J. Virol. 2006, 80, 2808–2814. [Google Scholar] [CrossRef] [Green Version]
- Zinzula, L.; Nagy, I.; Orsini, M.; Weyher-Stingl, E.; Bracher, A.; Baumeister, W. Structures of Ebola and Reston Virus VP35 Oligomerization Domains and Comparative Biophysical Characterization in All Ebolavirus Species. Structure 2019, 27, 39–54, e36. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Leser, G.P.; Kors, C.A.; Lamb, R.A. Structure of the Paramyxovirus Parainfluenza Virus 5 Nucleoprotein in Complex with an Amino-Terminal Peptide of the Phosphoprotein. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Banerjee, A.K. Phosphoprotein, P of human parainfluenza virus type 3 prevents self- association of RNA-dependent RNA polymerase, L. Virology 2009, 383, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Masters, P.S.; Banerjee, A.K. Complex formation with vesicular stomatitis virus phosphoprotein NS prevents binding of nucleocapsid protein N to nonspecific RNA. J. Virol. 1988, 62, 2658–2664. [Google Scholar] [CrossRef] [Green Version]
- Beer, B.; Kurth, R.; Bukreyev, A. Characteristics of Filoviridae: Marburg and Ebola viruses. Naturwissenschaften 1999, 86, 8–17. [Google Scholar] [CrossRef]
- Khalafallah, M.T.; Aboshady, O.A.; Moawed, S.A.; Ramadan, M.S. Ebola virus disease: Essential clinical knowledge. Avicenna J. Med. 2017, 7, 96–102. [Google Scholar] [CrossRef]
- Messaoudi, I.; Amarasinghe, G.K.; Basler, C.F. Filovirus pathogenesis and immune evasion: Insights from Ebola virus and Marburg virus. Nat. Rev. Microbiol. 2015, 13, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Luthra, P.; Jordan, D.S.; Leung, D.W.; Amarasinghe, G.K.; Basler, C.F. Ebola virus VP35 interaction with dynein LC8 regulates viral RNA synthesis. J. Virol. 2015, 89, 5148–5153. [Google Scholar] [CrossRef] [Green Version]
- Reid, S.P.; Cárdenas, W.B.; Basler, C.F. Homo-oligomerization facilitates the interferon-antagonist activity of the ebolavirus VP35 protein. Virology 2005, 341, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, N.E.; Leyrat, C.; Gerard, F.C.; Bourhis, J.M.; Blondel, D.; Jamin, M.; Barbar, E. The LC8-RavP ensemble Structure Evinces A Role for LC8 in Regulating Lyssavirus Polymerase Functionality. J. Mol. Biol. 2019, 431, 4959–4977. [Google Scholar] [CrossRef]
- Di Palma, F.; Daino, G.L.; Ramaswamy, V.K.; Corona, A.; Frau, A.; Fanunza, E.; Vargiu, A.V.; Tramontano, E.; Ruggerone, P. Relevance of Ebola virus VP35 homo-dimerization on the type I interferon cascade inhibition. Antivir. Chem. Chemother. 2019, 27, 2040206619889220. [Google Scholar] [CrossRef]
- Williams, J.C.; Roulhac, P.L.; Roy, A.G.; Vallee, R.B.; Fitzgerald, M.C.; Hendrickson, W.A. Structural and thermodynamic characterization of a cytoplasmic dynein light chain-intermediate chain complex. Proc. Natl. Acad. Sci. USA 2007, 104, 10028–10033. [Google Scholar] [CrossRef] [Green Version]
- Makokha, M.; Huang, Y.J.; Montelione, G.; Edison, A.S.; Barbar, E. The solution structure of the pH-induced monomer of dynein light-chain LC8 from Drosophila. Protein Sci. 2004, 13, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Dick, T.; Ray, K.; Salz, H.K.; Chia, W. Cytoplasmic dynein (ddlc1) mutations cause morphogenetic defects and apoptotic cell death in Drosophila melanogaster. Mol. Cell. Biol. 1996, 16, 1966–1977. [Google Scholar] [CrossRef] [Green Version]
- Barbar, E. Dynein light chain LC8 is a dimerization hub essential in diverse protein networks. Biochemistry 2008, 47, 503–508. [Google Scholar] [CrossRef]
- Rapali, P.; Szenes, A.; Radnai, L.; Bakos, A.; Pal, G.; Nyitray, L. DYNLL/LC8: A light chain subunit of the dynein motor complex and beyond. FEBS J. 2011, 278, 2980–2996. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, N.; Estelle, A.; Waugh, N.; Davey, N.E.; Blikstad, C.; Ammon, Y.C.; Akhmanova, A.; Ivarsson, Y.; Hendrix, D.A.; Barbar, E. Systematic identification of recognition motifs for the hub protein LC8. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; Nyarko, A.; Löhr, F.; Karplus, P.A.; Barbar, E. The Anchored Flexibility Model in LC8 Motif Recognition: Insights from the Chica Complex. Biochemistry 2016, 55, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Merino-Gracia, J.; Garcia-Mayoral, M.F.; Rodriguez-Crespo, I. The association of viral proteins with host cell dynein components during virus infection. FEBS J. 2011, 278, 2997–3011. [Google Scholar] [CrossRef] [Green Version]
- Jacob, Y.; Badrane, H.; Ceccaldi, P.E.; Tordo, N. Cytoplasmic dynein LC8 interacts with lyssavirus phosphoprotein. J. Virol. 2000, 74, 10217–10222. [Google Scholar] [CrossRef] [Green Version]
- Raux, H.; Flamand, A.; Blondel, D. Interaction of the rabies virus P protein with the LC8 dynein light chain. J. Virol. 2000, 74, 10212–10216. [Google Scholar] [CrossRef] [Green Version]
- Knabb, M.T.; Danielsen, C.A.; McShane-Kay, K.; Mbuy, G.K.; Woodruff, R.I. Herpes simplex virus-type 2 infectivity and agents that block gap junctional intercellular communication. Virus Res. 2007, 124, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Mebatsion, T. Extensive attenuation of rabies virus by simultaneously modifying the dynein light chain binding site in the P protein and replacing Arg333 in the G protein. J. Virol. 2001, 75, 11496–11502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergen, J.M.; Pun, S.H. Evaluation of an LC8-binding peptide for the attachment of artificial cargo to dynein. Mol. Pharm. 2007, 4, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo-distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [Green Version]
- Blocquel, D.; Habchi, J.; Durand, E.; Sevajol, M.; Ferron, F.; Erales, J.; Papageorgiou, N.; Longhi, S. Coiled-coil deformations in crystal structures: The measles virus phosphoprotein multimerization domain as an illustrative example. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1589–1603. [Google Scholar] [CrossRef]
- Tarbouriech, N.; Curran, J.; Ruigrok, R.W.; Burmeister, W.P. Tetrameric coiled coil domain of Sendai virus phosphoprotein. Nat. Struct. Biol. 2000, 7, 777–781. [Google Scholar] [CrossRef]
- Harding, S.; Rowe, A.J.; Horton, J.C. Computer-aided Interpretation of Sedimentation Data for Proteins. In Analytical Ultracentrifugation in Biochemistry and Polymer Science; Royal Society of Chemistry: London, UK, 1992; pp. 90–125. [Google Scholar]
- Schuck, P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Vaughan, T.G. IcyTree: Rapid browser-based visualization for phylogenetic trees and networks. Bioinformatics 2017, 33, 2392–2394. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30 (Suppl. S1), S162–S173. [Google Scholar] [CrossRef]
- Studer, G.; Tauriello, G.; Bienert, S.; Biasini, M.; Johner, N.; Schwede, T. ProMod3-A versatile homology modelling toolbox. PLoS Comput. Biol. 2021, 17, e1008667. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [Green Version]
- Forman-Kay, J.D.; Kriwacki, R.W.; Seydoux, G. Phase Separation in Biology and Disease. J. Mol. Biol. 2018, 430, 4603–4606. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, Y.; Tang, Q.; Yang, X.; Qin, Y.; Chen, M. Inclusion bodies of human parainfluenza virus type 3 inhibit antiviral stress granule formation by shielding viral RNAs. PLoS Pathog. 2018, 14, e1006948. [Google Scholar] [CrossRef]
- Zhang, S.; Cheng, Q.; Luo, C.; Yin, L.; Qin, Y.; Chen, M. An alanine residue in human parainfluenza virus type 3 phosphoprotein is critical for restricting excessive N(0)-P interaction and maintaining N solubility. Virology 2018, 518, 64–76. [Google Scholar] [CrossRef]
- Xie, J.; Wang, L.; Zhai, G.; Wu, D.; Lin, Z.; Wang, M.; Yan, X.; Gao, L.; Huang, X.; Fearns, R.; et al. Structural architecture of a dimeric paramyxovirus polymerase complex. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhang, S.; Jiang, Y.; Cheng, Q.; Zhong, Y.; Qin, Y.; Chen, M. Inclusion Body Fusion of Human Parainfluenza Virus Type 3 Regulated by Acetylated alpha-Tubulin Enhances Viral Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Pfaller, C.K.; Cattaneo, R.; Schnell, M.J. Reverse genetics of Mononegavirales: How they work, new vaccines, and new cancer therapeutics. Virology 2015, 479-480, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Byrappa, S.; Gupta, K.C. Human parainfluenza virus type 1 phosphoprotein is constitutively phosphorylated at Ser-120 and Ser-184. J. Gen. Virol. 1999, 80 Pt 5, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- De, B.P.; Lesoon, A.; Banerjee, A.K. Human parainfluenza virus type 3 transcription in vitro: Role of cellular actin in mRNA synthesis. J. Virol. 1991, 65, 3268–3275. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.; Plemper, R.K. The paramyxovirus polymerase complex as a target for next-generation anti-paramyxovirus therapeutics. Front. Microbiol. 2015, 6, 459. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez Galvan, J.; Donner, B.; Veseley, C.H.; Reardon, P.; Forsythe, H.M.; Howe, J.; Fujimura, G.; Barbar, E. Human Parainfluenza Virus 3 Phosphoprotein Is a Tetramer and Shares Structural and Interaction Features with Ebola Phosphoprotein VP35. Biomolecules 2021, 11, 1603. https://doi.org/10.3390/biom11111603
Rodriguez Galvan J, Donner B, Veseley CH, Reardon P, Forsythe HM, Howe J, Fujimura G, Barbar E. Human Parainfluenza Virus 3 Phosphoprotein Is a Tetramer and Shares Structural and Interaction Features with Ebola Phosphoprotein VP35. Biomolecules. 2021; 11(11):1603. https://doi.org/10.3390/biom11111603
Chicago/Turabian StyleRodriguez Galvan, Joaquin, Brianna Donner, Cat Hoang Veseley, Patrick Reardon, Heather M. Forsythe, Jesse Howe, Gretchen Fujimura, and Elisar Barbar. 2021. "Human Parainfluenza Virus 3 Phosphoprotein Is a Tetramer and Shares Structural and Interaction Features with Ebola Phosphoprotein VP35" Biomolecules 11, no. 11: 1603. https://doi.org/10.3390/biom11111603